
Abstract
We present the Raman and infrared spectra of different tourmaline species in the spectral range associated with the hydroxyl stretching modes, investigated through polarized Raman spectroscopy. Different lineshapes are observed for the OH spectra in uvite, fluor-uvite, magnesio-foitite, dravite and elbaite samples, and can be related to the coordination of OH in the two different structural V[O(3)]- and W[O(1)]-occupied sites. Local arrangements around the two different OH sites were assigned, and different ion substitutions for these five tourmaline species were identified. Our work with polarized Raman spectroscopy revealed that OH-stretching modes are described by totally symmetric, irreducible representations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The supergroup of tourmaline minerals forms a complex class of borosilicates with different compositions and colors, and occurrence in different geologic environments (Henry and Dutrow 1996). Tourmalines present a very complex chemical composition with the general chemical formula XY3Z6T6O18(BO3)3V3W (Hawthorne and Henry 1999). This supergroup of minerals presents a trigonal crystalline structure and belongs to the space group R3m (C 53v ). The crystal structure can be described by a ring formed by six tetrahedra of SiO4. Occasionally, Si on the site T can be replaced by boron (Rosenberg and Foit 1979; Gasharova et al. 1997; Henry et al. 2011). Triangular groups of BO3 are located in alternate positions above and below the tetrahedra of the ring. The tetrahedral rings are connected to each other by Z and Y octahedra. The Z-octahedron is formed by trivalent cations, such as Al3+, Cr3+ and V3+, or some bivalent cations, such as Mg2+ and Fe2+. The Y-site is in a nearly regular octahedron that can be occupied by many different cations, such as Li1+, Mg2+, Fe2+, Mn2+, Al3+, Cr3+, V3+, Fe3+ and Ti4+. The X-site is located in the center of the ring formed by 3-D framework of the ZO6 octahedra and is usually occupied by sodium, calcium, potassium, or it can also be unoccupied. The W [or O(1)] site in the structural formula is located along the c-axis in a central position in relation to the tetrahedral ring. This site can be occupied by OH1−, O2− or F1−. The three sites named V [or O(3)] are usually occupied by OH− but can also contain significant amounts of O2−. The general structure of tourmaline described above is represented schematically in Fig. 1, which indicates the different cationic sites: X (center), 3 Y (within the rings) and 6 Z sites (border), as well as the two different O sites: one O(1) (W, at the center) with C 3v symmetry and three O(3) sites (V, at the border) with C s symmetry. The octahedral Z-sites are smaller than the octahedral Y-sites. Due to the large number of possible substitutions in the different lattice sites, different types of tourmaline can be found in nature. Because of the many possible solid solutions, the tourmaline supergroup has more than 18 species according to the occupancy of the X, Y, Z and W sites (Henry et al. 2011).

Schematic representation of the general structure of tourmaline projected on the [001] basal plane [adapted from (Dietrich 1985)]
Raman spectroscopy is a well-established and widely applied experimental technique for the identification and study of structural properties of minerals (Griffith 1969; Alvarez and Coy-Yli 1978; Zhang and Scott 2007). However, to the best of our knowledge, there are few Raman studies on tourmaline (Alvarez and Coy-Yli 1978; Gasharova et al. 1997; McKeown 2008; Hoang et al. 2011; Zhao et al. 2012; Skoggby et al. 2012). Moreover, most Raman studies on tourmaline focus on the low-frequency features, associated with the metal ion–oxygen bond vibrations as they relate to their general crystal structure. Gasharova et al. (1997) investigated the Raman spectra of buerguerite–schorl, elbaite-species, and dravite–uvite, and proposed assignments for some of the Raman modes in accordance with the tourmaline structures. McKeown (2008) reported a detailed Raman analysis of the structure of heat-treated buerguerite, where the assignments of most of the low-frequency modes were obtained. Among the Raman studies on tourmaline, few papers have been devoted to the high-frequency features associated with the O–H stretching modes (3,300–3,900 cm−1). These features can provide important information about ionic substitutions in the different types of tourmaline, since OH vibration frequencies will be modified by the ion substitution in the X, Y and Z sites. Some infrared-spectroscopic studies investigating the O–H stretching modes for different tourmaline species have been reported and different spectral shapes for the OH-stretching band have been observed for different types of tourmaline. (Castañeda et al. 2000; Gonzalez-Carreno et al. 1988; Oliveira et al. 2002; Prasad and Sarma 2005; Reddy et al. 2007). However, the coordination of the V and W sites could not be completely established from the infrared studies. Once Raman and infrared spectroscopy are complementary techniques, the study of this region using Raman spectroscopy may clarify and provide complementary information about these minerals, such as the OH orientation and the more common cations substitutions around the V and W sites. Raman studies on the O–H stretching modes have been applied successfully, for example, in the investigation of F/OH substitution in topaz minerals (Pinheiro et al. 2002). Recently, a polarized Raman study of seven different natural tourmaline samples from Vietnam reported the symmetry assignment of the low-frequency metal ion-oxygen bonding vibrations and the O–H stretching vibrations (Hoang et al. 2011). In addition, Skoggby et al. (2012) reported a more detailed investigation of the O–H stretching mode where the short-range arrangements around the OH sites were assigned for an elbaite sample (Skoggby et al. 2012).
In this work, we present a systematic, polarized Raman and unpolarized infrared spectroscopy study of different species of natural tourmaline. We expanded the range of tourmaline compositions beyond those of Skoggby et al. (2012) to include eight different tourmaline samples from five different species: uvite [CaMg3(Al5Mg)(BO3)3Si6O18(OH)3OH], fluor-uvite [CaMg3(Al5Mg)(BO3)3Si6O18(OH)3F], dravite [NaMg3Al6(BO3)3Si6O18(OH)3OH], magnesio-foitite [□Mg3Al6(BO3)3Si6O18(OH)3(OH)] and elbaite [Na(Li,Al)3Al6(BO3)3Si6O18(OH)3OH].
