
Abstract
Two samples of bazzite, a very rare Sc analog of beryl, from Tørdal, Telemark, Norway and Kent, Central Kazakhstan were studied by electron microprobe, optical absorption, and Mössbauer spectroscopies; the latter sample was also studied by FTIR. Electron microprobe results show that the Norway bazzite is composed of two bazzites with slightly different FeO contents, viz. 5.66 and 5.43 wt%. The Kazakhstan sample consists of several varieties of bazzite displaying strong differences in iron, manganese, magnesium, and aluminum contents (in wt%): FeO from 2.02 to 6.73, MnO from 0.89 to 2.98, MgO from 0.37 to 1.86, and Al2O3 from 0.30 to 1.30. Mössbauer spectroscopy shows different degrees of iron oxidation. The Norway bazzite is completely Fe2+, while the Kazakhstan sample contains roughly equivalent Fe3+ and Fe2+ accommodated in the octahedral site. The difference in iron oxidation causes strong variations in the intensity of the broad optical absorption band around 13,850 cm−1, which is assigned to Fe2+ → Fe3+ IVCT; as a result, there are strong differences in the intensity of blue color. Dichroism (E||c ≫ E⊥c) is much stronger in the Kazakhstan sample than in the Norway one. Intensities of the electronic spin-allowed bands of [6]Fe2+ at ~8900 and ~10,400 cm−1 are somewhat higher in the latter than in the former. FTIR spectra of the sample from Kent show the presence of only water type II molecules with the H–H vector perpendicular to the c-axis, in contrast to more typical beryls that always show at least weak minor bands of H2O I. This result shows that trapped water molecules in structural channels of studied bazzite occupy only sites next to or between six-membered rings centered by Na atoms. Definite structure can be observed in the vicinities of ν2 and ν3 peaks. Peaks at 1621 and 3663 cm−1 are assigned to “doubly coordinated” H2O (IId), whereas maximums at 1633 and 3643 cm−1 likely represent “singly coordinated” H2O (IIs). Interpretation of the third components in complex ν2 and ν3 bands needs further investigations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bazzite, Be3(Sc,Al)2Si6O18, a very rare Sc-bearing accessory mineral, is the structural analog of beryl (Fig. 1a) (e.g., Peyronel 1956; Armbruster et al. 1995) with nearly half of the octahedral sites occupied by Sc3+. There is also extensive octahedral Fe2+, Fe3+, and Mg substitution, with minor (<10%) Al (Armbruster et al. 1995). Charge balance for Fe2+, Mn, and Mg in the octahedral site is mainly maintained by Na+, which likely enters into channels of the structure (Armbruster et al. 1995).

Fragments of the crystal structure of bazzite: a general view along c-axis; b a chain of octahedral sites and adjacent sixfold prismatic interstices stretched along c-axis of the structure
Light blue bazzite from granite pegmatites in Tørdal, Telemark, Norway was described by Bergstrøl and Juve (1988) as clear, well-formed crystals of fine hexagonal habit (2–5 mm across) or irregular plates in microcline feldspar. By a semi-quantitative electron microprobe determination, the scandium content was 20% Sc2O3.
Chistyakova et al. (1966) and Chistyakova (1968) described dark blue bazzite from an occurrence near Kent village in Central Kazakhstan, where it is found as inclusions in zoned crystals of quartz and fluorite. Its crystal chemical formula was calculated from wet chemical analysis as Be3.06 (Sc1.26, Fe0.17, Al0.03) (Fe0.31, Mn0.13, Mg0.12) (Si5.93, Be0.07) O18 [Na0.55, K0.03, Cs0.01·0.87H2O]. Platonov et al. (1981) studied two blue and dark blue bazzite crystals from this locality using optical absorption and Mössbauer spectroscopies. The actual compositions of the samples were not determined and, unfortunately, the crystals were subsequently lost. Mössbauer spectroscopy revealed a rather complicated distribution, interpreted as Fe2+ in the octahedral sites of the structure and Fe3+ entering both the octahedral structural sites and the adjacent trigonal prism interstitial sites to form Fe2+-Fe3+pairs elongated along c-axis of the structure (Fig. 1b). The electronic Fe2+ → Fe3+ IVCT absorption band at around 15,000 cm−1 was found to be well resolved from the spin-allowed dd-band of [6]Fe2+ at ~11,200 cm−1.
As in isostructural beryl, different types of water molecules in bazzite can be recognized in channel cavities using Raman and/or IR spectroscopy. Type I of H2O has its H–H vector parallel to c-axis, while in molecules H2O (II), the H–H vector is perpendicular to c (e.g., Wood and Nassau 1967). Hagemann et al. (1990) assumed on the basis of Raman spectroscopy study of bazzite from Furkabasistunnel (Switzerland) that water is represented mainly by type II H2O related to high Na content. A similar conclusion was reached by Armbruster et al. (1995) on the basis of FTIR spectra of bazzite from that locality. In the latter paper, IR spectra showed only bands caused by water II, while some bands were not assigned.
