
Abstract
Nefedovite, Na5Ca4(PO4)4F, has been investigated by in situ high-temperature powder (30–690 °C) and single crystal (27–827 °C) X-ray diffraction and Raman spectroscopy. Nefedovite is tetragonal, space group I-4, a = 11.6560(2), c = 5.4062(2) Å, V = 734.50(2) Å3 (R1 = 0.0149). Nefedovite is a 1D antiperovskite, since its crystal structure contains chains of corner-sharing anion-centered [FCa4Na2]9+ octahedra. The chains are parallel to the c direction. Nefedovite is stable up to 727 °C and undergoes a displacive phase transition in the temperature range 277–327 °C. With increasing temperature, the PO4 tetrahedra in the crystal structure of nefedovite gradually rotate around the imaginary fourfold inversion axes aligning the O2…O3 edge parallel to [110], which ultimately leads to the appearance of the mirror plane perpendicular to the c direction and the change of space group from I-4 (82) to I4/m (87). The crystal structure of nefedovite expands strongly anisotropically with the direction of the maximum thermal expansion oriented perpendicular to the chains of anion-centered octahedra. The information-based structural complexity analysis demonstrates that both low- and high-temperature modifications of nefedovite are structurally simple with the IG,total value less than 100 bits per unit cell. The structural complexity decreases along the phase transition, which is typical for displacive phase transitions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nefedovite, Na5Ca4(PO4)F, is a fairly typical mineral of ultra-agpaite pegmatites and hydrothermalites of the Khibiny alkaline massif (Kola Peninsula, Russia) along with other phosphates, such as apatite, Ca5(PO4)3F, and nacaphite, Na2CaPO4F. Initially, the mineral was reported by Khomyakov A.P. et al. (1983) in the pegmatites veins in urtite of the mine dumps at Mt. Yuksporr, and in the core of a drill hole in the Kuniok River valley. In the case of Mt. Yuksporr, nefedovite is associated with coarse-grained orthoclase, nepheline, aegirine, eudialyte, titanite, lamprophyllite, (SrNa)Ti2Na3Ti(Si2O7)2O2(OH)2, and djerfisherite, K6(Fe,Cu,Ni)25S26Cl. Powder-like aggregates of nefedovite replacing pale green apatite occur as poikilitic phenocrysts in the rocks along with nacaphite.
In the Kuniok River valley, nefedovite could be found in aegirine-orthoclase veinlets as colorless transparent isometric grains up to 0.5 mm in size. Nefedovite was also found associated with natrite, Na2CO3, and other alkaline minerals such as silicates and sulfides including pectolite, NaCa2Si3O8(OH), lobanovite, K2Na(Fe2+4Mg2Na)Ti2(Si4O12)2O2(OH)4, tinaksite, K2NaCa2TiSi7O18(OH)O, shcherbakovite, K2NaTi2O(OH)Si4O12, rasvumite, KFe2S3, etc. Powder-like nacaphite often replaces nefedovite. Undersaturation of the carbonate-phosphate solution with fluorine leads to the formation of nefedovite, whereas the supersaturation results in the crystallization of nacaphite.
In addition to its occurrence in nature, synthetic Eu2+- and Cu+-doped analogs of nefedovite, Na5Ca4(PO4)F, have been investigated by a number of authors for their interesting luminescence (Xiao et al. 2012; Yerpude et al. 2018). The Eu-doped synthetic nefedovite has been considered as a potential material for white light-emitting diodes (LEDs) and its Cu-doped analogue is of interest as a blue-emitting phosphor. The high thermal stability of synthetic nefedovite is an advantage from the viewpoint of its possible applications.
On the basis of the powder X-ray diffraction studies, Khomyakov et al. (1983) suggested that nefedovite is triclinic with marked pseudo-tetragonal cell. The crystal structure of nefedovite was solved and refined by Sebais (1984). The structural models for refinement were constructed on the basis of related compounds with the general formula Na5-xREE4[TO4]4A1-x, where T = Si, Ge, P and A = OH, F. Three synthetic silicates with rare-earth metals were selected as the most suitable candidates: NaSmSiO4 (space group I4/m), Na5Y4(SiO4)F (space group I-4) and [NaGdSiO4]4·xNaOH (space group I-4). Although the authors also noted small triclinic deviations in the dataset, the structure was determined to be tetragonal with space group I-4 and the unit cell parameters a = 11.644, c = 5.396 Å, V = 731.60 Å3 (R1 = 0.073). The slightly modified [NaGdSiO4]4·0.2NaOH crystal structure was used as a starting model.
