
Abstract
Iron-rich rocks of Orosirian Period in the Chilpi Group on the northern margin of the Bastar Craton, Central India, contain an association of hematite-magnetite-greenalite-chamosite-quartz in oxide-silicate facies. Additionally chert (quartz) and siderite occur in chert and carbonate facies. Presence of these mineral assemblages was investigated to infer the redox state of the depositional basin. The results have indicated formation temperature variation of 116–255 °C (average 198 °C) and log P(O2) between – 37 and – 60 (average –44). A ferruginous state of the shallow water depositional environment, having oxygen content of 10–2 to 10–5 times the present atmospheric level, is inferred. The variations in composition of greenalite-chamosite association indicate development of the mineral phases from the reaction involving kaolinite-illite and magnetite-siderite as end-members. Thermodynamic requirements for the formation of the rare association of magnetite-greenalite-cronstedtite indicate the precipitation of the mineral phases from seawater with enhanced activities of Fe2+, Al, Si, Mg and C compared to the level in the present day seawater. The results indicate a steep fall in the atmospheric oxygen content immediately after the Great Oxidation Event of 2400–2000 Ma.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Iron-rich chemical sedimentary rocks are commonly found in marine deposits of all ages, but differ in terms of textural and mineralogical features. The Precambrian iron-rich sediments are mostly cherty and granular iron-formations but the Phanerozoic deposits are dominantly pisolitic ironstones (Maynard 1986; Bekker et al. 2010). The distribution patterns of iron formation show changing states of oxygen content in the atmosphere and ocean. The iron formations in Archean and Proterozoic Eon are more voluminous and are temporally associated with the periods of rising oxygen content in the atmosphere and ocean. The Phanerozoic ironstones are relatively less voluminous and found in basins having records of marine anoxic events. In spite of voluminous data generated regarding the genesis of iron-rich chemical sediments, the deposition of precursor materials to form iron-rich rocks remains poorly resolved (Planavsky et al. 2010; Rasmussen et al. 2013, 2017; Schröder and Warke 2016). Mineralogically four Fe–Mg phyllosilicate minerals (amesite, chamosite, greenalite and cronstedtite), with a dominant XRD peak at 7 Å, are found in Banded Iron Formations (BIFs), granular iron formations (GIFs) and pisolitic ironstones. The term “septechlorite” was used in old literature for these minerals, but the term has not been approved by the AIPEA nomenclature committee (Bailey 1980). The members of this group have different crystal structures. For example, amesite, greenalite and cronstedtite are 1:1 type phyllosilicates, whereas clinochlore-chamosite are 2:1 phyllosilicates. Individual mineral phases of both the groups contain substantial amount of Fe2+ in their structure, and the stability of these minerals depends on the redox state of the microenvironment of their occurrences. The extremely low oxygen fugacity requirement for the stability of some of these phases has a major control on their formation and preservation. Theoretical and experimental studies to analyze the stability of these minerals provide tools to interpret the oxygen fugacity, pH, temperature and other chemical environments of ancient deposits (Inoue et al. 2009, 2018; Bourdelle et al. 2013). In this contribution we have analyzed compositions of the Fe2+-rich chlorites in Paleoproterozoic iron-rich rocks and carbonates of the Chilpi Group (2050–1850 Ma) of the Bastar Craton, Central India (Fig. 1) to infer the environmental parameters of the basin of deposition immediately before the “Proterozoic iron-formation gap” (Bekker et al. 2014; Mohanty and Mishra 2023).

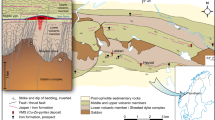
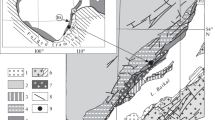
Regional geological map of a part of central India, showing distribution of the Chilpi Group (after Jain et al. 1991, with ages of stratigraphic units added in the present compilation). Framed area near Lohara is shown in Fig. 2. Inset (Top left): Outline map of Peninsular India showing positions of five cratonic nuclei (AC Aravalli Craton, BC Bastar Craton, BKC Bundelkhand Craton, DC Dharwar Craton, and SC Singhbhum Craton) and location of Fig. 1 indicated by a box in the Bastar Craton
Geological setting of the study area
Regional geology
The Bastar Craton in Central India is one of the five cratonic nuclei identified in the Peninsular India (inset in Fig. 1). The antiquity of the craton is indicated by the presence of tonalite-trondhjemite-granodiorite (TTG) gneisses of ~ 3610 Ma (Rb–Sr whole rock age; Sarkar and Gupta 1990) and K-rich granites of 3582 ± 4 Ma (U–Pb zircon age; Rajesh et al. 2009), constituting the Sukma Gneiss, in the southern part of the craton. Greenstone successions of metasedimentary rocks, metabasalts and banded iron formation (BIF) of four different ages, intruded by granites, occur in the eastern part of the craton. The oldest greenstones of Paleoarchean Era (the Sukma Group; 3550–3400 Ma) occur as enclaves within the Sukma Gneiss and the greenstone belt of Mesoarchean Era, occupying a WNW-ESE trending basin (the Bengpal Basin; 3200–3050 Ma) in the southeastern corner of the craton. The stratigraphic units of Neoarchean Era are found in a number of elongate basins aligned in N-S to NNE-SSW directions, imparting the dominant tectonic trend of the craton, referred as the “Mahanadi trend” (Krishnan 1943; Holmes 1955, 1965), reclassified as “Dharwarian trend” (Radhakrishna and Naqvi 1986). The Neoarchean greenstone belts of two different ages occur in different parts of the craton. The older greenstone sequence forming the Bailadila belt (2800–2700 Ma) is located in the southern part of the craton and the younger greenstone sequence constitute the Sonakhan-Abujhmar-Sakoli belts (2700–2500 Ma) in the eastern, central and western parts, respectively.
The northwestern part of the craton has a succession of volcanosedimentary rocks of Neoarchean-Paleoproterozoic Era, constituting the Dongargarh Supergroup (Sarkar 1957). The Amgaon Gneiss, having 3396 ± 47 Ma (Sm–Nd whole rock age; Ahmad et al. 2009) Paleoarchean components, is considered to be stratigraphic equivalent of the Sukma Gneiss (Mohanty 2015, 2021); it constitutes the basement of the Neoarchean-Paleoproterozoic successions in the northwestern part of the craton (Fig. 1). The northwestern boundary of the Amgaon Gneiss is marked by a ~ ENE-WSW striking regional shear zone (the Central Indian Shear), which defines the boundary between the Southern Province of the Bastar Craton and the Satpura Orogenic/Mountain Belt (SMB) on the margin of the Northern Province (Jain et al. 1991). The Neoarchean succession of the Dongargarh Supergroup is known as the Nandgaon Group, consisting of the Bijli Rhyolite Formation (2663 ± 14 U–Pb zircon age; Manikyamba et al. 2016) at the base, overlained by the Pitepani Andesite Formation. Granite plutons intruding the Nandgaon Group occur in a linear zone with ~ N-S alignment. The southernmost pluton (Mohala/Kotri Granite) is the largest one, with U–Pb zircon age of 2506 ± 50 Ma (Manikyamba et al. 2016). The Dongargarh Granite in the center has U–Pb zircon age of 2485 ± 7 Ma (Bickford et al. 2014). The Malanjkhand Granite on the northern part of the craton has three different variants; the oldest primary magmatic phase has U–Pb zircon age of 2478 ± 9 Ma (Panigrahi et al. 2002) and the younger components developed by partial melting during the Satpura orogeny (Mohanty 2021) have 2243 ± 217 Ma (Rb–Sr whole rock age; Panigrahi et al. 1993) and 2106 ± 102 Ma (Rb–Sr whole rock age; Panigrahi et al. 1993). The Paleoproterozoic succession of the Dongargarh Supergroup is known as the Khairagarh Group (2150–2100 Ma; Mohanty 2015, 2021). Detrital zircon ages from the sedimentary units of Khairagarh Group indicate a maximum depositional age of 2210 ± 35 Ma (Khanna et al. 2019) and the volcanic horizons of the succession have Rb–Sr whole rock age of 2120 ± 35 Ma (Sinha et al. 1998). The Khairagarh Group is unconformably overlained by the Chilpi Group (2050–1850 Ma; Mohanty 2015, 2021), comprising of coarse clastics, BIF, carbonates and shale. A suite of subalkaline mafic dykes with NNW-SSE trend intrude the Bastar Craton. These dykes have U–Pb baddeleyite ages of 1891 ± 1 Ma (French et al. 2008) and 1851 ± 3 Ma (Shellnutt et al. 2019). A Mesoproterozoic-Neoproterozoic sedimentary basin containing undeformed sediments of the Chhattisgarh Supergroup, having unconformable relation with the Chilpi Group, covers a large part in the northeast corner of the Bastar Craton. A tuff horizon in the lower part of the Chhattisgarh Supergroup has zircon U–Pb age of 1510 ± 18 Ma (Bickford et al. 2009). The lowermost depositional age of the Chhattisgarh Supergroup is reported to be ~ 1630 Ma (Patranabis-Deb et al. 2007; Mukherjee and Ray 2010). The Singhora Group, lowermost unit of the Chhattisgarh Supergroup, is intruded by a mafic dyke having Rb–Sr whole rock age of 1641 ± 120 Ma (Pandey et al. 2012). Therefore, the opening of the Chhattisgarh basin is inferred to be ~ 1700 Ma (Mohanty 2021). The closing age of the Chhattisgarh basin is not very well constrained, but a Neoproterozoic age from an equivalent basin indicates its existence during the Ediacaran Period (Mohanty 2021).