In this investigation, we undertake additional Mössbauer and optical absorption spectroscopic studies of light blue and blue bazzite from Tørdal (Norway) and Kent (Kazakhstan), respectively, with the aim of tracing the relationship of the broad Fe2+ → Fe3+ IVCT band to the oxidation state and structural positions of iron. We also use FTIR spectroscopy for further, more detailed documentation of the state of H2O in the structure of bazzite—a rare Na-rich beryl analogous with strongly dominated or even exclusive H2O (II) molecules.
Samples
Bazzite from Tørdal was available as a relatively large (around 5 mm long) light blue semi-transparent, homogeneously colored crystal. A properly oriented transparent sample ~0.5 mm across was prepared for optical spectroscopy study as a section parallel to the c-axis. The sample was glued with epoxy onto a glass plate parallel to a well-formed prism face and polished on both sides to a thickness of 0.12 mm, suitable for measuring optical absorption spectra in two polarizations, E||c and E⊥c. In transmitted polarized light, the sample displays a slight dichroism: bluish at E||c and nearly colorless at E⊥c.
A crystalline bazzite aggregate from Kent ca. 0.5 cm in size was also available for our study. Observation of the gently cracked aggregate under a binocular microscope revealed that most of the bazzite crystals were non-transparent and displayed heterogeneous color varying from light- to dark blue. A small prism (ca. 1.0 × 0.5 mm), elongated along c-axis and mostly transparent, was selected for optical absorption spectroscopy. It was prepared as an oriented thin (0.19 mm) section as described above. Under polarizing light, it shows strong dichroism that is dark blue at E||c and colorless at E⊥c. The color of the sample, especially well seen in E||c-polarization, is quite variable with an irregular distribution, varying from dark blue to light blue to colorless.
Three E||c-oriented polished self-supported platelets of the thickness ~0.1 mm were prepared from tiny blue grains of Kent bazzite for FTIR study. One small grain of Tørdal bazzite and two grains of Kent bazzite were selected for electron microprobe analysis. Orientations of all samples for optical absorption and FTIR studies were accomplished using the well-developed prismatic habit of the crystals and controlled by conoscopic observation under a polarizing light microscope.
All remnant materials were examined and manually cleaned under a binocular microscope from grains of other minerals (mostly of colorless quartz and brown grains of iron oxides in case of Kent bazzite), milled to fine powders, and used for Mössbauer spectroscopy.
Experimental methods
Quantitative analysis of bazzites was performed on the Brown University Cameca SX-100 electron microprobe. Operating conditions consisted of a point beam, 15 kV voltage, 20 nA current, and 30 s (on peak) counting times for all elements using the PAP correction procedures (Pouchou and Pichoir 1991). One-sigma standard deviations are <1% for major elements and 3–5% for minor elements. Standards used for bazzite analysis included Wakefield, Quebec diopside (Si, Ca); synthetic forsterite (Fo97), Univesity of Rhode Island (Mg), Amelia albite, Purdue University (Na), Kakanui hornblende NMNH 143,965 (Al), rhodonite, AMNH 104,738 (Mn), Rockport, Massachusetts fayalite, NMNH 85,276 (Fe), synthetic orthoclase OR-1, AMNH (K), ScPO4 NMNH 168,495 (Sc), rutile (Ti), and synthetic MgCr2O4 (Cr). The electron microprobe was equipped with extra-large diffracting crystals (LTAP, LLIF, LPET) that generate roughly 5 × the count rate of standard-sized diffracting crystals. Extra-large crystals were used in the analysis of Si, Mg, Mn, Fe, K, Sc, Ca, and Ti. Standard-sized crystals were used for Na, Al, and Cr. Na and K were analyzed using a loss routine to account for any volatilization of the elements under the beam. Beryllium was analyzed by difference, because its X-rays cannot be detected directly by most electron microprobes.
Mössbauer spectra were acquired at 295 K using a source of 40 mCi 57Co in Rh on a SEE Co. model WT302 spectrometer (Mount Holyoke College). Experimental time was 3–4 days, and results were calibrated against α-Fe foil. About 10 mg of sample were diluted with sucrose and deposited without packing into a holder backed by Kapton® polyimide film tape. Data were collected over a ±4 mm/s velocity range in 1024 channels. Spectra were corrected for nonlinearity via interpolation to a linear velocity scale, which is defined by the spectrum of the 25-µm Fe foil used for calibration. All data were corrected to remove the fraction of the baseline due to the Compton scattering of 122 keV gamma rays by electrons inside the detector.
Spectra were fitted with Lorentzian doublets using the MEX_FielDD program acquired from the University of Ghent courtesy of E. DeGrave. Isomer shifts (IS, or δ) and quadrupole splittings (QS, or ∆) of the doublets were allowed to vary, and widths (full width at half amplitude) of all peaks were coupled to vary in pairs. Widths were constrained to vary as pairs, and both isomer shift and quadrupole splitting were unconstrained. Errors are estimated as ±0.02 mm/s for δ, ∆, and peak width (Γ); and ±1–3% (absolute) for doublet areas.
