
Abstract
Familial aggregation of hematological malignancies has been reported highlighting inherited genetic predisposition. In this study, we targeted four candidate genes: JAK2 and RUNX1 genes assuring a prominent function in hematological process and CBL and NPM1 as proto-oncogenes. Their disruption was described in several sporadic hematological malignancies. The aim of this study is to determine whether JAK2, CBL, RUNX1, and NPM1 germline genes mutations are involved in familial hematological malignancies. Using direct sequencing, we analyzed JAK2 (exons 12 and 14); CBL (exons 7, 8 and 9); NPM1 (exon 12) and the entire RUNX1 in 88 independent families belonging to Tunisian and French populations. Twenty-one sporadic acute leukemias were included in this study. We reported a heterozygous intronic c.1641 + 6 T > C JAK2 variant (rs182123615) found in two independent familial cases diagnosed with gastric lymphoma and Hodgkin lymphoma. The in silico analysis suggested a potential impact on splicing, but the functional splicing minigene reporter assay on rs182123615 variant showed no aberrant transcripts. In one sporadic acute myeloblastic leukemia, we reported an insertion 846 in. TGTT in exon 12 of NPM1 gene that may impact the normal reading frame. The rs182123615 JAK2 variant was described in several contexts including myeloproliferative neoplasms and congenital erythrocytosis and was supposed to be pathogenic. Through this current study, we established the assessment of pathogenicity of rs182123615 and we classified it rather as rare polymorphism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer results from the accumulation of several gene alterations. Approximately 5 % of all cancers correspond to the inherited form among them blood cancer. Familial aggregations of hematological malignancies were described in several studies underlying heritable germline genes mutations [1, 2]. The investigation of this inherited predisposition leads to the identification of driver gene mutations responsible for the tumorigenesis process.
Until now, only a few genetic alterations leading to familial hematological malignancies have been reported, describing mutations with low penetrance susceptibility through several association studies. In our investigation, we have underlined four genes widely described in several hematological malignancies in familial and sporadic context namely JAK2, CBL, RUNX1, and NPM1 [3].
JAK2 and RUNX1 have a prominent function in hematological process. The encoded JAK2 protein activates the transcriptional JAK-STAT signaling pathway involved in proliferation and survival of hematopoietic, immune, cardiac, and other cells [4, 5]. While RUNX1, also known as AML1 gene, acts as a key regulator of the expression of various hematopoietic genes [6, 7]. The disruption of the normal process involving both JAK2 and RUNX1 genes may lead to tumorigenesis. Indeed, JAK2 mutations may activate the JAK-STAT pathway resulting in oncogenic events observed in myelodysplastic syndrome [8], in primary acute myeloid leukemia (AML) and Philadelphia-negative myeloproliferative disorders [9, 10], majorly in 95 % of polycythemia vera and in 50 % of essential thrombocythemia and primary myelofibrosis. The most prevalent mutation is JAK2 V617F which occurs within the pseudokinase domain leading to the inhibition of kinase activity [11]. Somatic RUNX1 mutations were reported in several forms of cancers involving myelodysplastic syndrome and AML [12, 13] while germline mutations were found in autosomal familial platelet disorder predisposing to myelodysplastic syndromes/AML development [14].
Both CBL and NPM1 genes were classified as proto-oncogenes. They may promote growth factor independence and cellular proliferation leading to a tumorigenesis effect in several cell lines [15]. The CBL gene encodes for E3 ubiquitin-protein ligase involved in cell signaling and protein ubiquitination. Most mutations occur in exons 7, 8 and 9 encoding the zinc binding Ring-finger and the linker domains [16]. Heterogeneous somatic CBL mutations were reported especially in chronic myelomonocytic leukemia [15, 17], while germline mutations were identified only in juvenile myelomonocytic leukemia [18, 19].