The Chilpi Group
The Chilpi Group in the northwestern part of the Bastar Craton occurs in three different linear subbasins with NNE-SSW alignments, which were parts of the Chilpi Basin on the southern margin of the SMB or the Central Indian Tectonic Zone (CITZ). The westernmost subbasin occurs along the western periphery of the Malanjkhand Granite (Fig. 1). The eastern subbasin extends from Mahali on the north through Chilpighat to Lohara on the south, and the southern subbasin is located near Kanhari and Chhuikhadan (Fig. 1). The Chilpi Group of the western subbasin has unconformable contact with the Malanjkhand Granite and the Amgaon Gneiss. In the eastern subbasin, the Chilpi Group has unconformable relationship with the Pitepani Formation of the Nandgaon Group on its western margin and an unconformable-cum-fault contact with the Chhattisgarh Supergroup on the eastern margin. The Chilpi Group has unconformable relationship with the Malanjkhand Granite, Pitepani Formation and the Khairagarh Group on the western margin, and an unconformable-cum-fault contact with the Bijli Formation, Pitepani Formation and the Chhattisgarh Supergroup on the eastern margin of the southern basin. On the basis of these stratigraphic relationships and available ages of the Nandgaon Group and the Chhattisgarh Supergroup (discussed in Sect. “Regional Geology”) the age of the Chilpi Group is suggested to be 2050–1850 Ma (Mohanty 2015, 2021; Mohanty and Mishra 2023). The development of the Khairagarh Group (2150–2100 Ma) and the Chilpi Group (2050–1850 Ma) can be considered to have a relationship with the tectonic uplift, erosion and extensional failure of the Satpura Mountain Belt, developing homologous linear basins running parallel to the alignment of the orogen (Mohanty 2023). All these basins (Bijawar, Gwalior and Mahakoshal basins on the North Indian Block and Chilpi Group on the South Indian Block) have a volcanosedimentary unit (2150–2100 Ma) at the base and a metasedimentary succession containing BIF (2050–1850 Ma) in the upper part. The rocks of these Paleoproterozoic basins show an overall low intensity deformation and low grade metamorphism (not exceeding greenschist facies) developed during the basin inversion at ~ 1850 Ma. The stratigraphic units of these basin have unconformable contact with the overlying undeformed sedimentary rocks of the Vindhyan Supergroup in the North Indian Block and the Chhattisgarh Supergroup in the South Indian Block, which show no effect of metamorphism. The later sedimentary successions have depositional age of 1700 Ma to 550 Ma.
The Chilpi Group comprises a basal conglomerate, yellowish brown gritty quartzite with interbanded chert, variegated interbanded quartzite and slaty shale (IBQS), banded hematite jasper (BHJ)/banded hematite quartzite (BHQ) with or without carbonate, massive quartzite (grey, white, light pink), cherty quartzite, dolomite/magnesian limestone, reddish pink slate, buff quartzite and shale, and splintery purple shale (Fig. 2). The shale unit in the IBQS in the lower part of the succession contains uranium mineralization in the Kanhari-Chhuikhadan subbasin in the south. Concentrated iron ore bands (mostly hematite with magnetite) are associated with BHJ and BHQ in the Mahali-Chilpighat-Lohara subbasin.

On the basis of facies analysis of sedimentary units in the Khairagarh Group, an active continental rift environment with shallow marine inner shelf, shore face, delta mouth to tidal flat conditions associated with aeolian and freshwater deposits are identified (Sinha et al. 1998; Sensarma and Mukhopadhyay 2003; Chakraborty and Sensarma 2008). The Chilpi Group is also reported to have records of a shallow marine environment of an upper shore-face (~ 15 m) in a rift basin in the lower part up to the carbonate unit and turbidity current deposits on the shelf margin (~ 120 m) in the upper part above the carbonate unit (Tripathi et al. 1981; Thorat et al. 1990).
The presence of BIF in a shallow marine depositional environment of the Chilpi Group of 2050–1850 Ma provides an opportunity to examine the oxidation state of the sea water immediately after the great oxidation event (GOE) and before the ‘boring billion period’ or ‘Proterozoic iron formation gap’ (Mohanty and Mishra 2023). Geochemical studies of the carbonate horizon above the BIF in the Lohara area of the eastern subbasin of the Chilpi Basin indicated an anoxic condition of the shallow sea (Mishra and Mohanty 2021). Petrographic studies combined with the electron probe micro-analysis (EPMA) studies indicated the presence of chamosite in the carbonate unit of the Chilpi Group. A detailed analysis of different chlorite group minerals, with a dominant 7 Å XRD peak, in the iron-rich band of the BIF and carbonate unit of the Chilpi Group was undertaken to determine the temperature of stability and oxygen fugacity conditions from the compositional variations.
Materials and methods
Field mapping was carried out in the Chilpi Basin in the area west of Lohara. About 55 samples were collected from freshly broken surface outcrops (Fig. 2) of the chemogenic sediments (iron-rich rocks of BIF and carbonate unit). Petrographic, XRD and EPMA studies were carried out on selected samples to identify different minerals present and their textural relationship (Figs. 3, 4, 5). Bulk rock geochemical analyses were carried out on a set of samples to infer the depositional condition in the basin (Mishra and Mohanty 2021; Mohanty and Mishra 2023).

XRD patterns of samples containing chamosite and greenalite. The sample numbers are annotated on the right-hand side of the XRD spectrum. K-60: quartz (65.2%), hematite (14.8%), greenalite (10.4%) and chamosite (9.6%); K-43: hematite (63.4%), magnetite (16.8%) and greenalite (19.8%); K-45-1: magnetite (51.1%), hematite (10.5%), greenalite (38.0%) and quartz (0.4%); and K-51: hematite (52.2%), magnetite (33.5%), chamosite (13.0%), greenalite (0.8%) and quartz (0.5%). The d-value of the peaks are labelled for the identified peak (having common annotation on the lower panel), alongwith the (hkl) value of crystal faces of chamosite (after Brindley 1951). Abbreviations used for identified minerals: Chm chamosite, Gre greenalite, Hem hematite, Ilt illite, Mag magnetite, and Qz quartz. Note: 3.34 Å peak (of quartz) in K-60 has been clipped

Photomicrographs of selected analyzed samples. a A carbonate rock (dolomite), M-41, showing pisolites of fine-grained dolomite bordered by a quartz-rich laminae and a medium-grained matrix between the pisolites under plane polarized light. b Oolites of chamosite and fine-grained magnetite with a border laminae of Fe-silicate phase within a fine-grained matrix of hematite-magnetite and chamosite in K-17 seen under dual mode microscopy (combined reflection and transmitted modes). c Retention of oolites within a fine-grained mixture of magnetite-hematite, showing fibro-radial grains of magnetite, cronstedtite and greenalite under dual mode microscopy of K-27. d An iron-rich rock K-45, preserving an oolite containing acicular grains of greenalite and magnetite, and micro-platy cronstedtite viewed under dual mode microscopy. e A banded iron-rich rock K-30, showing disrupted bands and preservation of green–brown chamosite in dual mode microscopy. f An oolite of magnetite (dark gray) surrounded by hematite (light gray) in K-27, indicating oxidation/replacement at the margin viewed in dual mode microscopy; chamosite oolites are present around the grain

BSE image of the textural context of the grains analyzed. a Carbonate rock M-41, showing micro-oolites of dolomite with chamosite inclusion with quartz encrustation and chamosite, ankerite and quartz in matrix. b Carbonate rock M-51, showing platy clinochlores within massive dolomite and radiation haloes around few grains. c Iron-rich rock K-45, showing needle-shaped grains of greenalite and magnetite within an oolite. d Iron-rich rock K-17, showing an elliptical oolite containing sudoite core surrounded by chamosite and maghemite bands; a hemi-spherical oolite of magnetite surrounded by chamosite is present on the lower part; radiation haloes around few grains may be noted. e A banded rock K-43 having magnetite – maghemite – greenalite bands containing platy greenalite. f A iron-rich rock K-27, showing elliptical oolites/pisolites with internal cracks and powdery grains of cronstedtite
The XRD analysis was carried out on an PANalytical Empyrean X-ray Diffractometer with Pixel 3D detector using Cu Kα radiation and nickel filter using a scan speed of 43.095 s/step. For bulk mineralogy, the random mount of less than 63 μm size fraction powder was used. The powdered samples were scanned from 2θ of 4° to 70° (step size 0.013°). Qualitative identification of mineral phases was carried out using Panalytical© HighScoreTM software. The XRD spectra were used for mineral identification by peak search and match using "HighScore plus" software and then manual confirmation of each high-intensity peak of suspected mineral. For the quantification of each identified mineral phase, Rietveld method was performed for refinement and quantification, which provided the volume percent of the mineral identified in the mixture (rock).
Electron Probe Micro Analyser (EPMA) analysis of BIF and carbonate samples was carried out by CAMECA SX-5 electron probe micro analyzer equipped with five wave length dispersive spectrometers and back-scattered electron (BSE) detectors. The EPMA analysis was carried out at 15 keV and 20 nA column condition, using standards of albite (Na), Al2O3 (Al), apatite (F, P), BaSO4 (Ba), CaSiO3 (Si), Co (Co), Cr2O3 (Cr), diopside (Mg, Ca), Fe2O3 (Fe), orthoclase (K), NaCl (Cl), Ni (Ni), rhodonite (Mn), TiO2 (Ti) and V (V) for silicate calibration.