This research work is a continuation of our previous investigations devoted to the thermal evolution of natural and synthetic antiperovskites, i.e. the compounds based on anion-centered octahedra (Avdontceva et al. 2015a, b, 2023a, b). Antiperovskites, the inverse analogues of perovskites with anions and cations interchanged, are extensively studied due to their attractive physical properties such as ion conductivity, energy storage, superconductivity, giant magnetoresistance, luminescence, negative thermal expansion, etc. (Sullivan et al. 2015; Li et al. 2011; Oudah et al. 2016; Clarke et al. 2021; Dawson et al. 2021).
Herein, we present and discuss the results of in situ powder high-temperature X-ray diffraction (HTXRD) and single-crystal X-ray diffraction (SCXRD) studies of nefedovite over a wide temperature range. The phase transition and the crystal structure of the resulting high-temperature modification are described for the first time. This makes a contribution to the understanding of the thermal behavior of compounds with anion-centered octahedra. In addition, we report on the Raman spectra and the structural complexity calculation, as well as the theoretical analysis of potential sodium-ion migration in nefedovite at room-temperature conditions.
Materials and methods
Sample
The sample of nefedovite from the collection of the last author originates from a microcline-pectolite-sodalite-aegirine vein in urtite at Mt. Koashva (Khibiny, Kola peninsula, Russia). The mineral occurs there as transparent, colorless isometric crystals up to 2 mm in diameter.
Raman spectroscopy
The Raman spectra of nefedovite were obtained by means of a Horiba Jobin–Yvon LabRam HR 800 system in the range 70–4000 cm−1 using a solid-state laser with λ = 532 nm. The spectrometer was calibrated against a silicon standard, the spectral resolution was 2 cm−1 The data were accumulated from 2 to 10 s and the 50 × objective was employed for the measurements that were performed at room temperature with the sample oriented randomly. Further visualization and analysis of the spectra were done by means of the OriginPro 2018 SR1 b9.5.1.195 software.
Chemical composition
The chemical composition of nefedovite was studied using a Hitachi S-3400N scanning electron microscope equipped with an Oxford Instruments Energy Dispersive Spectrometer X-Max 20 (20 kV/1.7 nA, the working distance is 10 mm). The spectra were recorded automatically using the AzTec Energy software package with the TruQ technique. The following standards were applied: CaSiO3 for Ca, GaP for P, albite for Na, BaF2 for F.
In situ powder and single crystal high-temperature X-ray diffraction
The thermal behavior of nefedovite was investigated by two complementary methods. First it was studied in air by powder high-temperature X-ray diffraction (HTXRD) using a Rigaku Ultima IV diffractometer with a high temperature attachment (40 kV/30 mA, CoKα1 + 2 radiation, Bragg–Brentano geometry and PSD D/Tex Ultra detector). A powder sample was deposited on a Pt holder (20 x12x2 mm3) and fixed using a hexane suspension. The temperature measuring range was 30–690 °C with 30 °C steps. The unit-cell parameters for all temperatures were refined by the Rietveld method using Topas 4.2 (Bruker AXS 2009). The background was modeled by a Chebyshev polynomial approximation of the 12th order. No significant changes were detected in the diffraction patterns analysis at all temperatures. X-ray diffraction patterns at different temperatures for nefedovite are shown in Fig. 7 in the Supplementary materials.