The NPM1 or nucleophosmin gene encodes a molecular chaperone moving between the nucleus and the cytoplasm, facilitating the transport of ribosomal proteins through the nuclear membrane [20]. NPM1 mutations result in aberrant cytoplasmic dislocation of the mutant protein. This event appears to be critical for leukemogenesis [21, 22]. NPM1 mutations often occur in exon 12 but occasionally are found in other exons [23].
Most CBL and NPM1 mutations were described in myeloid malignancies including AML [24–27] and myelodysplastic syndrome [28].
The occurrence of JAK2, CBL, RUNX1, and NPM1 mutations in several hematological neoplasms incite us to hypothesize that some patients with familial hematological malignancies might harbor mutations in these genes. The aim of this study is to establish the involvement of these four genes in 98 familial cases with aggregated hematological malignancies with or without solid tumors and 21 sporadic acute leukemia cases belonging to Tunisian and French populations.
Methods
Patients
JAK2 (exons 12 and 14); CBL (exons 7, 8 and 9); NPM1 (exon 12); and the entire RUNX1 genes were analyzed in 98 patients belonging to 88 independent families: 13 Tunisian and 75 French families recruited via a French national cooperative network focusing on familial hematological malignancies and the GenHem INSERM/DGRS Franco-Tunisian project.
The hematological malignancies cases included chronic or acute, lymphoid or myeloid leukemias, Hodgkin’s or non Hogdkin’s lymphomas, and myeloproliferative or myelodysplastic syndromes.
The studied cohort consists of 80 patients belonging to 71 familial forms of hematological malignancies (at least two cases of hematological malignancies with or without solid tumors in first, second or third degree relatives); 17 patients from 17 families with aggregation of tumors including one case of hematological malignancy in first, second or third degree relatives and 1 patient who had a multiple primitive tumor with hematological malignancy but without family history.
Thirteen Tunisian sporadic acute lymphoblastic leukemia (ALL) and 8 sporadic acute myeloblastic leukemia (AML) cases were included in this study only for JAK2, CBL, and NPM1 genes investigation.
A control Tunisian population was recruited consisting of 100 healthy blood donors. Blood samples were obtained after the donors had given their informed consent.
Gene sequencing
To perform the mutational analysis, genomic DNA was extracted from peripheral blood cells obtained during complete remission according to standard protocols of treatments. Informed consent was obtained from the patients, relevant family members (healthy relatives) or their legal guardian as required by the Helsinki Declaration. Genomic DNA was extracted from whole blood with the EZ1 DNA tissue kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The exon coding region and the intron-exon junction of hot spot regions: the JAK2 exons 12, 14; CBL exons 7,8,9; NPM1 exon 12 and the entire RUNX1 gene were amplified using standard PCR methods. The primer sequences are available upon request. The amplified PCR products were column-purified and both strands were sequenced using the BigDye Terminator Cycle Sequencing Ready Reaction Kit v1.1 (Applied Biosystems- Foster City, USA) and loaded onto an ABI Prism 3500 sequencer (Applied Biosystems). The sequence chromatograms obtained were compared with the published human JAK2, NPM1, CBL and RUNX1 gene sequence using the SeqScape software program v2.5 (Applied Biosystems).
In silico analysis
To predict the effects of non-synonymous SNP (nsSNP) at amino acid levels, bioinformatic tools provided in the Alamut pack V2.5 http://www.interactive-biosoftware.com and condel http://useast.ensembl.org/tools.html were used.
The SIFT (Sorting Intolerant from Tolerant) method predicts the effect of the substituted amino acid on protein function based on sequence homology and the physical properties of amino acids. Normalized probabilities of substitutions are calculated under default settings, and probabilities ≤0.05 are taken to be deleterious. While PolyPhen-2 (Polymorphism Phenotyping v2) tool predicts the impact of amino acids substitutions on the structure and function on protein based on phylogenetic and structural information characterizing the obtained genetic variation.