Results
The XRD patterns of bulk rock samples indicate presence of greenalite, having maximum peaks at 2.57 Å, followed by 7.2 Å and 3.56 Å peaks, and chamosite, having 14.22 Å, 7.08 Å, 4.7 Å, 3.54 Å, and 2.2 Å peaks (Fig. 3). Chamosite was further characterized by the flattened odd (00 l) peak (almost close to the background value) and sharp even (00 l) peaks (Reynolds et al. 1992). Presence of associated mineral phases and their relative proportion were determined from automatic detection software. A banded ferruginous chert sample (K-60) contains 65.2% quartz, 14.8% hematite, 10.4% greenalite and 9.6% chamosite. Three ironstone samples have 63.4% hematite, 19.8% greenalite, 16.8% magnetite and negligible amount of chamosite (in K-43); 51.1% magnetite, 38% greenalite, 10.5% hematite and 0.4% quartz (in K-45) and 52.2% hematite, 33.5% magnetite, 13% chamosite, 0.8% greenalite and 0.5% quartz (in K-51).
EPMA studies were carried out on carbon coated samples of both BIF (four samples) and carbonate (two samples) suite. Spot analyses of 16 points on these samples (Table 1) show variations in SiO2 (4.56–44.81%), Al2O3 (6.3–32.44%), FeO (5.79–68.27%), MgO (1–32.41%), and K2O (0.02–0.25%). The oxide total was found to be 85–89%, having Σ(CaO, Na2O, K2O) < 1% (i.e., Ca + Na + K < 0.5%) except one analysis of dolomite sample (K-51/6) in which it was 1.28%, possibly due to replacement of Mg by Ca. Based on these compositional analysis, the structural formula of each phase was calculated on the basis of 14 oxygen atoms and assuming an ideal anion framework of O10(OH)8. All the analyzed phases have < 4 Si ions at the tetrahedral site. The number of Al3+ ions at the tetrahedral site was determined from the number of ions required to fill the tetrahedral site, which remained unfilled for two samples containing cronstedtite-like phase in which the required number of ions were accommodated by Fe2+ ions (i.e., substitution of Si4+ + □ ↔ 2Fe2+). The Al3+ ions at the octahedral site was calculated from the difference between the total Al and Al3+ ions at the tetrahedral site. All the Mg ions were allocated to the octahedral site. The number of Fe3+ ions at the octahedral site was calculated from the difference between the Al3+ ions at the tetrahedral and octahedral sites for greater occupancy of Al at tetrahedral site compared to the octahedral site. The amount of Fe2+ ions at the octahedral site was determined from the difference between the total Fe and the Fe3+ ions present at tetrahedral and octahedral sites. The vacancy at the octahedral site was determined from the sum of ions at the octahedral site (Table 2). Although it is difficult to determine the Fe3+/Fe2+ ratio using most of the instrumental analytical techniques, the charge balancing technique for EPMA analytical results of chlorite in iron-rich rocks, mentioned here, provides an optimum solution for use in calculations of oxygen fugacity for the assemblage. The carbonate samples were found to contain relatively higher proportions of Mg compared to Fe at the octahedral site (except one). One carbonate sample (M-51) has Ca ion as a substitution for Mg (cf., Shannon and Wherry 1922; Shimoda 1970; Alysheva et al. 1977; Billault et al. 2002; Trincal and Lanari 2016 for Ca bearing chlorite analyses) and substitution of Fe3+ for Fe2+ at the octahedral site (in 2 analyses); the phase has been identified to be clinochlore. Five analyses of another carbonate sample (M-41) show higher proportion of Mg compared to Fe (all Fe2+) and presence of vacancy; these are identified to be chamosite. For the carbonate samples average Fe/(Fe + Mg + Mn) is 0.36 and Fe2+/ΣFe is 0.92. The BIF samples contain cronstedtite-like chlorite composition (in 2 analyses), greenalite-like phase (in 4 analyses), and sudoite (in 3 analyses). Analysis of M-17 has shown presence of higher amount of vacancy at the octahedral site; one having vacancy of 1.80 (indicating substitution of Si4+ + □ ↔ 2Fe2+ as well as substitution of 2Al3+ + □ ↔ 3Mg2+) has been interpreted to be sudoite-like phase and two others having vacancy of 0.67 and 1.17 are identified to be sudoite. All these three analyses have Fe2+ in the octahedral site, along with Si. Three analyses of K-43 and one analysis of K-45 indicated presence of > 4 Fe atoms at the octahedral site; these are identified as greenalite-like chlorite. Two analyses of K-27 have > 5 Fe atoms at the octahedral site and > 2 Fe atoms (less Si than Fe) in the tetrahedral site; these indicate the phase to be a cronstedtite-like chlorite. Greenalite-like chlorites have both Fe2+ and Fe3+ in the structure, the proportion of Fe2+ being high in all samples. The BIF samples have average Fe/(Fe + Mg + Mn) of 0.83 and Fe2+/ΣFe of 0.98. The compositional variations in the analyzed phases provided opportunity to estimate the temperature and oxygen fugacity for equilibrium of these phases. The methods proposed by Inoue et al. (2009, 2018) and Bourdelle et al. (2013) were used for these calculations. The relevant equations for geothermometry using half-formula unit in quartz-chlorite assemblage are:
[3.Mg-sudoite + Al-free chlorite = 3.Mg-corundophilite + 7.quartz + 4.water]
where R2+ = Fe2+ and/or Mg, R3+ = Fe3+ and/or Al, and □ = site vacancy (Inoue et al. 2009; Bourdelle et al. 2013).
Oxygen fugacity was calculated using the relationship:
[chamosite + ¼ oxygen = H-deficient chlorite + ½ water] (Walshe 1986).
The ideal activities (α) used for temperature and oxygen fugacity estimation for different mole fractions (X) of the reactions in Eqs. 1 and 2 are: αAl-free chlorite = 1 × [(XMg)6(Xsi)2], αsudoite = 1728 × [(XMg)2(X□,o)(XAl,o)3(Xsi)(XAl,t)], αcorundophilite = 45.563 × [(XMg)4(XAl,o)2(XAl,t)2)], αChamosite = 59.720 × [(XFe2+)5(XAl,o)(Xsi)(XAl,t)], and αH-deficient chlorite = 729 × [(XFe2+)4(XFe3+,o)(XAl,o)(Xsi)(XAl,t)] for Fe3+ in structural formula or 729 × [(XFe2+)4(X□,o)(XAl,o)(Xsi)(XAl,t)] for Fe3+ = 0 in formula (Walshe 1986; Inoue et al. 2009). For Bourdelle et al. (2013) method, the activity coefficients and proportions of different mole fractions were slightly different from this (see Bourdelle et al. 2013). The assumption of αqz = αwater = 1 is used for both these equations.
The equilibrium constant (K1) for the reaction in Eq. 1 was calculated for the determined atomic composition of the mineral phases present (Table 2) using the relationship:
The equilibrium constant (K2) for the reaction in Eq. 2, at a known temperature (T in °K), is:
(Walshe 1986; Inoue et al. 2018).
The formation temperature (T in °K) was estimated from the equilibrium constant (K1) for the reaction (Eq. 1) using the following empirical equations:
(Bourdelle et al. 2013).
(Inoue et al. 2009).
(Inoue et al. 2018).
The oxygen fugacity (PO2) was calculated from Eq. (4) for different formation temperatures using the relationship:
(Walshe 1986; Inoue et al. 2018).
The results for 16 analyses (Table 2) indicate average paleotemperature of 184.5 °C (Eq. 5; Bourdelle et al. 2013), 232.4 °C (Eq. 6; Inoue et al. 2009) and 198.1 °C (Eq. 7; Inoue et al. 2018). The method by Bourdelle et al. (2013) indicates a minimum temperature of 119 °C and a maximum temperature of ~ 270 °C. On the other hand, the minimum and maximum temperatures of ~ 116 °C and ~ 255 °C obtained by Inoue et al. (2018) method are in the acceptable range for the observed grade of metamorphism in the study area. Therefore, the use of Inoue et al. (2018) calibration equation having better statistical probability than Inoue et al. (2009) has been used for estimation of the oxygen fugacity. The average temperature value in Inoue et al. (2018) method does not change substantially and increases by an amount of 6 °C after excluding 5 samples with extreme compositions, i.e., two clinochlore, two cronstedtite-like phase and one sudoite with high vacancy (Table 2). When examined separately for different rock associations, the results provide different average temperatures for carbonate and iron-rich rocks (205.5 °C for carbonate rocks and 192.4 °C for iron-rich rocks using the method by Inoue et al. 2018, and 221.9 °C for carbonate rocks and 156.2 °C for iron-rich rocks using the method by Bourdelle et al. 2013). The estimations of average oxygen fugacities have small variations between carbonate and iron-rich rocks (− 43.3 in carbonate rocks and − 44.6 in iron-rich rocks, with an average value of − 44.0 for all the samples using the temperature from calculations of Inoue et al. 2018 method). Oxygen fugacity calculated for formation temperature of 25 °C provides average value of − 73.4 (the average value for carbonate and iron-rich rocks are − 73.4 and − 73.4, respectively).