A suitable single crystal of nefedovite was selected and placed into a quartz capillary with a 10 micron wall thickness and studied by the in situ single crystal X-ray diffraction (SCXRD) analysis using a Rigaku Xtalab Synergy-S diffractometer (MoKα radiation, 50 kV/1.0 mA, high-speed direct-action detector (HyPiX-6000HE)) in the temperature range 27–827 °C with 50 °C steps. The hemispheres of diffraction data (frame width 0.5°, counting time 0.8 s) were collected for all temperatures using the same strategy. The orientation of the crystal did not change during all the measurements. The CrysAlisPro software was used for the data processing (Rigaku Oxford Diffraction 2015). An absorption correction was applied using the SCALE3 ABSPACK algorithm. The crystal structure of nefedovite was refined for all temperatures using the SHELX program package included in the OLEX2 shell (Sheldrick 2015; Dolomatov, 2009). It was observed that in the temperature range 277–327 °C the crystal structure of nefedovite undergoes a reversible phase transition from the non-centrosymmetric space group I-4 to the centrosymmetric space group I4/m. The crystal structure of the high-temperature (HT) modification of nefedovite at 327 °C was initially refined in the I-4 space group. The ADDSYM program within the PLATON software package (Spek 2003) was used for the checking of missing symmetry operations, which led to the determination of the centrosymmetric I4/m space group. The similar missing symmetry checks up to 327 °C did not show any deviations from the I-4 space group. Since the systematic absences for the I-4 and I4/m space groups are identical, the inspection of the reciprocal space does not allow to monitor the transition critical point, which can be established on the basis of structural data only. It is well-probable that the exact temperature of the phase transition can be determined from the measurement of other physical properties associated with the absence of an inversion center in the symmetry group of low-temperature (LT) modification.
Nefedovite is stable up to 727 °C and loses its crystallinity completely in the temperature range 727–827 °C. The phase transition mechanism is discussed in detail below.
The crystal structure data and refinement parameters for both modifications are shown in Table 1, the atomic coordinates and equivalent isotropic displacement parameters (Å2) for the LT and HT modifications are given in Tables 2, 3, respectively. Tables 4, 5 provide selected bonds lengths for both modifications. The data for the HT modification are given for the temperature of 427 °C.
The main thermal expansion coefficients were calculated and the figures of thermal expansion tensor were visualized using the TTT program package (Bubnova et al. 2013). The Vesta software was used for crystal-structure visualization and analysis (Momma and Izumi 2011).
For the analysis of the bond-length changes with increasing temperature, the following rigid-body motion correction was used:
where L and l0 are the corrected and observed A1–A2 bond lengths, respectively, and Beq(A1) and Beq(A2) are equivalent temperature factors of the cation (A1) and anion (A2), respectively (Downs 2000).
Structural complexity
The structural complexity of both modifications of nefedovite was calculated on the basis of the Shannon information per atom (strIG) and per unit cell (strIG,total) using the following equations (Krivovichev 2012, 2013, 2014; Krivovichev et al. 2022):
where k is the number of crystallographic orbits (= the number of occupied Wyckoff sites in the crystal structure), pi is a random-choice probability for an atom from the ith crystallographic orbit, that is
where mi is the multiplicity of the ith crystallographic orbit relative to the reduced unit cell and ν is the number of atoms in the reduced unit cell. The TOPOS Pro software was used for the calculation (Blatov et al. 2014).
Bond-valence studies of Na-Ion Transport
The bond-valence sum (BVS) maps were calculated for the preliminary assessment of possible Na-ion migration pathways. The BVS maps at various bond-valence mismatches Zi–∑si (Zi = 1 for Na+, si = exp[(R0–R)/b], where R0 and b are empirical constants (Brown and Altermatt 1985; Brese and O’Keeffe 1991), R is the cation–anion distance, and ∑si is BVS) were calculated using the BondStr program (Rodríguez-Carvajal 2018) for Na+ sites in grids with steps of ∆x, ∆y, and ∆z = 0.1 Å in the unit cell with the fixed sites of other atoms. The calculated data were visualized using the VESTA software (Momma and Izumi 2011).