Bioinformatic predictions of splicing alterations
Four different algorithms were used to predict the effect of genetic variation affecting splicing sites in any human sequence, based on the evaluation of consensus values of potential splice sites and search for branch points. The used tools are the following: Splice Site Finder-like (SSF, see http://www.interactive-biosoftware.com), MaxEntScan(MES, http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html), splice site prediction by Neural Network (NNS, http://www.fruitfly.org/seq_tools/splice.html), and Human Splicing Finder (HSF, http://www.umd.be/HSF/). SSF, MES, NNS, and HSF were interrogated simultaneously using the integrated software Alamut v.2.5 (Interactive Biosofware; http://www.interactive-biosoftware.com). No thresholds were used for this analysis.
Splicing minigene reporter assay
The splicing minigene assay previously described [29] was performed to test the effect of the variant JAK2 c.1641 + 6 T > C on splicing of JAK2 exon 12. Wild-type and mutant genomic segments were amplified by PCR from patient genomic DNA using forward primer JAK2Ex12F-BamHI: (5′-GACCGGATCCTCAAAGTTCAATGAGTTGACCCC-3′) and reverse primer JAK2Ex12R-MluI: (5′-GACCACGCGTACATCTAACACAAGGTTGGCA-3′), carrying 5′ tails with BamHI and MluI restriction sites, respectively (underlined). The PCR-amplified genomic segments encompassed the exon 12 (128 bp) of JAK2 and part of the flanking intronic 5′ and 3′ regions (170 and 163 bp, respectively). After digestion with BamHI and MluI enzymes, the PCR products were inserted into the BamHI and MluI cloning sites in the intron of pCAS2, a two-exon splicing reporter minigene vector (Fig. 4b). The insert was then sequenced to identify the wild-type and variant minigene constructs and to ensure that no extra mutations were added during amplification or cloning. The wild-type and mutant minigene constructs were then transiently transfected into HeLa cells using the FuGENE 6 transfection reagent, according to manufacturer’s instructions (Roche Applied Science). Cells were collected 24 h post-transfection, and the splicing patterns of the wild-type and mutant minigenes were analyzed by RT-PCR, electrophoresis, and sequencing of gel-purified RT-PCR products, as previously described [29].
Results
I-Genes sequencing
RUNX1 gene analysis
The entire RUNX1 gene was sequenced in 98 familial cases of aggregated hematological malignancies (Table 1). Three variants were found, the first variant c.654 C > T p.Ser218Ser was found in three Tunisian familial cases: one independent patient diagnosed with AML at the age of 18, and two second degree related patients diagnosed with Hodgkin disease and CML at the age of 17 and 35, respectively.
The second variant c.167 T > C p.Leu56Ser was found in 2 unrelated female patients belonging to French familial cases. The first patient was diagnosed with breast cancer at the age of 66, the second one was diagnosed with breast cancer at the age of 53 and myeloproliferative disorders at the age of 54.
The third variant p.Pro463Pro was identified in 2 unrelated patients belonging to French familial cases. The first one was diagnosed with myeloma at the age of 35 and the second one was diagnosed with myelodysplastic syndrome at the age of 50. No concomitant mutations were observed in JAK2, CBL, and NPM1 genes in all these patients.
CBL gene analysis
CBL exons 7-8-9 were investigated in which most mutations occur. Only one intronic variant 1095 + 19 G > T (rs2510152) was detected at homozygous and heterozygous levels with high frequency of 85 % covering several forms of sporadic and familial hematological malignancies. The in silico analysis using Splice Site Finder-Like, MaxEntScan, NNSPLICE and Human Splicing Finder (HSF, http://www.umd.be/HSF) in Alamut V2.5 (http://www.interactive-biosoftware.com), did not predict any damaging effects.
NPM1 gene analysis
The exon 12 of NPM1 gene analysis did not reveal any mutation in all familial cases analyzed. One Tunisian patient diagnosed with AML at the age of 52 without any familial history, showed the presence of a nucleotide insertion occurring in exon 12, the 846 in. TGTT (Fig. 1, Table 1). This mutation may have a damaging effect on protein function. No concomitant mutations were observed in JAK2, CBL and RUNX1 genes investigated in this patient.