Discussion
Geothermometry and oxygen fugacity
During laboratory synthesis, clay-minerals (smectite, illite and kaolinite) are shown to be reactive with metallic iron, leading to the oxidation of metallic iron and the formation of new Fe-rich minerals such as greenalite, cronstedtite and magnetite (Fig. 6a; Pignatelli et al. 2013; Rivard et al. 2013, 2015). In the case of kaolinite, berthierine is shown to be the dominant product along with smaller amounts of greenalite and cronstedtite. The precipitation of these Fe-silicate minerals was associated with an increase in pH and decrease in Eh value to more negative value. Further, the anoxic conditions favoured the stability of berthierine, but increased O2 caused dissolution of berthierine to form iron oxides and oxyhydroxides (Rivard et al. 2013, 2015). On the other hand, smectite and illite as starting materials led to the formation of filamentous and needle shaped greenalite and cronstedtite as well as magnetite at all temperatures, and dissolution of quartz and clay-minerals (Pignatelli et al. 2014). These experiments provide valuable inputs regarding neoformation of cronstedtite and greenalite and stability of these minerals at low-temperature at the early stage in a neutral to alkaline condition (pH 6.5 to 7.8) under low Eh condition (Pignatelli et al. 2014).

Al–Fe-Si ternary diagram for Fe-silicate minerals (cf., Maynard 1983). a Synthesised products during reaction of clay and metallic iron: product 1 in oxic synthesis (Rivard et al. 2013), product 2 in anoxic synthesis (Rivard et al. 2013), product 3 at 50 °C (Pignatelli et al. 2014), and Fe-rich product 4 (Rivard et al. 2015). b Samples of the Chilpi Group, defining the iron-enrichment/depletion trend (purple arrow). Ideal compositions of kaolinite, illite, smectite, greenalite and cronstedtite are marked by brown rectangles along the atomic proportion axes. Fields of clinochlore, chamosite, greenalite-like chlorite and cronstedtite-like chlorite are indicated by colour-coded areas joining the atomic proportion axes of Al–Fe and Si-Fe
The EPMA data for the analyzed mineral compositions were examined in the ternary composition plot of Al-Si-Fe (Fig. 6b; cf., Maynard 1983). The plot shows distribution of all the data along a line joining the composition field between illite-kaolinite and magnetite-hematite. Textural relations of the analyzed phases could identify the presence of chamosite-greenalite-cronstedtite as filamentous, acicular, platy and dusty grains associated with magnetite-hematite as inclusions within a single grain, pisolites as well as with the matrix (Figs. 4, 5). Primary layering defined by Fe-rich and silicate-rich laminae in which chamosite-greenalite-cronstedtite occur indicate the precipitation of Fe-silicate phases and subsequent modification later during low-grade metamorphism. Geothermometry and oxygen fugacity determined from the composition of the analyzed phases provide the temperature of 116–255 °C and log fO2 of – 37 to – 60. For the formation temperature of 25 °C log fO2 of iron-rich rock is – 74 and carbonate rock is – 74. The carbonate rock occurs stratigraphically above the iron-rich rock. The results of oxygen fugacity, therefore, indicate no change in the level of dissolved oxygen in the seawater after deposition of the iron-rich rocks. Chamosite precipitation from seawater by reduction of Fe3+ to Fe2+ requires the amount of oxygen to be significantly lower than 0.2% of the present atmospheric level (cf., Tang et al. 2016; Rasmussen et al. 2019). The formation of Fe2+-rich corrensite (a green mineral having similar XRD patterns as chamosite-greenalite, but having more OH in the structure) in the laboratory at 1 atm of PCO2 and ≤ 3 × 10–5 atm of PO2 (Murakami et al. 2004) indicates the level of oxygen in the atmosphere during deposition of the Chilpi Group was between 10–2 and 10–5 PAL.
Formation of greenalite-chamosite
Greenalite and chamosite are relatively rare minerals confined to anoxic basins of few places. A sediment starved depositional environment with ferruginous water column having alkaline condition has been suggested to be the primary requirement for formation of these minerals (Beukes and Gutzmer 2008). However, the exact mechanisms remain cryptic and a diagenetic modification of percussor minerals is favoured in most of the models. These interpretations arise out of low and variable concentrations of Al and Si in modern seawater and nonavailability of data regarding secular variations of concentrations of these elements (particularly in Archean and Proterozoic Eons). The thermodynamic controls and the stability of different Fe-bearing minerals (associated with BIFs) at low temperature condition are analyzed here in the backdrop of updated information available regarding laboratory synthesis, solubility and distribution of different minerals and elements to provide insights to evaluate the origin of greenalite and chamosite in Archean and Proterozoic basins.
The inorganic precipitation of Fe from the water column can take place by two primary mechanisms: (a) upwelling and oxidation of Fe2+ in the surface ocean and (b) the chemical precipitation of Fe2+ from anoxic water (Ewers 1983; Beukes and Klein 1992; Tosca et al. 2016, and references therein). In the first model, amorphous ferrihydrites, which can be thermodynamically analyzed in simple composition of ferric hydroxides, Fe(OH)3, are initial products in oxic and suboxic settings; these convert into hematite during diagenesis. The second model explains the development of magnetite and siderite in anoxic facies, in which greenalite is a common association in Archean and Proterozoic iron formations. The Gibbs free energy (ΔG) at 25 °C and 1 bar for formation of different phases for formation from the elements (Table 3) provides clue regarding the relative order of precipitation in aqueous media based on the energy requirement. Some of the relevant reactions discussed below provide the information regarding the factors controlling the stability and preparing the phase diagrams (Figs. 7, 8, 9, 10).

Pourbaix diagram for iron-water system at 25 °C and 1 bar: a for aFe = 100, 10–2, 10–4 and 10–6, with no sulphur, CO2 and NH4 activities, having Fe, Fe(OH)2 and Fe(OH)3 as solid substances (after Pourbaix 1966), b for same conditions as in (a), having Fe, Fe3O4 and Fe2O3 as solid substances (after Pourbaix 1966), and c Fe-H2O-CO2-NH4-S-O2 system for aFe = 10–2, 10–3, 10–4 and 10–5; PCO2 = 10–1 bar; aNH4+ = aSO42− = 10–2, having Fe-siderite-magnetite-goethite-ammoniojarosite as solid substances (after González et al. 2015). Blue lines mark the limit of stability for water. A scale of pE + pH has been provided in (c)

Effect of pH on saturation of Si, Al, Fe3+, Fe2+, Ca and Mg for precipitation of selected minerals/phases, based on solubility studies at 25 °C and 1 bar: a iron minerals/phases and b non-iron minerals/phases. Solubility conditions: greenalite – a[SiO2, aq] = 10–2.7 and 10–4 M (Klein and Bricker 1977); magnetite (cr) calculated for pe + pHm = 4 (Kobayashi et al. 2013) and pe + pHm = 7 (Yalçintaş et al. 2016); Fe(OH)2 and siderite – PCO2 = 10–1.5 atm and SO4 (total) = 10–1.68 M (Cravotta 2008); Fe(OH)3 – Ksp = 10–37.1 (Langmuir 1997); Al(OH)3, goethite and kaolinite – SO4 (total) = 10–1.68 M and H4SiO4 = 10–3.52 M (Cravotta 2008); gibbsite – Ksp = 10–33.9 (Langmuir 1997); calcite and dolomite – closed to atmospheric CO2 (Somasundaran et al. 1985)

Controls of selected variables on the stability fields of different Al-Si-minerals and Fe-mineral associations in Fe-Si-water systems at 25 °C. a Activities of dissolved silica (H4SiO4) and total Al on Al-Si-minerals and chert association (after Maynard 1983); b activities of dissolved silica (H4SiO4) and Al complexes on greenalite-chamosite-chert precipitation (after Maynard 1983), showing the region of higher activities derived from metalliferous hot geothermal waters and brines (from Damyanov and Vassileva 2001); c activity of dissolved silica (H4SiO4) and CO2 partial pressure on chert-greenalite-(stilpnomelane)-siderite assemblage (from Klein and Bricker 1977); d CO2 partial pressure and pH controls for kaolinite-berthierine-siderite association (from Maynard 1986); e partial pressures of CO2 and O2 on precipitation of chamosite-siderite-Fe(OH)3 (after Maynard 1983); and f PCO2 and PO2 relationship for greenalite-magnetite-dolomite-siderite-Fe(OH)3 stability (from Klein and Bricker 1977). Note: Figures b–f have a[Fe2+, aq] = l0−6. Activities in average present-day seawater have a[Mg2+] = l0−1.77 (Thompson 1966), a[Ca2+] = l0−2.67 (Thompson and Ross 1966), a[Al, total] = l0−6.75 (Sackett and Arrrenius 1962) and a[H4SiO4] = l0−3.75 (Goldberg 1965; Klein and Bricker 1977). a[SiO2, aq] = l0−2.7 is saturation with respect to amorphous silica (Siever 1962, 1992), implying precipitation of chert

Eh–pH diagrams depicting the stability fields of some minerals (or their precursors) in the system Fe-H2O-O2-SiO2 at 25 °C and 1 atm total pressure (from Klein and Bricker 1977; Klein 2005). Contours show partial pressure of oxygen (from Walker et al. 1983). Boundaries between aqueous species and solids at a[Fe, aq] = 10–6. a[H4SiO4] = l0−2.7 represents seawater saturated with respect to amorphous silica (Siever 1962, 1992) and a[H4SiO4] = l0−4 represents average present-day sea water (Goldberg 1965; Klein and Bricker 1977). a[Mg2+] = l0−1.77 (Thompson 1966) and a[Al,total] = l0−6.4 (Goldberg 1965) represent activities in average present-day seawater. a Phase stability of magnetite, siderite, greenalite, minnesotaite, and stilpnomelane-like composition (with Fe2+/Fe3+ = 2:1) for two different dissolved C content. b Conditions as in (a) but stilpnomelane not considered. c Greenalite stability for two different activities of Si. d Stability of greenalite having composition representative of natural occurrences. Box with red line in (b–d) indicates mean with standard deviation of the oxygen fugacity range of the studied samples at 25 °C, assuming pH of 7–8
Thermodynamic and laboratory studies have shown that in simple aqueous system at normal temperature (25 °C) and pressure (1 bar), ferric hydroxide (Fe(OH)3) and hematite are stable products in oxidizing environment (Fig. 7a, b) and ferrous hydroxide (Fe(OH)2) and magnetite are stable under reducing conditions (Fig. 7a, b). However, the precipitation and stability is pH dependent (Fig. 7); low pH is the region of solution activities and high pH is the region of stability of solid phases. The stability fields of different iron oxide and hydroxide phases are controlled by the concentration of iron in aqueous media (aFe) as well as the redox potential (pE or Eh); high iron concentration increases the stability/precipitation field amorphous/solid phases over dissolved components (Fe2+ and Fe3+), and decreasing redox potential favours stability and precipitation of Fe2+ phases over Fe3+ phases (Fig. 7a, b). The presence of reductant components, such as S, CO2, CH4, NH4, etc., increases the stability field of magnetite, reducing the stability field of ferric hydroxide and hematite, and appearance of new phases, such as siderite, ammoniojarosite, pyrite, etc., depending upon the concentrations of reductants (Fig. 7c). Concentrations of other soluble components, Si, Al, Mg, Ca, etc., control the formation and stability of other mineral phases (Fig. 8).