Optical absorption spectra were measured in the range 350–2000 nm (ca. 28,570–5000 cm−1) with a single-beam microspectrophotometer constructed on basis of a SpectraPro-275 triple grating monochromator, highly modified polarizing mineralogical microscope MIN-8, and PC. Ultrafluars 10× serve as objective and condenser. Two changeable photoelectric multiplying tubes and cooled PbS cells were used as photodetectors. A mechanical highly stabilized 300-Hz chopper and lock-in amplifier were applied to improve the signal/noise ratio. Spectra were scanned with steps Δλ= 1, 2, 5 nm in the ranges 330–450, 450–1000, and 1000–2000 nm, respectively. The spectral slit width did not exceed 1 nm in the whole range studied. The diameter of the measuring spot was less than 200 µm. The spectra were normalized to 1.0 cm thickness. The resultant linear absorption coefficient was then plotted versus the wavenumber.
The spectrum of Kent bazzite was analyzed by a curve-fitting procedure using the Peakfit 4.11 (Jandel Scientific) software. The high-energy absorption edge was approximated by a combination of Gaussian and Lorenzian forms, whereas for the fitting of component absorption bands, pure Gaussians were applied.
Polarized, single-crystal IR absorption spectra of Kent bazzite were measured at room temperature in the near- and middle-IR region in the spectral range 9000–1000 cm−1 using a Bruker FTIR spectrometer IFS 66 equipped with an IR-microscope at TU Berlin. Spectra were scanned using a 60-µm-diameter spot at a spectral resolution of 2 cm−1. The time-averaged signal was collected over 200 scans. Reference spectra were measured in air. The spectra obtained were normalized to 1.0 cm thickness.
Results
Bazzite from Tørdal, Norway
Electron microprobe data suggest that the sample is homogeneous. However, there are two types of bazzite, discerned in backscattered images (Fig. 2), seen as light and dark zones. The compositions of these two varieties are found to be rather close, except for a slight difference in iron content: the mean FeO concentrations in the light and dark bazzites are 5.66 and 5.43 wt%, respectively. The crystal chemical formula calculation, averaged over 10 points for light [Be is taken as 3.00 atoms per formula unite (a.p.f.u.)], gives Be3.00 (Sc1.32, Al0.10, \({\text{Fe}}_{0.33}^{3+},\;\) \({\text{Fe}}_{0.16}^{2+}\), Mn0.13, Mg0.02)Σ = 2.06 Si5.94O18 [Na0.32, K0.02]. The dark area, averaged over 15 points, gives Be3.00 (Sc1.33, Al0.10, \({\text{Fe}}_{0.34}^{3+},\;\) \({\text{Fe}}_{0.14}^{2+},\)Mn0.13, Mg0.02)Σ = 2.06 Si5.94O18 [Na0.32, K0.02]. These electron microprobe compositions, with the exception of Cs that was not determined in our samples,Footnote 1 are nearly identical to another Tordal occurrence of blue bazzite, also from Tørdal, Norway. That cesian bazzite (ca. 3 wt% Cs2O) was found by Juve and Bergstøl (1990) as tiny sky-blue crystals grown on faces of larger beryl crystals. Our sample seems similar.

Backscattered electron microprobe image of bazzite from Tørdal, Norway. It contains bazzites of two different compositions, light and dark, slightly differing by the mean iron content, 5.66 and 5.43 wt% FeO, respectively
The Mössbauer spectra was fitted with a variety of doublet combinations until the fit, shown in Fig. 3 and Table 1, was chosen as most suitable. No evidence of the highly distorted Fe2+/Fe3+ hybrid distribution was found. Contrary to the crystal chemical formula, which shows a predominant Fe3+ content (see above), the final fit consists of three Fe2+ octahedral distributions and none of Fe3+. Although beryl normally has a quite high isomer shift (IS) value for Fe2+, δ, near 1.4 mm/s (e.g., Viana et al. 2002), this is seen in only one distribution, viz. [6]Fe2+(II) (Table 1). The other two distributions have smaller parameters, where the lowest IS value is paired with the highest QS distribution. It is unclear whether they represent three distinct sites, or if this is a part of the asymmetry, as is typical for beryl spectra (e.g., Price et al. 1976; Platonov et al. 1979; Viana et al. 2002). Although this sample can also be additionally fitted with 4.5% of octahedral Fe3+, it is difficult to justify this feature because the un-accounted area at the upper right side of the low velocity peak is mirrored on its right side (Fig. 3), and may just be a remnant of the typical asymmetric broadening.

Mössbauer spectrum of the bazzite from Tørdal, Norway, fitted by three octahedral Fe2+ distributions. Parameters of the fit are given in Table 1
The polarized optical absorption spectrum of bazzite from Tørdal, recalculated to the thickness of 1 cm, is shown in Fig. 4 in the spectral range from 350 to 1800 nm (ca. 28,500–5600 cm−1). It consists of a nearly isotropic high-energy absorption edge partly superimposing a broad dichroic (E||c > E⊥c) envelope, labeled as c, with a maximum around 14,500 cm−1. On its low-energy wing, there is a distinct doublet of absorption bands, labeled as a and b (E||c > E⊥c), with maxima at around 10,510 and 9000 cm−1. A weak but distinct narrow doublet peak of absorption is clearly seen at ~7040 and 7190 cm−1 in both polarizations (E⊥c > E||c). A stronger single E⊥c-polarized absorption line appears at around 5260 cm−1. No signs of spin-forbidden absorption bands of Fe2+ are seen in the visible or near UV ranges.