846 in. TGTT in NPM1 gene (sporadic AML case)
JAK2 gene analysis
The sequencing of JAK2 exons 12 and 14 and its flanking intronic regions revealed the presence of only one heterozygous variant c.1641 + 6 T > C (Fig. 2), in two unrelated patients (Table 1) belonging to Tunisian familial cases.

c.1641 + 6 T > C JAK2 variant
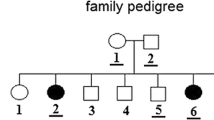
The first patient was a female diagnosed with gastric lymphoma at the age of 29 and the second one was a 32-year-old male (proband IV2, Fig. 3) diagnosed with nodular sclerosis Hodgkin lymphoma type II stage I. After being cured, he relapsed 5 years and a half later, showing stage III Bb Hodgkin lymphoma (HL), he developed bone metastases and died 1 year later. His cousin was diagnosed with HL stage IV B at the age of 36 and. After the failure of the treatment, she relapsed twice, 2 and 3 years later, and died due to the disease progress, unfortunately she was not included in this study since the DNA was not available.

Family pedigree underlined: analyzed relatives for JAK2 variant. *heterozygous form of JAK2 c.1641 + 6 T > C; **homozygous form of c. 1641 + 6 T > C; +Variant PRF1: Ala211Val
When permissions were available, we also screened the patient’s healthy relatives: 11 members (III2, IV1, IV3, IV4, IV5, IV7, IV8, V1, V2, V3 and V4) were tested for c.1641 + 6 T > C JAK2 variant (Fig. 3). It was carried by 5/11 relatives: the healthy brother, wife and son (IV1, IV3, V2) carry the c.1641 + 6 T > C JAK2 variant at heterozygous form, and his healthy son and daughter (V1, V3) carry it at homozygous form. In order to investigate the occurrence of the c.1641 + 6 T > C JAK2 variant, 198 control chromosomes recruited among consent Tunisian healthy blood donors were screened for the exon 12 JAK2 and its adjacent introns regions. We detected the variant in two control cases at homozygous form and one case at heterozygous state.
II-Evaluation of the effect of the variant JAK2 c.1641 + 6 T > C on splicing
We evaluated the effect of the variant JAK2 c.1641 + 6 T > C on the strength of the natural 5′ splice site of exon 12 as well as on the potential creation of splice site, by using four bioinformatic prediction algorithms (SSF, MES, NNS, HSF), interrogated simultaneously using the integrated software Alamut V2.3 (http://www.interactive-biosoftware.com). All 4 algorithms predicted a decrease (−7.6 % for SSF, −47.3 % for MES, −66.7 % for NNS, −2.7 % for HSF) of the strength of the natural 5′ splice site of JAK2 exon 12 (Fig. 4a). These bioinformatics predictions suggested a potential impact of the variant JAK2 c.1641 + 6 T > C on splicing.

Analysis of the effect of JAK2 c.1641 + 6 T > C on splicing. a Bioinformatics predictions of the effect on exon 12 splicing of JAK2 c.1641 + 6 T > C. Four bioinformatics prediction algorithms were interrogated simultaneously using the integrated software Alamut v2.3 (http://www.interactive-biosoftware.com). b Schematic representation of the pCAS2-JAK2-exon 12 minigene used in the functional splicing minigene reporter assay. Boxes represent exons and lines in between indicate introns. The minigene was generated by inserting the exon 12 of JAK2 and flanking intronic sequences into the intron of pCAS2. Arrows above the exons indicate the positions of primers used in RT-PCR analysis. c 2 % Agarose gel electrophoresis of RT-PCR transcript as splicing pattern of wild-type (WT) and mutant (MT) JAK2 c.1641 + 6 T > C exon 12 minigenes. The minigene carrying no insert (V) was used as control. The gel was stained with ethidium bromide
Then, we tested the effect of this variant on JAK2 exon 12 splicing by performing an ex vivo functional assay that relies on the use of a minigene vector (Fig. 4b). HeLa cells were transfected with pCAS2-JAK2-exon12-derived minigenes and the splicing pattern of the mutant minigene was compared to the wild-type by RT-PCR and sequencing analysis. As expected, the wild-type minigene produced transcripts containing JAK2 exon 12 (Fig. 4c). On the other hand, the corresponding mutant minigene generated a similar splicing pattern to the wild-type one, and no aberrant transcripts with JAK2 exon 12 skipping were detected (Fig. 4c).