The formation of pure greenalite by primary precipitation can be explained by the reactions:
(Tosca et al. 2016; Rasmussen et al. 2017).
The main thermodynamic controls for formation of pure greenalite in aqueous system (assuming ideal activity, a, for water as 1) at 25 °C and 1 bar are: aFe2+, aSiO2(aq), and pH (defined as –log aH+). Natural occurrences of greenalites differ from the ideal compositions in having Al and Mg in their crystal structure (Mg–Al-greenalite):
Therefore, activities of constituent elements (Mg2+ and Al3+) are other controls on the stability and precipitation of greenalite. The primary precipitation of chamosite has thermodynamic controls similar to Mg–Al-greenalite and can be analyzed for an ideal and a natural compositions as:
The thermodynamic conditions discussed for greenalite and chamosite (Eqs. 9, 10, 11, 12) are also suitable for explaining the precipitation of kaolinite (Eq. 13), which has requirement of higher Gibbs free energy (Table 3).
(Walshe 1986; Devidal et al. 1997).
Although natural concentrations of Si and Al at surface seawater pH of 7–8 are suitable for stability/precipitation of kaolinite/gibbsite (Figs. 8b, 9a; cf., Langmuir 1997; Cravotta 2008; vanLoon and Duffy 2010), having same requirements of aSiO2(aq), and pH (Eqs. 9, 10, 11, 12), a ferruginous low pE condition at pH of 7–8 will favour precipitation of greenalite, having low requirement of Al (Fig. 8a, b). The formation of pure greenalite, Mg–Al-greenalite and chamosite compositions given in Eqs. 9, 10, 11, 12 has identical aSiO2(aq), and pH, but these involve decreasing activity of Fe2+, and increasing activities of Mg and Al for precipitation of chamosite compared to ideal greenalite. All these conditions for the formation of greenalite have a major control of pH (6th order activity of H+), favouring alkaline condition for formation of greenalite from solutions with low concentration of the constituent elements (Fig. 8). The presence of chert bands in many BIF deposits of Archean and Paleoproterozoic age as well as chert-greenalite-chamosite association can be explained using Eqs. 9, 10, 11, 12 for basins with dissolved silica above the solubility limit of log aSiO2(aq) > − 2.7 (Fig. 9b, c).
Despite a large number of studies on aluminium and silica concentration in seawater, the reports on solubility, kinematics of precipitation and adsorption of these elements are limited. The reported solubility limit of Al and silica for pH of 7–8 is log aSiO2 = − 2.7 (Drever 1988) and log aAl,total = – 6.3 (Goldberg 1965; Klein and Bricker 1977). These solubility data provide the limits for precipitation of pure greenalite, Fe-chamosite and chert (Fig. 9b, c). The activities of Si and Al in seawater are much lower (log aSiO2 = – 3.75 and log aAl,total = – 6.75; Damyanov and Vassileva 2001). The higher concentration of Al and Si in seawater, compared to the limit defined by the solubility, required for precipitation of chamosite and chert has been the main argument for diagenetic and hydrothermal source models for the precipitation of chert and chamosite (more evaluation later).
The associations of magnetite and siderite with greenalite and chamosite in chemical precipitates (BIF and dolomite) are sensitive to oxygen fugacity (PO2) and partial pressure of CO2 (PCO2). The primary precipitation of these two minerals and dolomite in Fe(Mg)-H2O system can be written as:
(Condie 1967; Rivard et al. 2013)
(Condie 1967)
(Lindsay 1979).
The reactions in Eqs. 14 and 15 in aqueous media can lead to the formation of Fe2+ and Fe3+ ions and release of H+, OH− and e− as intermediate reactions, which are governed by the pH and Eh of the media. The appearance of dolomite in the assemblage has additional requirements regarding concentrations of Mg and Ca (Eq. 16). For the pH range of 7–8, dolomite precipitation can take place when aMg is 10–0.5–10–3 and aCa is > 10–3 (Fig. 8b). The present day seawater has aMg of 10–1.77 (Thompson 1966) and aCa of 10–2.62 at pH = 7 (Thompson and Ross 1966), which are lower than the solubility limit of aMg = 10–0.5 for dolomite at pH of 7 (Fig. 8b). These requirements for primary precipitation of dolomite not being fulfilled in present day seawater, a diagenetic model is always invoked for occurrence of dolomite. Presence of voluminous dolomite deposits in Precambrian rocks, requiring high concentration of Mg and Ca, cannot be explained by a diagenetic source. These indicate a possibility of higher concentration of Mg and Ca in Precambrian seawater compared to the present day level of concentration.
The occurrence of siderite in iron-formations can be analyzed in terms of its occurrence in euxinic seas throughout the geological history and abundance of large massive siderite-rich beds in pre-1800 Ma sedimentary sequences (Ohmoto et al. 2004). The phase boundary defining the stability relations of greenalite-siderite, and siderite-kaolinite-berthierine can be evaluated from the following reactions:
(Rye et al. 1995).
(Iijima and Matsumoto 1982; see also Curtis 1985).
The partial pressure of CO2 and the concentration of aqueous silica in solution control the stability relation of these minerals (Fig. 9c, d). For H4SiO4 activity of 10–3.4 to 10–4 in solution, greenalite is a stable phase compared to siderite, at PCO2 < 10–1.7 atm at 30 °C (Rye et al. 1995). The occurrence of stilpnomelane in some of the BIFs can be explained in terms of Si activity higher than greenalite and chamosite (Fig. 9c) as well as higher Al and Fe3+ activities and lower H+ activity compared to greenalite, but absence of stilpnomelane extends the limit of greenalite-chamosite field towards the field of chert. Fe oxidation/reduction is controlled by the activity of CO2; a low value favours precipitation of stilpnomelane, minnesotaite, chamosite and greenalite compared to siderite. For the solubility limit of Si at log aSi = – 2.7 to – 3, the lower limit of PCO2 for siderite precipitation has a value of log PCO2 = 0 at pH of 7 (Fig. 9c).
Presence of siderite in many of the Precambrian iron formations provides the upper limit of PCO2 for stability of chamosite as well as berthierine; high value (log PCO2 > − 1 at pH of 7) favours siderite formation but a low to moderate value (log PCO2 < – 1 at pH of 7) favours berthierine preservation (Fig. 9d; Maynard 1986). The PCO2 value of surface seawater for pH of 7.5–8 is much lower (log aCO2 = – 3.5) falls in the stability field of greenalite and berthierine (Fig. 9c, d), fulfilling the condition required for precipitation and stability of these minerals, but the precipitation and preservation of siderite requires higher partial pressure of CO2. The experimental investigations and phase relationships indicate stability of these minerals in Precambrian anoxic basins with pH of 7.7–8.3 having upper limit of silica solubility (log aSiO2) = – 2.7 and log aFe2+ of – 4.5 to – 5.6 (Tosca et al. 2016). Under these conditions, the phase relations of greenalite-chamosite in association with Fe minerals and kaolinite require extremely low partial pressure of O2, having log aO2 ≈ – 55 to – 65 at pH of 7–8 (Fig. 9e, f). Rasmussen et al. (2015) have shown that direct precipitation of greenalite from seawater as colloidal nanoparticles takes place in nature. Based on a comparison of the natural greenalite and synthetic products, Tosca et al. (2016) suggested that the presence of Fe2+ and SiO2(aq) in anoxic condition favours precipitation of a hydrated disordered Fe2+-silicate (gel), compositionally identical but structurally different from greenalite, which undergoes dehydration (structural rearrangement) as nanoparticles of greenalite and grows by linking along the c-axis. The petrographic observations of dusty grains and filamentous grains of chamosite-greenalite in the observed samples possibly indicate their development by this mechanism.