Polarized optical absorption spectrum of bazzite from Tørdal, Norway. The colored labels display the HTML colors, calculated from the spectra measured at the actual thickness of the sample (0.12 mm) for illumination by polarized light of the standard CIE illuminant C
Bazzite from Kent, Kazakhstan
Electron microprobe data obtained on two grains show strong variations of bazzite composition within each samples studied. The backscattered image of sample #1 (Fig. 5) shows that the bazzite crystal consists of at least three types of bazzite of different composition, A, B, and C. In addition, there are inclusions of other phases including quartz, Ca-La-Ce-Th phosphates, and bertrandite or phenakite. Each type of bazzite displays strong variations of iron, manganese, magnesium, and aluminum contents (in wt%): FeO from 2.02 to 6.73, MnO from 0.89 to 2.98, MgO from 0.37 to 1.86, and Al2O3 from 0.30 to 1.30. The titanium content scarcely amounts to ~0.2, i.e., 0.01 a.p.f.u. The average composition was recalculated to the following crystal chemical formula:

Backscatter electron microprobe image of Kent bazzite grain #1. It contains bazzite of at least three different compositions, A, B, and C, Ca-La-Ce-Th phosphates, quartz and what appears to be bertrandite or phenakite
bazzite A: Be3.00 (Sc1.24, Al0.11, \({\text{Fe}}_{0.37}^{3+}\), \({\text{Fe}}_{0.06}^{2+ }\), Mn0.13, Mg0.13)Σ = 2.04 Si5.95O18 [Na0.36, K0.01];
bazzite B: Be3.00 (Sc1.22, Ti0.01, \({\text{Fe}}_{0.28}^{3+ }\), Al0.13, Mn0.24, Mg0.20)Σ = 2.08 Si5.95O18 [Na0.39];
bazzite C: Be3.00 (Sc1.37, \({\text{Fe}}_{0.15}^{3+ }\), Al0.04, Mn0.11, Mg0.33)Σ = 2.00 Si6.00O18 [Na0.38].
Grain #2 consists of bazzite only. Intergrowths of two bazzites of different average compositions, D and E are seen:
bazzite D: Be3.00 (Sc1.39, \({\text{Fe}}_{0.14}^{3+ }\), Al0.04, Mn0.15, Mg0.32)Σ = 2.04 Si5.88O18 [Na0.45];
bazzite E: Be3.00 (Sc1.22, \({\text{Fe}}_{0.26}^{3 + }\), Al0.16, Mn0.24, \({\text{Fe}}_{0.07}^{2 + }\), Mg0.17)Σ = 2.12 Si5.90O18 [Na0.44].
The Mössbauer spectrum and its fit chosen as the most suitable one after the fitting with a variety of doublet combinations, is shown in Fig. 6. Again, as in case of Norwegian sample (see above), there is no evidence of highly distorted Fe2+/Fe3+ hybrid distribution. All distributions are iron in octahedral coordination and the final fit consists of three Fe2+and one Fe3+ distribution (Table 1). The Fe3+ distribution is very broad, but has standard octahedral parameters. No set of IS and QS values of [6]Fe2+ in either of the samples studied (Table 1) corresponds to the quadrupole doublet of octahedral Fe2+, IS = 1.47 mm/s, and QS = 2.55 mm/s, derived by Platonov et al. (1981), who assigned the only defined Fe2+ quadrupole doublet in the bazzite spectrum to \({\text{Fe}}_{{\text{oct}}}^{2 + }\). Moreover, they estimated the parameters of quadrupole doublet of \({\text{Fe}}_{{\text{oct}}}^{{\text{3}} + }\) as IS = 0.61 mm/s and QS = 0.82 mm/s, which are also quite different, especially the IS value, from what is compiled in Table 1 for [6]Fe3+ distribution in the sample studied here. Note also that although the electron microprobe data suggest a rather low Fe2+ content (see above), the Fe3+: Fe2+ ratio of around 1:1 (Table 1) from Mössbauer spectroscopy is close to that measured by Platonov et al. (1981).

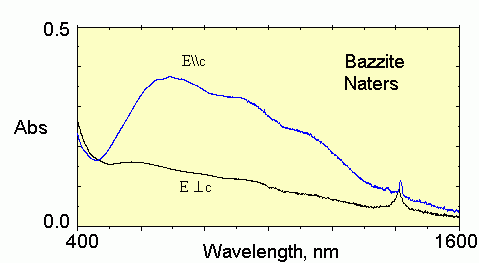
The polarized optical absorption spectrum of bazzite from Kent is shown in Fig. 7. Like the bazzite from Tørdal, it consists of a practically isotropic high-energy absorption edge. The edge is partly superimposed upon a broad, strongly dichroic (E||c > > E⊥c) absorption envelope with a maximum c at around 14,500 cm−1. On the low-energy wing of the envelope, two shoulders, a and b, at ~9200 and ~11,000 cm−1 can be distinguished. Also as in the sample from Norway, there is a doublet absorption peak at 7040 and 7190 cm−1 in both polarizations, E||c and E⊥c, and a stronger E⊥c-polarized absorption line appears at ~5260 cm−1. On the whole, the polarized spectrum of this bazzite is quite similar to that of the pale blue bazzite from Natters, Switzerland studied by Rossman (2016), but significantly differs from those described by Platonov et al. (1981).