Results
Raman spectroscopy
The Raman spectrum of nefedovite is shown in Fig. 1. Generally, it agrees well with the spectrum obtained for other anhydrous phosphates (Litasov and Podgornykh 2017; Frezzotti et al. 2012). The main bands correspond to stretching and bending vibrations of PO4 tetrahedra: the most intense band at 955 cm−1 can be attributed to symmetric (ν1) vibrational modes of P-O, whereas the anti-symmetric stretching vibrations are reflected in middle-intensity and low intensity modes at 1069 cm−1 and 1037 cm−1, 1056 cm−1, respectively. Bands at 590 cm−1 and 431 cm−1 are connected with in-plane (ν2) and out-of-plane (ν4) bending modes of PO4 tetrahedra. The low-intensity band at 313 cm−1 can be attributed to Ca-O,F stretching vibrations. The bands up to 260 cm−1 are lattice modes.

Raman spectrum of nefedovite
Chemical composition
The chemical formula of nefedovite was calculated on the basis of P = 4. The average composition is (wt %): CaO 33.2, Na2O 22.59, K2O 0.45, P2O5 41.79, SrO 0.25, F 2.94, total 101.22. The empirical formula, (Na4.95K0.06)Ʃ5.01(Ca4.02Sr0.01)Ʃ4.03(PO4)4F1.05, is in good agreement with the ideal formula Na5Ca4(PO4)4F.
Crystal structure description
The results of the crystal structure refinement at 27 °C demonstrate a good agreement with the data obtained previously (Sebais et al. 1984). As it was mentioned above, nefedovite crystallizes in the I-4 space group and, according to Sebais et al., its crystal structure can be described on the basis of clusters consisting of four CaO7F polyhedra located at z = 0 and z = ½ (< Ca–O > = 2.449 Å). The bond-valence calculations reveal that the addition of the O3 atom to the Ca coordination polyhedron and the O2 atom to the Na1 polyhedron is appropriate, despite the relatively high bond- lengths (Brese and O’Keeffe 1991) (Tables 3, 4). The clusters of Ca polyhedra are connected via corners to form a three-dimensional framework (Fig. 2a). The Na atoms occupy two non-equivalent sites with the columns of edge-sharing Na1O7 polyhedra (< Na1–O > = 2.583 Å) and chains of corner-sharing Na2O8F2 polyhedra (< Na2–O,F > = 2.751 Å) oriented along the c axis (Fig. 2b). The columns and the chains are linked together by face-sharing with Ca-centered polyhedra of the clusters. The structure contains four independent O sites in general positions. The bond lengths in the PO4 tetrahedra have standard values and deviations (Table 4).

The fragments of the crystal structure of nefedovite described in terms of cation-centered polyhedra: Ca cluster in the center of the unit cell (projection onto the (001) plane (a), chains and columns of Na1 and Na2 polyhedra elongated along the c axis (b)
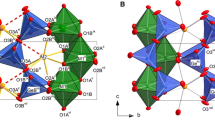
The crystal structure of nefedovite can also be described in terms of anion-centered polyhedra. The main structural subunits in this case are F-centered octahedra [FCa4Na2]9+, which share trans-corners to form chains parallel to the c axis (< Ca―F > = 2.457, < Na2―F > = 2.703 Å). The PO4 tetrahedra and Na1 atoms are located between the chains (Fig. 3a, b). Although nefedovite is not a classical antiperovskite, the chains can be considered to be derived from the antiperovskite framework of corner-sharing anion-centered octahedra (Krivovichev 2024).

The crystal structure of nefedovite described in terms of anion-centered octahedra: projection onto the (001) plane (a), and the chain of anion-centered octahedra (b)
The HT-modification of nefedovite crystallizes in the centrosymmetric space group I4/m. It has the same overall structural topology and differs slightly from the crystal structure of nefedovite at 27 °C. In particular, the O3 site can no longer be considered to be bond to the Ca atom, owing to the rather long Ca―O3 distance of 2.982 Å. In contrast to the room-temperature structure, the high-temperature one contains three independent O sites with two of them (along with the P site) located on the mirror symmetry plane with the z coordinate equal to ½. This difference is the result of the re-orientation of phosphate tetrahedra as the temperature increases. This is clearly reflected in the progressively decreasing value of the angle between the O2–P1–O3 and (110) planes from 7.01° (27 °C) to 1.7° (277 °C) before the phase transition occurs at 277–327 °C (Table 6). Thus, the detailed inspection of the crystal structure demonstrates that PO4 tetrahedra rotate around one of its local (imaginary) fourfold inversion axes gradually to align the O2…O3 edge parallel to [110], which ultimately leads to the appearance of the mirror plane perpendicular to the c axis and the space-group change from I-4 to I4/m (Fig. 4a,b).