Thermodynamic stability relations favour ferrihydrite precipitation under oxidizing condition of seawater vis-à-vis precipitation of crystalline Fe3+ oxide phases such as maghemite (γ-Fe2O3), lepidocrocite (γ-FeOOH), hematite (α-Fe2O3), and goethite (α-FeOOH) for lower free energy of formation (ΔG; Table 3; cf., Cornell and Schwertmann 2003; Cravotta 2008). On the other hand, under reducing conditions and with decreasing oxidation potential, ferrihydrites become thermodynamically unstable, leading to the precipitation of siderite and magnetite as stable phases, depending upon the pH, redox potential (pE or Eh) and CO2 partial pressure or carbonate concentration (Eqs. 13, 14, 15; Fig. 10a, b).
The stability and precipitation of greenalite/cronstedtite and magnetite association can be evaluated in terms the reaction:
(Walshe 1986).
The stability fields of phases related to this relationship are controlled by oxygen fugacity and silica saturation in the system; higher oxygen partial pressure favours magnetite, but a lower value expands the field of greenalite, and higher silica saturation expands the field of greenalite (Fig. 10c, d). For a constant concentration of Fe and Si in the system, the phase boundary will be parallel to the contour of oxygen partial pressure (Fig. 8). Saturation of other components, such as Al and Mg, leads to the precipitation and stability of Mg–Al-greenalite and reduces the field of magnetite (Fig. 10d). The oxygen fugacity data for the analyzed samples fall in the greenalite stability field for the saturation of Si, extending into the magnetite boundary (Fig. 10c). For high CO2 partial pressure, with appearance of siderite and greenalite, the determined oxygen fugacity range of the samples of the Chilpi Group also fall in the siderite field, near the phase boundary of magnetite-siderite-greenalite (Fig. 10b). For saturation field of Si, Mg and Al, the observed oxygen fugacity range falls in the stability field of greenalite (Fig. 10d). The thermodynamic stability for greenalite-magnetite-siderite association in pH of 7–8 is possible for a lower saturation of Si, Al and Mg (Fig. 8a) at a lower oxygen fugacity condition. Hence, we infer that the association of greenalite–cronstedtite–magnetite–siderite in the Chilpi Group represents original chemical precipitation at extremely low anoxic state of the ocean, and some of the magnetite – siderite were converted to chamosite-greenalite through metamorphic reaction between illite-kaolinite and siderite-magnetite-hematite (Fig. 6b). The relevant equation for siderite-clay reaction is given in Eq. 18, and for magnetite-hematite the following reactions may be considered for the formation of berthierine as a precursor for chamosite.
(Sheldon and Retallack 2002).
The release of oxygen from the reactions in Eqs. 19 and 20 can cause oxidation of magnetite to hematite, and shift the phase boundary of magnetite-greenalite towards lower Eh. Textural evidence of oxidation of a single magnetite to form hematite at the border region has been documented (Fig. 4f; see also Mohanty and Mishra 2023).
Conclusions
Iron-rich rocks of the Chilpi Group, Bastar Craton, Central India, were deposited in a shallow sea during the Orosirian Period. Preservation of pisolitic-oolitic texture indicate wave action during the depositional process. Record of rare association of magnetite-greenalite-hematite-chert association has been confirmed from XRD analyses of the samples. Compositional variations determined from EPMA analyses provided details of the phases, and identification of clinochlore, sudoite, chamosite, greenalite-like chlorite and cronstedtite-like chlorite. Temperature and oxygen fugacity conditions for the stability of different chlorite phases indicate values of ~ 116–255°C and average P(O2) of 10–44 bar, respectively. The association of magnetite-greenalite-cronstedtite is inferred to be primary precipitation in anoxic condition of seawater with P(O2) of 10–73 bar at 25 °C, when atmospheric oxygen content was 10–2–10–5 PAL, similar to the Archean value.
References
Ahmad T, Kaulina TV, Wanjari N, Mishra MK, Nitkina EA (2009) U-Pb zircon chronology and Sm–Nd isotopic characteristics of the Amgaon and Tirodi Gneissic Complex, Central Indian Shield: constraints on Precambrian crustal evolution. In: Singh VK, Chandra R (eds) Precambrian Continental Growth and Tectonism. Excel India Publishers, New Delhi, pp 137–138
Alysheva EI, Rusinova OV, Chekvaidze VB (1977) On sudoites from the polymetal deposits of Rydnyy Altai. Doklady Academii Nauk SSSR 236:722–724
Bailey SW (1980) Summary of recommendations of AIPEA nomenclature committee on clay minerals. Am Miner 65:1–7
Bekker A, Slack JF, Planavsky N, Krapež B, Hofmann A, Konhauser KO, Rouxel OJ (2010) Iron formation: the sedimentary product of a complex interplay among mantle, tectonic, oceanic and biospheric processes. Econ Geol 105:467–508. https://doi.org/10.2113/gsecongeo.105.3.467
Bekker A, Planavsky NJ, Krapež B, Rasmussen B, Hofmann A, Slack JF, Rouxel OJ, Konhauser KO (2014) Iron Formations: their origins and implications for ancient seawater chemistry. In: Holland HD, Turekian KK (eds) Treaties on Geochemistry, 2nd edn. Elsevier, Amsterdam, pp 561–628. https://doi.org/10.1016/B978-0-08-095975-7.00719-1
Beukes NJ, Gutzmer J (2008) Origin and paleoenvironmental significance of major iron formations at the Archean-Paleoproterozoic boundary. In: Hagemann S, Rosière CA, Gutzmer J, Beukes NJ (eds) Banded Iron Formation-Related High-Grade Iron Ore. Reviews in Economic Geology, 15. Society of Economic Geologists, Colorado (USA), pp 5–47. https://doi.org/10.5382/Rev.15.01
Beukes NJ, Klein C (1992) Models for iron-formation deposition. In: Schopf JW, Klein C (eds) The Proterozoic Biosphere: A Multidisciplinary Study. Cambridge University Press, Cambridge, pp 147–151
Bickford ME, Basu A, Kamenov GD, Mueller PA, Patranabis-Deb S, Mukherjee A (2014) Petrogenesis of 1000 Ma felsic tuffs, Chhattisgarh and Indravati Basins, Bastar Craton, India: Geochemical and Hf isotope constraints. J Geol 122:43–54. https://doi.org/10.1086/674802
Bickford ME, Basu A, Patranabis-Deb S, Dhang P (2009) Depositional history of the Mesoproterozoic Chhattisgarh basin, Central India: constraints from new SHRIMP zircon ages. In: Geological Society of America Annual Meeting (18–21 October 2009), Portland. Geological Society of America Abstract with Programs 41, pp 541
Billault V, Beaufort D, Patrier P, Petit S (2002) Crystal chemistry of Fe-sudoites from uranium deposits in the Athabasca basin (Saskatchewan, Canada). Clays Clay Miner 50:70–81
Blanc P, Gailhanou H, Rogez J, Mikaelian G, Kawaji H, Warmont F, Gaboreau S, Grangeon S, Grenèche J-M, Vieillard P, Fialips CI, Giffaut E, Gaucher EC, Claret F (2014) Thermodynamic properties of chlorite and berthierine derived from calorimetric measurements. Phys Chem Miner 41:603–615. https://doi.org/10.1007/s00269-014-0683-z
Bourdelle F, Parra T, Chopin C, Beyssac O (2013) A new chlorite geothermometer for diagenetic to low-grade metamorphic conditions. Contrib Miner Petrol 165:723–735. https://doi.org/10.1007/s00410-012-0832-7
Brindley GW (1951) The crystal structure of some chamosite minerals. Miner Mag 29:502–525. https://doi.org/10.1180/minmag.1951.029.212.04
Chakraborty T, Sensarma S (2008) Shallow marine and coastal eolian quartz arenites in the Neoarchean-Palaeoproterozoic Karutola Formation, Dongargarh volcano-sedimentary succession, central India. Precambr Res 162:284–301. https://doi.org/10.1016/j.precamres.2007.07.024
Condie KC (1967) Oxygen, carbon dioxide, and sulfur fugacities during diagenesis and low-grade metamorphism of late Precambrian subgraywackes from northern Utah. Am Mineral 52:1153–1160
Cornell RM, Schwertmann U (2003) The iron oxides: structure, properties, reactions, occurrences and uses, 2nd ed. Wiley-VCH, Weinheim (Germany), 664 pp. https://doi.org/10.1002/3527602097
Cravotta CA III (2008) Dissolved metals and associated constituents in abandoned coal-mine discharges, Pennsylvania, USA. Part 2: Geochemical controls on constituent concentrations. Appl Geochem 23:203–226. https://doi.org/10.1016/j.apgeochem.2007.10.003
Curtis CD (1985) Clay mineral precipitation and transformation during burial diagenesis. Philos Trans Royal Soc London Series A Math Phys Sci 315(1531):91–105