Discussion
A positive family history of blood cancer is recognized as one of the most important risk factors predisposing to potential development of hematological malignancies. In this study, we targeted the JAK2, CBL, RUNX1, and NPM1 genes, their prominent function is well defined in hematological process and were described in several hematological malignancies. The aim was to search germline genes mutations involved in familial hematological malignancies. For that reason, we have screened these four genes in 98 patients belonging to 88 independent Tunisian and French families.
The RUNX1 gene alterations have been commonly described in AML and less frequently in other hematological malignancies. We reported here several variants: a germline missense substitution c.167 T > C p.Leu56Ser (rs111527738) that occurred in exon 4, was found in 2 unrelated female patients 2/98 (2 %) diagnosed with breast cancer and breast cancer/thrombocythemia respectively. According to ExAC database, this variant was found in European (Non-Finnish) population with the frequency of (1.6 %). This substitution was previously described in cytogenetically normal AML [30]. A recent study targeting a novel AML cell line with a normal karyotype (CG-SH cells) showed the presence of this variant with a frequency of 10 % of analyzed cells. The in silico analysis via PolyPhen and condel predicts a potential damaging effect while SIFT predicts a tolerated effect [31]. These three algorithms present a high sensitivity but low specificity for analyzing genetic variants, further evidence should be established to support pathogenicity of p.Leu56Ser substitution. Two silent nucleotide changes, Ser218Ser and Pro463Pro, were found in 5 familial cases. They were previously classified as polymorphism with low frequency. Despite the fact of RUNX1 gene was described in several familial contexts as AML and myelodysplastic syndrome, we do not report any mutation in our 98 familial cohort. Recent studies correlate the RUNX1 gene expression to a poor prognosis in breast cancer [32]. Since we reported the p.Leu56Ser variation in two unrelated patients diagnosed with breast cancer and a deleterious effect predicted by in silico approaches, it will be interesting to establish an association study of this variant and its prognosis impact on breast cancer.
The NPM1 gene was described as potential high penetrance gene in heredity breast cancer and myelogenous leukemia. In our investigation, we did not detect genetic variantion in all analyzed familial cases with hematological malignancies and cosegregated solid tumors especially breast cancer. However, in one patient diagnosed with sporadic AML without karyotype abnormality, we identified a C terminal frameshift NPM1 mutation: 846 in. TGTT which may impact the normal reading frame of the gene. More than 30 different 4pb insertions or insertions/deletions in NPM1 were described leading to a longer protein with a new nuclear export signal motif. These insertions/deletions may harbor the normal protein function, which appear to be critical for leukemogenesis, since NPM1 is involved in crucial cell functions and its disruption result in aberrant cytoplasmic dislocation of the mutant protein [33, 34]. In this patient, no concomitant mutations were found in CBL, JAK2, GATA2, PRF1 and P53 genes investigated (data not shown), empathizing the pathogenicity of this NPM1 insertion.
The analysis of the proto-oncogene CBL did not reveal any mutation in familial or sporadic hematological malignancies cases despite the involvement of this gene in a variety of myeloid neoplasms, including de novo AML [27]. Only one intronic CBL variant 1095 + 19 G > T (rs2510152) was detected with high frequency at homozygous and heterozygous state. It was classified as polymorphism found in several populations at a frequency of 10 to 50 % according to NCBI database. The Human Splicing Finder software analysis showed no impact of this variant on the splice site. Several proto-oncogenes were reported in familial cancer context, the absence of CBL mutation in 98 familial cases of hematological malignancies may suppose its non involvement.