The arrangement of the PO4 tetrahedra in the crystal structures of LT- (a) and HT- (b) modifications of nefedovite. The projections onto the (010) plane
Thermal behavior
The temperature dependence of the unit-cell parameters were analyzed using the data of in situ single crystal and thermal powder X-ray diffraction analysis. The data correlate well with each other. Their behavior can be described by a linear polynomial approximation using the following equations:
a = 11.6458 + 0.03 × 10–3 T, R2 = 0.9993;
c = 5.4083 + 0.02 × 10–3 T, R2 = 0.9573;
V = 733.14 + 0.04 × T, R2 = 0.9983 (the HTXRD data).
and.
a = 11.6475 + 0.03 × 10–3 T, R2 = 0.9993;
c = 5.4048 + 0.02 × 10–3 T, R2 = 0.9746);
V = 733.16 + 0.04 × T, R2 = 0.9989 (the SCXRD data).
The absence of the visible changes in the dependencies at the critical point (Fig. 5) can be explained by the displacive character of the phase transition.

The temperature dependencies of the unit cell parameters of nefedovite according to SCXRD data (red squares) and HTXRD data (black diamonds) and the section of the figure of thermal expansion coefficients
The calculation of the thermal expansion coefficients indicates that the crystal structure of nefedovite expands strongly anisotropically: α11 = 27.8(2), α33 = 4.5(3), αV = 60.1(1) (HTXRD); α11 = 27.7(2), α33 = 4.9(2), αV = 60.4(6) (SCXRD). The anisotropy is almost unchanged with the increasing temperature with the ratio αmax/αmin ≈ 6 (30 °C) being slightly higher for the powder HTXRD data. The maximum thermal expansion corresponds to the direction perpendicular to the chains of [FCa4Na2]9+octahedra, indicating that these units correspond to the strongest cation–anion bonds in the structure. It worth noting that, according to our previous studies of the crystal structures based on the chains of face-sharing F-centered octahedra (Avdontceva et al. 2015a, b, 2023a, b), the situation is exactly the opposite. The direction of the maximum thermal expansion in those cases is parallel to the chains, which can be explained by the F-F repulsion across the shared edge. In the case of nefedovite, the chains are built up by the sharing corners with the distance between the F− anions being significantly greater than in the case of a face linkage.
The rotation of PO4 tetrahedra obviously leads to the fact that thermal expansion is maximum in the ab plane. If we describe the crystal structure of nefedovite as a heteropolyhedral framework consisting of PO4 tetrahedra and CaO7F polyhedra, then the rotation of PO4 tetrahedra leads to the removal of the corrugation of the ab plane of the framework.
Structural complexity
The structural complexity of both modifications of nefedovite was estimated using the Shannon information per atom and Shannon information per unit cell. The results are provided in Table 7.
Sodium-ion migration in nefedovite
Using the structural data for nefedovite, the BVS maps have been created in order to calculate the bond-valence energy landscape (BVEL) and to predict the probability of the migration of Na+ ions (Fig. 6). The calculations show the probability of the Na+ migration in nefedovite, which can occur within the channels along the c direction at 0.54 eV.

The BVS map isosurfaces (0.30) for sodium ions in nefedovite. Projection onto (001) plane
Discussion
According to our data, the crystal structures of LT and HT modifications of nefedovite are fully ordered. The analysis of the temperature-induced bond-length evolution indicates that PO4 tetrahedra are almost invariant with respect to the changing temperature. The relative elongation of the average < P–O > bond length in both modifications is less than 0.6% and is close to the errors of determination (< P–O > = 1.542 Å (27 °C) and 1.550 Å (727 °C)). As mentioned above, the crystal structure of nefedovite is characterized by the strongly anisotropic thermal expansion with minimal α33 expansion parallel to the chains of F-centered octahedra. This is clearly reflected in the practically total absence of the relative elongation of the F–Na2 bond (0.01 Å) oriented parallel to the c axis. At the same time, the F–Ca bond located within the (001) plane, increases slightly with a relative elongation of nearly 2.5% (< F1–Ca > = 2.461 Å (27 °C) –2.522 Å (727 °C)).