Polarized optical absorption spectrum of bazzite from Kent, Kazakhstan. The colored labels as in Fig. 4, calculated for the actual thickness of the sample 0.19 mm
Polarized IR spectra of bazzite from Kent in the range 6000–1000 cm−1 are shown in Fig. 8. The spectrum measured in polarization E||c is dominated by two very intense narrow bands at 3594 and 1621 cm−1. The former is surrounded by two weak satellites at 3663 and 3519 cm−1, while the latter is a complex envelope that reveals three peaks at 1631, 1621, and 1614 cm−1. In this polarization, a strong broad band at ~1900 cm−1, bands of medium intensity at 4003 and 3228 cm−1, and very weak bands at 5605 and 3327 cm−1 are observed. The spectrum measured in polarization E⊥c consists of very strong and broad envelope centered at ca. 3665 cm−1, a strong broad band near 1890 cm−1, and medium and weak bands at 5266, 2352, 1697, 1633, 1620, and 1548 cm−1. The main envelope near 3665 cm−1 includes narrow peaks at 3681, 3663, and 3643 cm−1, its long wavelength slope is complicated by smaller bands and shoulders at 3620, 3594, 3545, and 3519 cm−1 (Fig. 8).

Polarized single-crystal FTIR spectra of bazzite from Kent
In general, the main features of single-crystal IR spectra of Kent bazzite, namely the strongest bands in both polarizations, are similar to spectra of bazzite from Switzerland (Armbruster et al. 1995). At the same time, many important details of these spectra including the main band’s structure and intensity, number and positions of smaller peaks, are different. Bands at 3735, 3650, 3475, and 3270 cm−1, reported in Switzerland sample, are absent in spectra of Kent bazzite. Absorption maxima centered at 3681, 3643, 3620, 3519, 2352, 1697, 1633, 1614, and 1548 cm−1 (Fig. 8), as seen in the Kent sample, were not detected in bazzite from Switzerland. This can be caused by differences in crystal chemistry of the two samples as well as by the high noise/signal ratio and distortion of intensity proportions between main bands in the case of bazzite from Switzerland, for which spectra were measured in an unpolished, relatively thick crystal (Armbruster et al. 1995).
Discussion
The optical absorption spectrum of the Norway bazzite (Fig. 4) is typical of Fe2+-bearing silicates with Fe2+ in octahedral structural position that is consistent with the Mössbauer spectroscopy data (Fig. 3; Table 1). The doublet bands a and b at ~10,510 and ~9100 cm−1 should be assigned to split electronic 5 T 2g → 5 E g dd-transition of octahedral [6]Fe2+. As seen in beryl, they are polarized in E||c. Considerable red shifts of these bands, when compared with those in iron-bearing blue beryls (cf., e.g., Wood and Nassau 1968; Taran and Rossman 2001), are undoubtedly caused by a relatively large octahedral site, because the M–O distances in bazzite and beryl are 2.080 and 1.904 Å, respectively (Armbruster et al. 1995). Because by theory \({\text{Dq}}\frac{1}{{{{\bar R}^5}}}\), where Dq is crystal field strength and \(\bar R\) is average metal–oxygen distance in Fe2+O6 octahedron (e.g., Burns 1993), this causes a lower value of Dq of [6]Fe2+ in bazzite. Thus, energies of the spin-allowed crystal field bands of [6]Fe2+ are lower in the bazzite spectrum compared with beryl. The splitting of the electronic 5 E g state, which is not expected by the selection rules for D 3 symmetry of the octahedral site in both beryl and bazzite, may be due to the dynamic Jan–Teller effect on the degenerated ground state. Theory suggests that the 5 T 2g level splits to two sublevels, 5 A and 5 E at the point symmetry D 3 (e.g., Marfunin 1979). The observed splitting of the excited 5 E g level (bands a and b) requires that the lower (ground) state of Fe2+ in the distorted [compressed along c-axis (Armbruster et al. 1995)] octahedral site is 5 E, evoking the Jan Teller effect.
A very broad and relatively weak absorption envelope with a maximum at around 14,500 cm−1 (b and c) is most likely caused by electronic intervalence charge-transfer (IVCT) transition of Fe2+ + Fe3+ → Fe3+ + Fe2+ type. Its low intensity, compared with the spin-allowed bands a and b, is consistent with very low Fe3+ content in the sample, as it observed in the Mössbauer results (see above).
The high-energy absorption edge is usually assigned as a wing of very intense UV absorption bands, caused by electronic ligand–metal charge-transfer transitions mostly of O2− → Fe3+, Fe2+ type (e.g., Burns 1993). It is much higher in the Kentthan in the Tørdal bazzite, and is also consistent with much higher oxidation degree of iron in the former comparing with the latter. The polarization of the Fe2+/Fe3+ IVCT c-band and to a lesser extent, the near-infrared spin-allowed 5 T 2g → 5 E g bands a and b, E||c ≫ E⊥c, cause the distinct dichroism of the sample, bluish at E||c and nearly colorless at E⊥c (Fig. 4).
The relatively weak absorption lines at ~7040 and 7190 cm−1, seen in spectra of both samples studied in both polarizations, E||c and E⊥c, are obviously those tabulated by Armbruster et al. (1995) as combined 2ν1 and v1v3 vibrations, respectively. Accordingly, the much more intense single E⊥c-polarized line at ~5260 cm−1 must be a v2v3 vibration (all H2O type II).