As the V617F mutation in JAK2 gene was the most recurrent mutation described in several cancers, we did not detect it in 98 hematological malignancies included in our investigation. We reported an intronic JAK2 variant c.1641 + 6 T > C found in two Tunisian independent familial cases diagnosed with gastric lymphoma and Hodgkin lymphoma. This variant (rs182123615) was previously described in several pathologies including myeloproliferative neoplasms [35] and congenital erythrocytosis [36]. No functional assay to establish the assessment of pathogenicity of this variant has been reported until now.
Because of the degenerative nature of the splice sites, intronic variants outside conserved GT/AG positions at the 5′ and the 3′ intron boundaries of the acceptor and donor splice sites have been usually classified as variants of unknown significance, unless there is some functional evidence of their pathogenicity. In the clinical setting, alterations involving regulatory signals may lead to a pathogenic effect. In this study we investigated the intronic variant c.1641 + 6 T > C (rs182123615) to determine the functional impact on splicing. A familial segregation analysis of this JAK2 variant was enrolled in relatives of patient diagnosed with Hodgkin lymphoma and carrying this identified variant. Among 11 healthy relatives, 3/11 carried c.1641 + 6 T > C at heterozygous form and 2/11 at homozygous form. In a previous study, we have reported in the same Tunisian family a segregation of perforine PRF1 Ala211Val missense substitution carried by the proband (IV-2) diagnosed with Hodgkin Lymphoma, his cousin (IV-6) and 3 healthy relatives (III-2, IV-1, V-2). We have shown that the functional assay on Ala211Val variant has no impact on lytic function of perforin [37]. The absence of functional implication of the PRF1 and JAK2 variants may not avert suspicion of their implication as low risk factors in hematological malignancies. In proband IV2 carrying the JAK2 variant, we have also sequenced ASXL1, NPM1, CBL, IDH1, IDH2, YY1, CASP8, CASP10, FAS, FASL, CEBPA, AML1, CRAF, BRAF, TP53, ARTLS1, and GATA2 genes and also in several familial cases including in this study to check potential concomitant mutations. No mutations were found in these candidate genes (data not shown).
The in silico analysis of JAK2 variant was in favor of a deleterious splicing effect. The MaxEntScan predicted a 47 % decrease of the score of natural splice sites which may affect consensus splice sites and leads to a possible pathogenic effect. Despite in silico prediction, the functional splicing minigene reporter assay did not reveal any effect on splicing domain. Hence, the minigene constructs and the used cell line did not show any effect on mRNA processing. The presence of c.1641 + 6 T > C JAK2 variant in healthy probands and controls with a frequency of 1.5 % may suppose that it is not pathogenic and can be classified as rather a rare polymorphism. The in silico prediction does not reflect the in vivo situation. This may be explained by the degenerate nature of sequences which bind the splice sites. That is why Alamut classified the rs182123615 as a deleterious variant.
We aimed through this study to identify genetic inherited factors contributing in the background of familial hematological malignancies through molecular screening of four candidate genes widely evoked in several familial cancers and hematological malignancies. The investigation of RUNX1, CBL, and NPM1 genes does not revealed potential driver mutation leading to oncogenic processes, except the c.1641 + 6 T > C JAK2 variation found in Tunisian familial cases. This variant was reported in several studies and was described as potentially deleterious. Through this study, we establish its clinical assessment and we classified as rather a polymorphism than as a mutation. Despite the few genetic variations found in familial aggregation of hematological malignancies, they cannot be used alone as prognostic factors, they might contribute to the background of genetic factors which have to be revealed. Identification of a pathogenic mutation in clinically affected individuals allows to perform genetic counseling specially in familial context disease. We expect to enlarge our investigation in other genes reported in hematological malignancies such as IDH1, IDH2, and ASXL1.