Considering the crystal structure of nefedovite in terms of cation-centered polyhedra, we should note the continuous shortening of the Na1–O4, Na2–O4 and Na2–O1 bonds compared to the LT-modification. This tendency is related to the gradual rotation of PO4 tetrahedra before the phase transition described above. The Na1–O2 bond in the HT-modification is located almost parallel to the b axis and demonstrates the largest relative elongation (approximately 2%), whereas another Na1-O2 bond located parallel to the c axis remains practically unchanged within the experimental errors.
Thus, in the crystal structure of nefedovite the antiperovskite chains are the most robust extended units and the maximum expansion occurs within the plane perpendicular to the chains.
The crystal structure of nefedovite can also be described as a heteropolyhedral framework consisting of PO4 tetrahedra and CaO7F polyhedra with wide channels elongated along [001] and filled with Na+ cations. The channels are one-dimensional and provide low-barrier pathways for the migration of Na+ ions. The paths involve Na1 sites only, whereas the migration of Na2 site is very limited. Considering the percolation energy of 1.6 eV reported for the mobility of Na+ in polyanionic compounds (Boivin et al. 2017), the nefedovite structure type may be regarded as promising for the solid-state electrolytes.
According to Krivovichev (2024), antiperovskites can be divided into different topological types according to the D values, where D is the dimensionality of the unit formed by anion-centered octahedra. Nefedovite is a 1D antiperovskite with chains of trans-corner sharing [FCa4Na2]9+ octahedra that can be considered as a one-dimensional fragment of a classical [XA3] antiperovskite framework (A = cation; X = anion). For more information on the related minerals and inorganic compounds see Krivovichev (2024).
The crystal structures of the LT- and HT-modifications of nefedovite differ in their information-based parameters of structural complexity, but both belong to the structurally simple compounds with the total complexity less than 100 bit per unit cell (Krivovichev 2013). The HT-modification of nefedovite is simpler and has the lower value of physical density, which is in a good agreement with the general concept that, in the majority of inorganic structures, structural complexity decreases with the increasing temperature (Krivovichev 2013; Krivovichev et al. 2022). The phase transition is translationsgleiche (the unit cell remains the same), whereas the point symmetry increases, in agreement with the statistical principle that, for the displacive phase transitions, the symmetry increases with the increasing temperature (Landau, Lifshitz, 1980; Tomaszewski 1992; Filatov 2011).
References
Avdontceva MS, Krzhizhanovskaya MG, Krivovichev SV, Yakovenchuk VN (2015a) High-temperature order-disorder phase transition in nacaphite, Na2CaPO4F. Phys Chem Min 42:671–676. https://doi.org/10.1007/s00269-015-0753-x
Avdontceva MS, Zolotarev AA, Krivovichev SV (2015b) Order-disorder phase transition in the antiperovskite-type structure of synthetic kogarkoite, Na3SO4F. J Solid State Chem 23:42–46. https://doi.org/10.1016/j.jssc.2015.07.033
Avdontceva MS, Krzhizhanovskaya MG, Krivovichev SV, Zolotarev AA, Yakovenchuk VN (2023a) Polymorphism of Na2CaPO4F: crystal structures, thermal stability and structural complexity. J Solid State Chem 319:23779. https://doi.org/10.1016/j.jssc.2022.123779
Avdontceva MS, Zolotarev AA, Shablinskii AP, Bocharov VN, Kasatkin AV, Krivovichev SV (2023b) Galeite, Na15(SO4)5ClF4, and schairerite, Na21(SO4)7ClF6: phase transitions. Therm Expansion Therm Stab Symmetry 15(10):1871. https://doi.org/10.3390/sym15101871
Blatov VA, Shevchenko AP, Proserpio DM (2014) Applied topological analysis of crystal structures with the program package ToposPro. Cryst Growth Des 14:3576–3586. https://doi.org/10.1021/cg500498k
Boivin E, Chotard J-N, Bamine T, Carlier D, Serras P, Palomares V, Rojo T, Iadecola A, Dupont L, Bourgeois L (2017) Vanadyl-type defects in tavorite-like NaVPO4F: from the average long range structure to local environments. J Mater Chemi A5:25044–25055. https://doi.org/10.1039/C7TA08733K
Brese NE, O’Koeffe M (1991) Bond-valence parameters for solids. Acta Crystallogr B47:192–197. https://doi.org/10.1107/S0108768190011041
Brown ID, Altermatt D (1985) Bond-valence parameters obtained from a systematic analysis of the inorganic crystal structure database. Acta Crystallogr B 41:244–247. https://doi.org/10.1107/S0108768185002063
Bruker AXS Topas 4.2 (2009): General profile and structure analysis software for powder diffraction data Karsruhe, Germany.