By the curve-fitting procedure, the envelope in E||c-polarized spectrum of bazzite from Kazakhstan can be well approximated by three Gaussians a, b, and c (Fig. 9), similar to those in the Norwegian sample. This can be seen in the parameters’ linear intensity α, energy ν and half-width ω½ data compiled in Table 2. A noticeable deviation of the fitted curve from the experimental one in the spectral range from ca. 17,000 to 27,000 cm−1 might be due to a complex character of the absorption edge, which can be poorly represented as a combination of Gauss and Lorentz functions. We assume that this is mainly caused by electronic ligand–metal charge-transfer transitions of O2− → Fe3+, Fe2+ type. If several different structural types of Fe3+ and Fe2+ participate [octahedral, tetrahedral, or even channel (Armbruster et al. 1995)] to cause a set of UV bands of various intensities, energies, and widths, then the shape of the edge may be significantly influenced, causing its deviation from the combination of single Lorentz and Gauss forms.

Results of the curve-fitting analysis of the E||c-polarized spectrum of bazzite. The dots are experimental data obtained by the single-beam procedure of the spectra registration
The values of half-width, ω½, of a and b components of the curve fitting are typical for spin-allowed dd-transition of Fe2+ (Table 2). In Tørdal bazzite, their intensities α are somewhat higher, ~10 and ~15 cm−1, respectively, than in bazzite from Kent (Table 2). This is an evidence that in the actual samples studied by optical absorption spectroscopy, the iron content in the former is very likely somewhat higher than in the latter. This, in principle, fairly agrees with the electron microprobe data, which show that bazzite from Tørdal contains more Fe2+ than that from Kent (see the crystal chemical formulae above).
The ω½ value of the c component in Kent bazzite is much higher, about 8000 cm−1, than those of a- and b-bands (Table 2). This is an evidence in favor of the electronic intervalence charge-transfer (IVCT) nature of the c-band (e.g., Burns 1993). In fact, its ω½ value better corresponds to Fe2+ + Ti4+ → Fe3+ + Ti3+ IVCT transition, though the very low Ti content in the samples studied (see above), as well as the energy of the band, which is more appropriate for Fe2+/Fe3+, than Fe2+/Ti4+ IVCT (Burns 1993), does not support such interpretation. Therefore, we assume that the c-band is caused by Fe2+ + Fe3+ → Fe3+ + Fe2+ IVCT transition. However, much stronger intensity of the IVCT bands in the Kent rather than the Tørdal bazzite agrees with the much higher oxidation state of iron admixture in the former, than in the latter. The probability of formation of IVCT Fe2+, Fe3+ pairs in the case of disordered distribution of the cations is proportional to the product of Fe2+ and Fe3+ concentrations in samples. Judging from Mössbauer data, it should be much higher in Kent bazzite than in the Tørdal one. In beryls, this band is certainly related to [6]Fe2+ content (Goldman et al. 1978). Its spectral positions, shape, and width are typical of IVCT transition. Moreover, temperature dependence of it also evidences in favor of such interpretation (Taran et al. 1989; Taran and Rossman 2001).
The polarization of the c-band, E||c ≫ E⊥c, suggests that the IVCT transition may take place between Fe2+ and Fe3+ accommodated in positions aligned along the crystal axis c. As assumed for beryls (e.g., Platonov et al. 1979; Taran and Rossman 2001; Groat et al. 2010), these may be the structural octahedral sites and trigonal prism interstitions that alternate with each other along c-axis (Fig. 1b). Because the exact position of Fe3+ accommodated in the interstitial site is not known, we can only assume that Fe2+–Fe3+ vectors may have weak E⊥c components that are quite in accord with the polarization properties of the c-band (Fig. 1b). Taking the same calculation that Groat et al. (2010) applied for blue beryl, we evaluate the amount of ~1.5 wt% Fe involved in the electronic IVCT transition in the dark blue part of Kent bazzite. This is, probably, too low in concentration to confirm that c-band does relate to iron in the trigonal interstition of the structure by other methods, first of all by structure refinement. As mentioned by Groat et al. (2010), structure refinements on Fe-containing beryl-group minerals have never shown a residual electron density at the supposed trigonal site. Also, the repulsive cation–cation distances for the trigonal site of bazzite seem to be too short for interstitial Fe2+ or Fe3+. All this makes the probability Fe2+ and Fe3+ in the interstitial sites relatively low. Nevertheless, it is difficult to figure out any structural position that can satisfy all above-mentioned properties of the optical absorption band c: relation to [6]Fe2+ content, energy, width, polarization, and temperature response, assuming that it is indeed the Fe2+/Fe3+ IVCT band. Therefore, we still assume that a small part of Fe enters the trigonal interstices. The local charge balance in this case may be maintained by substitutions such as 3[6]Al3+ → 3[6]Fe2+ + [6i]Fe3+, where 6i denotes the trigonal interstition. This involves only a small portion of the overall amount of [6]Fe2+. The larger part of Al → [6]Fe2+ substitution must be balanced by Na+ and K+ cations in the channels.