Damyanov Z, Vassileva M (2001) Authigenic phyllosilicates in the Middle Triassic Kremikovtsi sedimentary exhalative siderite iron formation, Western Balkan, Bulgaria. Clays Clay Miner 49:559–585. https://doi.org/10.1346/CCMN.2001.0490607
Devidal J-L, Schott J, Dandurand J-L (1997) An experimental study of kaolinite dissolution and precipitation kinetics as a function of chemical affinity and solution composition at 150°C, 40 bars, and pH 2, 6.8, and 7.8. Geochim Cosmochim Acta 61:5165–5186. https://doi.org/10.1016/S0016-7037(97)00352-9
Drever JI (1988) The Geochemistry of Natural Waters, 2nd edn. Prentice-Hall, Englewood, p 437
Ewers WE (1983) Chemical factors in the deposition and diagenesis of banded iron formation. In: Trendall AF, Morris RC (eds) iron formation: facts and problems. Elsevier, Amsterdam, pp 491–512. https://doi.org/10.1016/S0166-2635(08)70054-9
French JE, Heaman LM, Chacko T, Srivastava RK, Singh RK (2008) 1891–1883 Ma Southern Bastar-Cuddapah mafic igneous events, India: a newly recognized large igneous province. Precambr Res 160:308–322. https://doi.org/10.1016/j.precamres.2007.08.005
Garrels RM, Christ CL (1965) Solutions. Minerals and Equilibria. Harper & Row, New York, p 450
Goldberg ED (1965) Minor elements in sea water. In: Riley JP, Skirrow G (eds) Chemical Oceanography (volume 1). Academic Press, London, pp 163–196
González E, Espada A, Muñoz JÁ, Blázquez ML, González F, Ballester A (2015) Reductive dissolution of magnetite and jarosite by Acidiphilium cryptum JF-5. Hydrometallurgy 157:292–297. https://doi.org/10.1016/j.hydromet.2015.08.003
Holmes A (1955) Dating the Precambrian of Peninsular India and Ceylon. Proc Geol Assoc Can 7:81–106
Holmes A (1965) Principles of Physical Geology. Thomas Nelson and Sons Ltd., London, p 1288
Iijima A, Matsumoto R (1982) Berthierine and chamosite in coal measures of Japan. Clays Clay Miner 30:264–274
Inoue A, Meunier A, Patrier-Mas P, Rigault C, Beaufort D, Vieillard P (2009) Application of chemical geothermometry to low-temperature trioctahedral chlorites. Clays Clay Miner 57:371–382. https://doi.org/10.1346/CCMN.2009.0570309
Inoue A, Inoué S, Utada M (2018) Application of chlorite thermometry to estimation of formation temperature and redox conditions. Clay Miner 53:143–158. https://doi.org/10.1180/clm.2018.10
Jain SC, Yedekar DB, Nair KKK (1991) Central Indian Shear Zone: a major Pre-Cambrian crustal boundary. J Geol Soc India 37:521–531
Khanna TC, Bizimis M, Barbeau DL Jr, Keshav Krishna A, Sesha Sai VV (2019) Evolution of ca. 2.5 Ga Dongargarh volcano-sedimentary Supergroup, Bastar craton, Central India: Constraints from zircon U-Pb geochronology, bulk-rock geochemistry and Hf-Nd isotope systematic. Earth Sci Rev 190:273–309. https://doi.org/10.1016/j.earscirev.2018.11.014
Klein C (2005) Some Precambrian banded iron-formations (BIFs) from around the world: Their age, geologic setting, mineralogy, metamorphism, geochemistry, and origin. Am Miner 90:1473–1499. https://doi.org/10.2138/am.2005.1871
Klein C, Bricker OP (1977) Some aspects of the sedimentary and diagenetic environment of Proterozoic banded iron-formations. Econ Geol 72:1457–1470. https://doi.org/10.2113/gsecongeo.72.8.1457
Kobayashi T, Scheinost AC, Fellhauer D, Gaona X, Altmaier M (2013) Redox behavior of Tc(VII)/Tc(IV) under various reducing conditions in 0.1 M NaCl solutions. Radiochim. Acta 101(5):323–332. https://doi.org/10.1524/ract.2013.2040
Krishnan MS (1943) Geology of India and Burma. Higginbothams, Madras, p 518
Langmuir D (1997) Aqueous environmental chemistry. Prentice-Hall, New Jersey, p 600
Lindsay WL (1979) Chemical Equilibrium in Soils. John Wiley & Sons, New York, p 449
Manikyamba C, Santosh M, Chandan Kumar B, Rambabu S, Tang L, Saha A, Khelen AC, Ganguly S, Singh TD, Subba Rao DV (2016) Zircon U-Pb geochronology, Lu-Hf isotope systematics, and geochemistry of bimodal volcanic rocks and associated granitoids from Kotri Belt, Central India: Implications for Neoarchean-Paleoproterozoic crustal growth. Gondwana Res 38:313–333. https://doi.org/10.1016/j.gr.2015.12.008
Maynard JB (1983) Geochemistry of Sedimentary Ore Deposits. Springer-Verlag, New York, p 305
Maynard JB (1986) Geochemistry of oolitic iron ores, an electron microprobe study. Econ Geol 81:1473–1483. https://doi.org/10.2113/gsecongeo.81.6.1473
Mishra PK, Mohanty SP (2021) Geochemistry of carbonate rocks of the Chilpi Group, Bastar Craton, India: Implications on ocean paleoredox conditions at the late Paleoproterozoic Era. Precambr Res 353:106023. https://doi.org/10.1016/j.precamres.2020.106023
Mohanty SP (2015) Palaeoproterozoic supracrustals of the Bastar Craton: Dongargarh Supergroup and Sausar Group. In: Mazumder R, Eriksson PG (eds) Precambrian Basins of India: Stratigraphic and Tectonic Context. The Geological Society, London, Memoirs 43:151–164. https://doi.org/10.1144/M43.11
Mohanty SP (2021) The Bastar Craton of Central India: Tectonostratigraphic evolution and implications in global correlations. Earth Sci Rev 221:103770. https://doi.org/10.1016/j.earscirev.2021.103770
Mohanty SP (2023) Proterozoic basins of the Bundelkhand Craton, India: Correlations and significance in understanding the tectonic evolution. Geosyst Geoenviron 2(2):100155. https://doi.org/10.1016/j.geogeo.2022.100155
Mohanty SP, Mishra PK (2023) Petrography and geochemistry of the iron-rich rocks in the banded iron formation of the Chilpi Group, Central India: Implications on the level of oxygen in the Paleoproterozoic atmosphere before the “Proterozoic iron ore gap.” Geochemistry 83(2023):125943. https://doi.org/10.1016/j.chemer.2022.125943
Mukherjee A, Ray RK (2010) An alternate view on the stratigraphic position of the ~1-Ga Sukhda Tuff vis-à-vis chronostratigraphy of the Precambrians of the Central Indian Craton. J Geol 118:325–332. https://doi.org/10.1086/651502
Murakami T, Ito J, Utsunomiya S, Kasama T, Kozai N, Ohnuki T (2004) Anoxic dissolution processes of biotite: implications for Fe behavior during Archean weathering. Earth Planet Sci Lett 224:117–129
Ohmoto H, Watanabe Y, Kumazawa K (2004) Evidence from massive siderite beds for a CO2-rich atmosphere before ~1.8 billion years ago. Nature 429:395–399. https://doi.org/10.1038/nature02573
Pandey UK, Sastry DVLN, Pandey BK, Roy M, Rawat TPS, Ranjan R, Shrivastava VK (2012) Geochronological (Rb-Sr and Sm-Nd) studies on intrusive gabbros and dolerite dykes from parts of Northern and Central Indian Cratons: implications for the age of onset of sedimentation in Bijawar and Chattisgarh Basins and uranium mineralisation. J Geol Soc India 79:30–40. https://doi.org/10.1007/s12594-012-0007-1
Panigrahi MK, Mookherjee A, Pantulu GVC, Gopalan K (1993) Granitoids around Malanjkhand copper deposit: types and age relationship. Proc Indian Acad Sci (Earth and Planet Sci) 102:399–413. https://doi.org/10.1007/BF02861511
Panigrahi MK, Misra KC, Bream B, Naik RK (2002) Genesis of the granitoid affiliated copper–molybdenum mineralization at Malanjkhand central India: facts and problems. In: Proceedings of the 11th Quadrennial IAGOD Symposium and Geocongress, Windhoek, Namibia (electronic extended abstract)
Patranabis-Deb S, Bickford ME, Hill B, Chaudhuri AK, Basu A (2007) SHRIMP ages of zircon in the uppermost tuff in Chattisgarh basin in central India require ~500-Ma adjustment in Indian Proterozoic stratigraphy. J Geol 115:407–415. https://doi.org/10.1086/518049
Pignatelli I, Mugnaioli E, Hybler J, Mosser-Ruck R, Cathelineau M, Michau N (2013) A multi-technique characterization of cronstedtite synthesized by iron-clay interaction in a step-by-step cooling procedure. Clays Clay Miner 61:77–289. https://doi.org/10.1346/CCMN.2013.0610408
Pignatelli I, Bourdelle F, Bartier D, Mosser-Ruck R, Truche L, Mugnaioli E, Michau N (2014) Iron–clay interactions: Detailed study of the mineralogical transformation of claystone with emphasis on the formation of iron-rich T-O phyllosilicates in a step-by-step cooling experiment from 90 °C to 40 °C. Chem Geol 387(2014):1–11. https://doi.org/10.1016/j.chemgeo.2014.08.010
Planavsky N, Bekker A, Rouxel OJ, Kamber B, Hofmann A, Knudsen A, Lyons TW (2010) Rare Earth element and yttrium compositions of Archean and Paleoproterozoic Fe formations revisited: new perspectives on the significance and mechanisms of deposition. Geochim Cosmochim Acta 74:6387–6405. https://doi.org/10.1016/j.gca.2010.07.021