Bubnova RS, Firsova BA, Filatov SK (2013) Software for determining the thermal expansion tensor and the graphic representation of its characteristic surface(Theta to Tensor-TTT). Glass Phys Chem 39:347–351. https://doi.org/10.1134/S108765961303005X
Clarke MJ, Dawson JA, Mays TJ, Islam MS (2021) Atomistic insights into the effects of doping and vacancy clustering on Li-Ion conduction in the Li 3 OCl antiperovskite Solid electrolyte. ACS Appl Energy Mater 4:5094. https://doi.org/10.1021/acsaem.1c00656
CRYSALISPRO Software System (2015), version 1.171.39.44; Rigaku Oxford Diffraction: Oxford, UK.
Dawson JA, Famprikis T, Johnston KE (2021) Anti-perovskites for solid state batteries: recent developments, current challenges and future prospects. J Mater Chem a 9:18746–18772. https://doi.org/10.1039/D1TA03680G
Dolomatov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) OLEX2: A complete structure solution, refinement and analysis program. J Appl Cryst 42:339–341. https://doi.org/10.1107/S0021889808042726
Downs RT (2000) Analysis of harmonic displacement factors. Rev Mineral Geochem 41:61–187. https://doi.org/10.2138/rmg.2000.41.3
Filatov SK (2011) General concept of increasing crystal symmetry with an increase in temperature. Crystallogr Rep 56:953–961. https://doi.org/10.1134/S1063774511060083
Frezzotti ML, Tecce F, Casagli A (2012) Raman spectroscopy for fluid inclusion analysis. J Geochem Explor 112:1–20. https://doi.org/10.1016/j.gexplo.2011.09.009
Khomyakov AP, Nechelyustov GN, Dorokhova GI (1983) Nefedovite, Na5Ca4(PO4)4F, a new mineral. Zap Vsesoyuznogo Mineral Obschchestva 112:479–483 (in Russian)
Krivovichev SV (2012) Topological complexity of crystal structures: quantitative approach. Acta Crystallogr A 68:393–398. https://doi.org/10.1107/S0108767312012044
Krivovichev SV (2013) Structural complexity of minerals: information storage and processing in the mineral world. Miner Mag 77:275–326. https://doi.org/10.1180/minmag.2013.077.3.05
Krivovichev SV (2014) Which inorganic structures are the most complex? Angew Chem Inter Ed 53:654–661. https://doi.org/10.1002/anie.201304374
Krivovichev SV (2024) Structural diversity and complexity of antiperovskites. Coord Chem Rev 498:215484. https://doi.org/10.1016/j.ccr.2023.215484
Krivovichev SV, Krivovichev VG, Hazen RM, Aksenov SM, Avdontceva MS, Banaru AM, Gorelova LA, Ismagilova RM, Kornyakov IV, Kuporev IV, Panikorovskii TL, Starova GL (2022) Structural and chemical complexity of minerals: an update. Miner Mag 86:183–204. https://doi.org/10.1180/mgm.2022.23
Landau LD, Lifshitz EM (1980) Statistical Physics. Course of Theoretical Physics vol 5 part 1. Pergamon Press Oxford. p 440
Li CC, Wang BS, Lin S, Lin JC, Tong P, Lu WJ, Sun YP (2011) Magnetic/structural phase diagram and enhanced giant magnetoresistance in Zn-doped antiperovskite. J Magn Magn Mater 323(17):2233–2237. https://doi.org/10.1016/j.jmmm.2011.03.038
Litasov KD, Podgornykh NM (2017) Raman spectroscopy of various phosphate minerals ans occurrence of tuite in the Elga IIE iron meteorite. J Raman Spectrosc 48(11):1518–1527. https://doi.org/10.1002/jrs.5119