Platonov et al. (1981) found a well-resolved Fe2+/Fe3+ IVCT band in bazzites from Kent at around 15,000 cm−1. However, at such fixed energy of the Gaussian component, the curve fitting of the E||c-polarized spectrum in Fig. 9 gives an even higher, unrealistic ω½ value ~9800 cm−1 for it. Use of two IVCT bands (one of them at 15,000 cm−1) also does not improve the situation, still giving a large ω½ value (~9000 cm−1) for the latter band. Perhaps strong overlap of the components composing the envelope and the complicate shape of the absorption edge, which cannot be well approximated by the functions available in the software applied, prevents any considerable improvement of the curve-fitting result.
Aside intense high-energy edge, spectra of both samples studied in E⊥c-polarization (Figs. 4, 7) display weak broad absorption features, which appear to be E⊥c-polarized components of the above-described bands a, b, and c, strong in polarization E||c. In the E⊥c-polarization, there is no evidence of a broad band at around 12,000 cm−1, which by analogy with beryl (e.g., Taran and Rossman 2001) could have been assigned to electronic spin-allowed dd-transitions of tetrahedral Fe2+. This result is in accord with the conclusions of Platonov et al. (1981) and with our Mössbauer spectroscopy data, which, as shown above, revealed the quadrupole doublets of only sixfold coordinated Fe2+ and Fe3+ ions (Figs. 3, 6; Table 1).
Maxima of the detected absorption bands of H2O molecules in the FTIR spectra measured in three separate grains of bazzite from Kent, their polarizations, and assignments are listed in Table 3. Broad bands near 1900 cm−1 in both polarizations represent first overtones of Si-O vibrations, they are always present also in beryl. The same result was reported for bazzite from Furkabasistunnel, Switzerland (Armbruster et al. 1995; Hagemann et al. 1990). Infrared spectra of all samples from Kent show the presence of only water type II molecules with the H–H vector perpendicular to the c-axis (Fig. 10). Bands of three fundamental modes of H2O II at ~1620 cm−1 (bending ν2), ~3660 cm−1 (antisymmetric stretching ν3), both in polarization E⊥c, and 3594 cm−1 (symmetric stretching ν1, E||c) are the main features in the bazzite spectra (Fig. 8). In all bazzite samples studied so far by IR spectroscopy, the Na content was less than 0.5 a.p.f.u., namely 0.32 Na in the sample from Switzerland (Armbruster et al. 1995) and 0.36–0.45 in the studied grains of bazzite from Kent. Thus the absence of any signals from H2O I molecules in any IR spectra leads to an important conclusion about ordered distribution of trapped water molecules in structural channels. They should occupy only sites next to or between six-membered rings centered by Na atoms. To our knowledge, in contrast to the bazzites, there have been no published spectra of natural beryl without at least weak minor bands of H2O I.

General schematic of the bazzite channel showing structural oxygen atoms surrounding possible channel constituents (modified after Fukuda and Shinoda 2008). The constituents in bazzite are II H2O (type I is shown for comparison). The cation in this figure is assumed to be Na+ or K+
Definite structure can be observed in the vicinities of ν2 and ν3 peaks. Both maxima are represented by envelopes formed from at least three overlapped separate narrow bands. Their maxima are located at 1614, 1621 and 1633 cm−1 in the case of ν2 (Fig. 11) and 3643, 3663, 3681 cm−1 for ν3. For the symmetric stretching ν1 band, we detected four-peak structure in spectra measured at low temperature (80 K). Fukuda and Shinoda (2008) found that Na-associated molecules H2O (II) may exist in beryl channels in two configurations, namely as H2O-Na-OH2 (“doubly coordinated,” IId) and H2O-Na (“singly coordinated,” IIs) (Fig. 10). H2O (IId) molecules are characterized by a ν2 band at 1620–1624 cm−1 and ν3 at 3660–3664 cm−1, while the same bands of configuration H2O (IIs) are centered at 1633–1637 and 3643 cm−1, respectively (Fukuda and Shinoda 2008; Fridrichova et al. 2016). The vibrations of H2O (IId) and H2O (IIs) in bazzite are very close to those in beryl, as is the case for all other bands of water molecules in IR spectra of these two minerals. Thus, we can assign peaks at 1621 and 3663 cm−1 to “doubly coordinated” H2O (IId), whereas maxima at 1633 and 3643 cm−1 can be assigned to “singly coordinated” H2O (IIs) (Table 3). Peaks of H2O (IId) in spectra of Kent bazzite are a bit more intense than those of H2O (IIs) (Figs. 8, 10). We conclude that configuration H2O-Na-OH2 is most abundant (but not predominant) for water molecules in structural channels of the samples studied. This is in general agreement with chemical compositions of the samples. Interpretation of the third components in the complex ν2 and ν3 bands needs further investigations. From their polarization, it is clear that they also belong to water type II.

Fine structure of the absorption envelope caused by ν2 bending vibration of type II H2O molecules
In addition to the discussed fundamental bands of type II H20, their overtones and combination bands as well as satellites due to combining with libration modes are present in the spectra (Table 3). Assignments of these bands were done on the basis of interpretation given by Wood and Nassau (1967).
Notes
Cs in the EDS peaks were not certainly discerned at preliminary scans of the sections of both bazzites, Norwegian and Kazakh. It might be present at the less than 1 wt% level.