Pourbaix M (1966) Atlas of electrochemical equilibria in aqueous solutions (First English Edition). Pergamon Press, Oxford, p 644
Radhakrishna BP, Naqvi SM (1986) Precambrian continental crust of India and its evolution. J Geol 94:145–166
Rajesh HM, Mukhopadhyay J, Beukes NJ, Gutzmer J, Belyanin GA, Armstrong RA (2009) Evidence for an early Archaean granite from Bastar Craton, India. J Geol Soc London 166:193–196. https://doi.org/10.1144/0016-76492008-089
Rasmussen B, Meier DB, Krapež B, Muhling JR (2013) Iron silicate microgranules as precursor sediments to 2.5-billion-year-old banded iron formations. Geology 41:435–438. https://doi.org/10.1130/G33828.1
Rasmussen B, Krapež B, Muhling JR, Suvorova A (2015) Precipitation of iron silicate nanoparticles in early Precambrian oceans marks Earth’s first iron age. Geology 43:303–306. https://doi.org/10.1130/G36309.1
Rasmussen B, Muhling JR, Suvorova A, Krapež B (2017) Greenalite precipitation linked to the deposition of banded iron formations downslope from a late Archean carbonate platform. Precambr Res 290:49–62. https://doi.org/10.1016/j.precamres.2016.12.005
Rasmussen B, Muhling JR, Tosca NJ, Tsikos H (2019) Evidence for anoxic shallow oceans at 2.45 Ga: Implications for the rise of oxygenic photosynthesis. Geology 47:622–626. https://doi.org/10.1130/G46162.1
Reynolds RC Jr, DiStefano MP, Lahann RW (1992) Randomly interstratified serpentine/chlorite: its detection and quantification by powder X-ray diffraction methods. Clays Clay Miner 40:262–267. https://doi.org/10.1346/CCMN.1992.0400302
Rivard C, Pelletier M, Michau N, Razafitianamahavaro A, Bihannic I, Abdelmoula M, Ghanbaja J, Villiéras F (2013) Berthierine-like mineral formation and stability during the interaction of kaolinite with metallic iron at 90 °C under anoxic and oxic conditions. Am Miner 98:163–180. https://doi.org/10.2138/am.2013.4073
Rivard C, Pelletier M, Michau N, Razafitianamahavaro A, Abdelmoula M, Ghanbaja J, Villiéras F (2015) Reactivity of Callovo-Oxfordian claystone and its clay fraction with metallic iron: role of non-clay minerals in the interaction mechanism. Clays Clay Miner 63:290–310. https://doi.org/10.1346/CCMN.2015.0630404
Robie RA, Hemingway BS (1995) Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar (105 pascals) pressure and at higher temperatures. In: U.S. Geological Survey Bulletin 2131. United States Government Printing Office, Washington, pp 1–461
Rye R, Kuo PH, Holland HD (1995) Atmospheric carbon dioxide concentrations before 2.2 billion years ago. Nature 378:603–605. https://doi.org/10.1038/378603a0
Sackett W, Arrrenius G (1962) Distribution of aluminum species in the hydrosphere-I: Aluminum in the ocean. Geochim Cosmochim Acta 26:955–968
Sarkar SN (1957) Stratigraphy and tectonics of the Dongargarh System: a new system in the Pre-cambrians of Bhandara-Drug-Balaghat area, Bombay and Madhya Pradesh. J Sci Eng Res 1(2):237–268
Sarkar G, Gupta SN (1990) Dating of early Precambrian granite complex of Bastar district, Madhya Pradesh. Records Geol Survey India 123(2):31–33
Schröder S, Warke MR (2016) Termination of BIF deposition in the Paleoproterozoic: the Tongwane Formation, South Africa. S Afr J Geol 119:329–346. https://doi.org/10.2113/gssajg.119.2.329
Sensarma S, Mukhopadhyay D (2003) New insights on stratigraphy and volcanic history of Dongargarh Belt, Central India. In: Roy A, Mohabey DM (eds) Advances in Precambrian of Central India. Gondwana Geological Society, Nagpur. Gondwana Geological Magazine 7:129–136.
Shannon EV, Wherry ET (1922) Notes on white chlorites. J Washington Acad Sci 12:239
Sheldon ND, Retallack GJ (2002) Low oxygen levels in earliest Triassic soils. Geology 30:919–922
Shellnutt JG, Hari KR, Liao AC-Y, Denyszyn SW, Vishwakarma N, Deshmukh SD (2019) Petrogenesis of the 1.85 Ga Sonakhan mafic dyke swarm, Bastar Craton, India. Lithos 334–335:88–101. https://doi.org/10.1016/j.lithos.2019.03.015
Shimoda S (1970) An expandable chlorite-like mineral from the Hanaoka Mine, Akita Prefecture, Japan. Clay Miner Bull 8:352–359
Siever R (1962) Silica solubility, 0–200°C, and the diagenesis of siliceous sediments. J Geol 70:127–150. https://doi.org/10.1086/626804
Siever R (1992) The silica cycle in the Precambrian. Geochim Cosmochim Acta 56:3265–3272. https://doi.org/10.1016/0016-7037(92)90303-Z
Sinha DK, Rai AK, Parihar PS, Hansoti SK (1998) Khairagarh basin, central India: its geology, geochemistry, geochronology and uranium metallogeny. In: International Seminar on ‘Precambrian crust in Eastern and Central India’, 29–30 October 1998, Bhubaneswar, India: Abstracts. Geological Survey of India, Kolkata, pp 135–137
Somasundaran P, Amankonah JO, Ananthapadmabhan KP (1985) Mineral-solution equilibria in sparingly soluble mineral systems. Colloids Surf 15:309–333. https://doi.org/10.1016/0166-6622(85)80081-0
Tang D, Shi X, Wang X, Jiang G (2016) Extremely low oxygen concentration in mid-Proterozoic shallow seawaters. Precambr Res 276:145–157. https://doi.org/10.1016/j.precamres.2016.02.005
Tardy Y, Garrels RM (1974) A method of estimating the Gibbs energies of formation of layer silicates. Geochim Cosmochim Acta 38:1101–1116. https://doi.org/10.1016/0016-7037(74)90007-6
Thompson ME (1966) Magnesium in sea water: an electrode measurement. Science 153:866–867. https://doi.org/10.1126/science.153.3738.866
Thompson ME, Ross JW (1966) Calcium in sea water by electrode measurement. Science 154:1643–1644. https://doi.org/10.1126/science.154.3757.1643
Thorat PK, Natrajan A, Guha K, Chandra S (1990) Stratigraphy and sedimentation of the Precambrians in parts of Bilaspur and Rajnandgaon districts, Madhya Pradesh. In: Precambrian of Central India. Geological Survey of India, Nagpur. Geological Survey of India Special Publication 28:167–180
Tosca NJ, Guggenheim S, Pufahl PK (2016) An authigenic origin for Precambrian greenalite: Implications for iron formation and the chemistry of ancient seawater. GSA Bull 128:511–530. https://doi.org/10.1130/B31339.1
Trincal V, Lanari P (2016) Al-free di-trioctahedral substitution in chlorite and a ferri-sudoite end-member. Clay Miner 51:675–689. https://doi.org/10.1180/claymin.2016.051.4.09
Tripathi C, Ghosh PK, Thambi PI, Rao TV, Chandra S (1981) Elucidation of the stratigraphy and structure of Chilpi Group. In: Archean of Central India. Geological Survey of India, Nagpur. Geological Survey of India Special Publication 3:17–30
vanLoon GW, Duffy SJ (2010) Environmental chemistry, a global perspective (Third Edition). Oxford University Press, New York, p 560
Walker JCG, Klein C, Suchodolski M, Schopf JW, Stevenson DJ, Walter MR (1983) Environmental evolution of the Archean-Early Proterozoic Earth. In: Schopf JW (ed) Earthʼs Earliest Biosphere, its Origin and Evolution. Princeton University Press, New Jersey, pp 260–290
Walshe JL (1986) A six-component chlorite solid solution model and the conditions of chlorite formation in hydrothermal and geothermal systems. Econ Geol 81:681–703. https://doi.org/10.2113/gsecongeo.81.3.681
Yalçintaş E, Scheinost AC, Gaonaa X, Altmaier M (2016) Systematic XAS study on the reduction and uptake of Tc by magnetite and mackinawite. Dalton Trans 45:17874. https://doi.org/10.1039/c6dt02872a
Acknowledgements
The EPMA facility at CRF of IIT (ISM), Dhanbad and XRD facility of Department of Earth Sciences at IIT Bombay were used for analyzing the samples. The authors are thankful to Prof. S. Sarangi and Dr. R. M. Pradhan for help in analysis of samples. Comments received from three anonymous reviewers and the editor Larissa Dobrzhinetskaya helped us to improve the quality of the manuscript.
Author information
Authors and Affiliations
Contributions
Sarada Prasad Mohanty: Conceptualization, Validation, Visualisation, Writing – review & editing, Supervision. Prasanta Kumar Mishra: Investigation, Formal analysis, Validation, Visualisation and discussion.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mohanty, S.P., Mishra, P.K. Greenalite-Chamosite composition, geothermometry and oxygen fugacity variations in pisolitic ironstone and carbonates of the Chilpi Group: implication on Paleoproterozoic seawater chemistry. Phys Chem Minerals 50, 34 (2023). https://doi.org/10.1007/s00269-023-01258-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00269-023-01258-1