References
Segel GB, Lichtman MA (2004) Familial (inherited) leukemia, lymphoma, and myeloma: an overview. Blood Cells Mol Dis 32(1):246–261
Goldin LR, Landgren O, McMaster ML, Gridley G, Hemminki K, Li X et al (2005) Familial aggregation and heterogeneity of non-Hodgkin lymphoma in population-based samples. Cancer Epidemiol Biomarkers Prev 14(10):2402–2408
Langabeer SE, Haslam K, Linders J, Percy MJ, Conneally E, Hayat A et al (2014) Molecular heterogeneity of familial myeloproliferative neoplasms revealed by analysis of the commonly acquiredJAK2, CALR and MPL mutations. Fam Cancer 13(4):659–663
Ghoreschi K, Laurence A, O’Shea JJ (2009) Janus kinases in immune cell signaling. Immunol Rev 228(1):273–287
Lv J, Wang X, Liu SY, Liang PF, Feng M, Zhang LL, Xu AP (2015) Protective effect of Fenofibrate in renal ischemia reperfusion injury: involved in suppressing kinase 2 (JAK2)/transcription 3 (STAT3)/p53 signaling activation. Pathol Biol 63(6):236–242
Ito Y (2004) Oncogenic potential of the RUNX gene family: ‘overview’. Oncogene 23(24):4198–4208
Kurokawa M (2006) AML1/Runx1 as a versatile regulator of hematopoiesis: regulation of its function and a role in adult hematopoiesis. Int J Hematol 84(2):136–142
Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL et al (2005) The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood 106(4):1207–1216
Berger R (2006) A recurrent mutation of the JAK2 gene in chronic myeloproliferative disorders. Pathol Biol 54(4):182–186
Inami M, Yamaguchi H, Hasegawa S, Mitamura Y, Kosaka F, Kobayashi A et al (2008) Analysis of the exon 12 and 14 mutations of the JAK2 gene in Philadelphia chromosome-positive leukemia. Leukemia 22(1):216
Morgan KJ, Gilliland DG (2008) A role for JAK2 mutations in myeloproliferative diseases. Annu Rev Med 59:213–222
Preudhomme C, Warot-Loze D, Roumier C, Grardel-Duflos N, Garand R, Lai JL et al (2000) High incidence of biallelic point mutations in the Runt domain of the AML1/PEBP2 alpha B gene in Mo acute myeloid leukemia and in myeloid malignancies with acquired trisomy 21. Blood 96(8):2862–2871
Harada H, Harada Y, Niimi H, Kyo T, Kimura A, Inaba T (2004) High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood 103(6):2316–2324
Owen CJ, Toze CL, Koochin A, Forrest DL, Smith CA, Stevens JM et al (2008) Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood 112(12):4639–4645
Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S et al (2009) Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 460:904–912
Swaminathan G, Tsygankov AY (2006) The Cbl family proteins: ring leaders in regulation of cell signaling. J Cell Physiol 209(1):21–43
Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, Szpurka H et al (2008) 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygousmutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res 68(24):10349–10357
Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S et al (2009) Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114(9):1859–1863
Pérez B, Mechinaud F, Galambrun C, Ben Romdhane N, Isidor B, Philip N et al (2010) Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet 47(10):686–691
Cordell JL, Pulford KA, Bigerna B, Roncador G, Banham A, Colombo E et al (1999) Detection of normal and chimeric nucleophosmin in human cells. Blood 93(2):632–642
Grisendi S, Mecucci C, Falini B, Pandolfi PP (2006) Nucleophosmin and cancer. Nat Rev Cancer 6(7):493–505
Bolli N, De Marco MF, Martelli MP, Bigerna B, Pucciarini A, Rossi R et al (2009) A dose-dependent tug of war involving the NPM1 leukaemic mutant, nucleophosmin, and ARF. Leukemia 23(3):501–509
Albiero E, Madeo D, Bolli N, Giaretta I, Bona ED, Martelli MF et al (2007) Identification and functional characterization of a cytoplasmic nucleophosmin leukaemic mutant generated by a novel exon-11 NPM1 mutation. Leukemia 21(5):1099–1103