References
Armbruster Th, Libowitzky E, Diamond L, Auernhammer M, Bauerhansl P, Hoffmann Ch., Irran E, Kurka A, Rosenstingl H (1995) Crystal chemistry and optics of bazzite from Furkabasistunnel (Switzerland). Mineral Petrol 52:113–126
Bergstøl S, Juve G (1988) Scandian ixiolite, pyrochlore and bazzite in granite pegmatite in Tørdal, Telemark, Norway. A contribution to the mineralogy and geochemistry of Scandium and Tin. Mineral Petrol 38:229–243
Burns RG (1993) Mineralogical application of crystal field theory. Cambridge University Press, Cambridge, p 550
Chistyakova MB (1968) Beryl and bazzite from crystal-bearing cavities of the granite pegmatites of Kazakhstan. In: The new data on minerals from the USSR, vol 18. Nauka, Moscow, pp 140–153 (in Russian)
Chistyakova MB, Moleva VA, Rasmanova ZB (1966) The first finding of bazzite in the USSR. Dokl Akad Nauk SSSR 169:1421–1424 (in Russian)
Fridrichova J, Bacik P, Bizovska V, Libowitzky E, Škoda R, Uher P, Ozdin D, Števko M (2016) Spectroscopic and bond-topological investigation of interstitial volatiles in beryl from Slovakia. Phys Chem Miner 43:419–437
Fukuda J, Shinoda K (2008) Coordination of water molecules with Na+ cations in a beryl channel as determined by polarized IR spectroscopy. Phys Chem Miner 35:347–357
Goldman SD, Rossman GR, Parkin KM (1978) Channel constituents in beryl. Phys Chem Minerals 3:225–235
Groat LA, Rossman GR, Dyar MD, Turner D, Piccoli PMB, Schultz AJ, Ottolini L (2010) Crystal chemistry of dark blue aquamarine from the True Blue showing, Yukon Territory, Canada. Can Mineral 48:597–613
Hagemann H, Lucken A, Bill H, Gysler-Sanz J, Stalder HA (1990) Polarized Raman spectra of beryl and bazzite. Phys Chem Minerals 17:395–401
Juve G, Bergstøl S (1990) Caesian Bazzite in Granite pegmatite in Tørdal, Telemark, Norway. Mineral Petrol 43:131–136
Marfunin AS (1979) Physics of minerals and inorganic materials: an introduction. Springer-Verlag, Berlin Heidelberg, p 340
Peyronel G (1956) The crystal structure of Baveno bazzite. Acta Cryst 9:181–186
Platonov AN, Taran MN, Pol’shin EV, Min’ko OY (1979) On the nature of colour of iron-bearing beryls. Izvestiya Akademii Nauk SSSR. Seriya Geologicheskaya 54–68 (in Russian)
Platonov AN, Pol’shin EV, Chistyakova MB, Taran MN (1981) Isomorphism of iron in bazzite from Kazakhstan pegmatites. Geokhimiya 393–398 (in Russian)
Pouchou JL, Pichoir F (1991) Quantitative analysis of homogeneous or stratified microvolumes applying the model “PAP”. In: Heinrich K, Newbury D (eds) Electron probe quantitation. Plenum Press, NewYork, pp 31–76
Price DC, Vance ER, Smith G, Edgar A, Dickson BL (1976) Mössbauer effect studies of beryl. J Physique 37:C6-811–C6-817
Rossman GR (2016) http://minerals.gps.caltech.edu/FILES/Visible/Beryl/Bazzite819_DHy-38_Naters.gif
Taran MN, Rossman GR (2001) Optical spectroscopic study of tuhualite and a re-examination of the beryl, cordierite, and osumilite spectra. Am Mineral 86:973–980
Taran MN, Klyakhin VA, Platonov AN, Pol’shin EV, Indutny VV (1989) Optical spectra of natural and artificial iron-containing beryls at 77–297 K. Soviet Physics. Crystallography 34:882–884
Viana RR, da Costa GM, De Grave E, Jordt-Evangelista H, Stern WB (2002) Characterization of beryl (aquamarine variety) by Mössbauer spectroscopy. Phys Chem Minerals 29:78–86
Wood DL, Nassau K (1967) Infrared spectra of foreign molecules in beryls. J Chem Phys 47:2220–2228
Wood DL, Nassau K (1968) The characterization of beryl and emerald by visible and infrared absorption spectroscopy. Am Mineral 53:777–800
Acknowledgements
The authors are thankful to Ievgen Naumenko (Kiev, Ukraine) who generously presented the sample of Norway bazzite for investigations. Also we are grateful to Elizabeth Sklute (South Hadley, MA, USA) for her assistance with Mössbauer data processing and Gerhard Franz (Berlin, Germany) for support with FTIR measurements. We thank two official reviewers, Thomas Armbruster and Lee A. Groat, for very reasonable comments and suggestions, which considerably improved the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

{kind=link}
Cite this article
Taran, M.N., Dyar, M.D., Khomenko, V.M. et al. Optical absorption, Mössbauer, and FTIR spectroscopic studies of two blue bazzites. Phys Chem Minerals 44, 497–507 (2017). https://doi.org/10.1007/s00269-017-0877-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-017-0877-2