Momma K, Izumi F (2011) VESTA 3for three-dimensional visualization of crystal, volumetric and morphology data. J Appl Crystallogr 44:1272–1276. https://doi.org/10.1107/S0021889811038970
Oudah M, Ikeda A, Hausmann JN, Yonezawa S, Fukumoto T, Kobayashi S, Sato M, Maeno Y (2016) Superconductivity in the antiperovskite Dirac-metal oxide Sr3-xSnO. Nat Commun 7:13617. https://doi.org/10.1038/ncomms13617
Rodríguez-Carvajal J. The program BondStr (or Bond_Str) and its GUI GBond_Str Version December 2018.
Sebais M, Dorokhova GI, Pobedimskaya EA, Khomyakov AP (1984) The crystal structure of nefedovite and its typomorphism. Dokl Akad Nauk SSSR 278:353–357 (in Russian)
Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Crystallogr 71:3–8. https://doi.org/10.1107/S2053229614024218
Spek AL (2003) Single-crystal structure validation with the program PLATON. J Appl Crystallogr 36:7–13. https://doi.org/10.1107/S0021889802022112
Sullivan E, Avdeev M, Blom DA, Gahrs CJ, Green RL, Hamaker CG, Vogt T (2015) Structure, stability, and photoluminescence in the anti-perovskites Na3W1−xMoxO4F (0≤x≤1). J Solid St Chem 230:279–286. https://doi.org/10.1016/j.jssc.2015.07.018
Tomaszewski PE (1992) Structural phase transitions in crystals. II Statistical Analysis Phase Trans 38:221–228
Xiao H, Xia Z, Liao L, Zhou J, Zhuang J (2012) Luminescence properties of a new greenish blue emitting phosphor Na5Ca4(PO4)4F:Eu2+. J Alloys Compd 534(5):97–100. https://doi.org/10.1016/j.jallcom.2012.04.042
Yerpude V, Nair GB, Ghormare KB, Dhoble SJ (2018) Electron-vibrational interaction in the 3d states of Cu+ ions activated in Na5Ca4(PO4)4F. Optik 161:266–269. https://doi.org/10.1016/j.ijleo.2018.02.050
Acknowledgements
The X-ray diffraction studies were performed in the X-ray Diffraction Resource Centre of St. Petersburg State University. The spectroscopic studies were performed in the “Geomodel” Resource Centre of St. Petersburg State University.
Funding
This research was funded by the Russian Science Foundation, grant № 22–77-00042, https://rscf.ru/project/22-77-00042/.
Author information
Authors and Affiliations
Contributions
Conceptualization, M.S.A., S.V.K., and A.A.Z.; methodology, M.S.A. and A.A.Z.; validation, S.V.K. and M.S.A.; formal analysis, A.P.S., M.S.A., V.N.B., N.S.V; V.N.Y, E.Y.A; investigation, M.S.A., A.A.S., M.G.K; writing—original draft preparation, M.S.A. and writing—review and editing, S.V.K., A.A.Z., M.G.K., A.P.S., V.N.B., N.S.V.; visualization, M.S.A., A.P.S., E.Y.A; supervision, project administration and funding acquisition, M.S.A. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Avdontceva, M.S., Shablinskii, A.P., Krzhizhanovskaya, M.G. et al. Nefedovite, Na5Ca4(PO4)4F: thermal evolution, phase transition and crystal structure refinement. Phys Chem Minerals 51, 13 (2024). https://doi.org/10.1007/s00269-024-01276-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00269-024-01276-7