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L et al (2005) Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 352(3):254–266
Sargin B, Choudhary C, Crosetto N, Schmidt MH, Grundler R, Rensinghoff M et al (2007) lt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood 110(3):1004–1012
Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, Arnoczky KJ et al (2007) Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood 110(3):1022–1026
Rau R, Brown P (2009) Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: towards definition of a new leukaemia entity. Hematol Oncol 27(4):171–181
Bains A, Luthra R, Medeiros LJ, Zuo Z (2011) FLT3 and NPM1 mutations in myelodysplastic syndromes: frequency and potential value for predicting progression to acute myeloid leukemia. Am J Clin Pathol 135(1):62–71
Gaildrat P, Killian A, Martins A, Tournier I, Frébourg T, Tosi M (2010) Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol Biol 653:249–257
Greif PA, Konstandin NP, Metzeler KH, Herold T, Pasalic Z, Ksienzyk B et al (2012) RUNX1 mutations in cytogenetically normal acute myeloid leukemia are associated with a Poor prognosis and upregulation of lymphoid genes. Haematologica 97(12):1909–1915
Gosse G, Celton M, Lamontagne V, Forest A, Wilhelm BT (2015) Whole genome and transcriptome analysis of a novel AML cell line with a normal karyotype. Leuk Res 39(7):709–718
Browne G, Taipaleenmäki H, Bishop NM, Madasu SC, Shaw LM, van Wijnen AJ, Stein JL, Stein GS, Lian JB (2015) Runx1 is associated with breast cancer progression in MMTV-PyMT transgenic mice and its depletion in vitro inhibits migration and invasion. J Cell Physiol 230(10):2522–32
Thiede C, Creutzig E, Reinhardt D, Ehninger G, Creutzig U (2007) Different types of NPM1 mutations in children and adults: evidence for an effect of patient age on the prevalence of the TCTG-tandem duplication in NPM1-exon 12. Leukemia 21(2):366–373
Marcinkowska-Swojak M, Handschuh L, Wojciechowski P, Goralski M, Tomaszewski K, Kazmierczak M et al (2016) Simultaneous detection of mutations and copy number variation of NPM1 in the acute myeloid leukemia using multiplex ligation-dependent probe amplification. Mutat Res 4:14–26
dos Santos MT, Mitne-Neto M, Miyashiro K, Chauffaille Mde L, Rizzatti EG (2014) Molecular genetic tests for JAK2V617F, Exon12_JAK2 and MPLW515K/L are highly informative in the evaluationof patients suspected to have BCR-ABL1-negative myeloproliferative neoplasms. J Clin Pathol 67(2):176–184
Bento C, Almeida H, Maia TM, Relvas L, Oliveira AC, Rossi C et al (2013) Molecular study of congenital erythrocytosis in 70 unrelated patients revealed a potential causal mutation in less than half of the cases (Where is/are the missing gene(s)?). Eur J Haematol 91(4):361–370
El Abed R, Bourdon V, Voskoboinik I, Omri H, Youssef YB, Laatiri MA et al (2011) Molecular study of the perforin gene in familial hematological malignancies. Hered Cancer Clin Pract 9(1):9
Acknowledgments
This work was supported by la Société Française d’Hématologie, le groupe Génétique et Cancer and Institut National du Cancer (INCa) and the Ministère de l’Enseignement Supérieur et de la Recherche Scientifique Tunisie. It is a part of the GenHem INSERM/DGRS project. We are grateful for the English correction and assistance provided by Mouna Bouali.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Hamadou, W.S., Bourdon, V., Gaildrat, P. et al. Mutational analysis of JAK2, CBL, RUNX1, and NPM1 genes in familial aggregation of hematological malignancies. Ann Hematol 95, 1043–1050 (2016). https://doi.org/10.1007/s00277-016-2678-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-016-2678-y