Results related to the symmetry of the OH-stretching modes and the short-range environment around the OH sites for these five species of tourmaline samples were obtained from the polarized Raman experiments.
Materials and methods
Eight different tourmaline samples from three different Brazilian states were investigated. Two elbaite samples, green and pink, are from Paraíba and Minas Gerais states, respectively, and the other samples are from the Brumado district in Bahia state. The chemical compositions of all samples were determined by electron microprobe analysis in the wavelength dispersive mode (WDS) using a Jeol JXA8900RL electron microprobe. Analyses were made with an acceleration voltage of 15 kV and a current of 10 nA. For each sample, 10 points were measured and averaged. Table 1 shows the results obtained from the microanalysis of the samples. Analytical standards were: Si, quartz; Al, corrundum; Ti, rutile; Na, jadeite; Ca, anortite; K, microcline; Mg, periclase; F, fluorite; Fe, magnetite; Mn, rhodonite; V, V2O3; Cr, Cr2O3 and Zn, ZnO. Copper was analyzed, but was below the detection limit. Mn and Fe have been assumed to be divalent, while Cr and V are assumed to be trivalent. Li and B were not analyzed.
From the electron microprobe results, the atomic proportions in each sample were calculated based on 31 anions (O, OH and F). B2O3 and H2O were calculated by stoichiometry, with B = 3 apfu and (OH + F) = 4 apfu. Li2O was included in the calculation only for elbaite with the condition of Li = 15 − (T + Z + Y). Table 2 shows the atoms per unit formula, the occupancy in the X, Y and Z sites in each sample and also the proportion of F in the W site. All eight samples are thus classified according to the atomic proportions in Table 2 (Henry et al. 2011). Uvites are characterized by the dominant amount of Ca in the X site, Mg in the Y site, Al in the Z site, and OH in the W site, and fluor-uvites are distinguished from uvites by the presence of fluorine in the W site. Elbaite is distinguished from the other species by the absence of Mg in the Y site, and it is also distinguished from schorl by having a lower Fe concentration and high Li (calculated in Table 2) and Al. Dravite and magnesio-foitite are distinguished from each other according to the dominance of Na or vacancies, respectively, in the X site and magnesio-foitite by the dominance of Mg in the Y site. The fluor-uvite samples studied here present strong pleochroism, a dependence of the optical absorption on the crystallographic direction. One of them (sample F-U1) is pink in the crystallographic direction c and appears to be green on the ab plane. The other one (sample F-U2) is green in the ab plane and almost colorless when viewed from the c direction. The red color of samples D and U2 indicate the presence of Fe3+ (Taran and Rossman 2002). Actually, red and pink can also be related to the presence of Mn2+, but significant amounts of Mn were not observed from the microprobe analysis.
Polarized micro-Raman measurements were carried out on a Dilor XY spectrometer equipped with a liquid N2-cooled CCD detector. The samples were focused on an optical microscope by a 50 × (N.A. = 0.55) objective and excited with an argon-ion laser at two different wavelengths (λ = 514.5 and 488.0 nm). Half-wave plates placed in front the laser were used to rotate the incident light polarization to the appropriate direction. Infrared (IR) transmission measurements were performed on a Fourier transform (FT) Spectrometer (Nicolet, model Nexus 470), equipped with a globar (SiC) source, a Ge-coated KBr beamsplitter, and a liquid N2-cooled HgCdTe detector. The FTIR spectra were collected under nitrogen purge, by averaging 64 scans, with a pre-selected resolution of 2 cm−1. For these measurements, we used 3.0 mg of finely ground crystals in 300 mg of vacuum-pressed potassium bromide (KBr) pellets. Blank KBr pellets were used as reference spectra.
Results and discussion
Raman spectroscopy measurements
The tourmaline samples, characterized by electron microprobe analysis, were investigated by polarized Raman spectroscopy in two frequency ranges: 200–1,000 and 3,400–3,800 cm−1. The lower frequency range is related to the general features of the mineral associated with the metal–oxygen bonds, and can be used to characterize the uvite, F-uvite Mg-foitite, dravite and elbaite species. The higher frequency region is associated with the OH− stretching modes in the two different lattice sites, O(1) and O(3) (shown in Fig. 1).
Figure 2 shows the Raman spectra of the green magnesio-foitite, green uvite, and pink elbaite, in the spectral range 200–1,000 cm−1. All the spectra were obtained with incident and scattered light, polarized perpendicular to the c-axis direction. Many features associated with the different vibrational modes of the structure are observed. The most striking feature that distinguishes uvite/Mg-foitite/dravite from the elbaites series are the two peaks at 215 ± 2 and 244 ± 2 cm−1, originating mainly from the Mg–O bond stretching on the Y site. The substitution of Mg by Al and Li in the Y site in elbaites is responsible for the different shapes on these features. The feature at 374 ± 2 cm−1, present in both spectra is associated with the Al–O stretching vibration on the Z site (Gasharova et al. 1997). The features observed between 600 and 750 cm−1 are mainly associated with Si–O vibrations of the Si6O18 ring. In the case of the elbaite, these vibrations are influenced by the presence of Al3+ cations on the Y site. The interactions of the Si6O18 ring atoms with these cations increase the frequencies of the Si–O vibrations (Mihailova et al. 1996).

Raman spectra, in the low-frequency range, of the elbaite (E2), magnesio-foitite (Mg-F), and uvite (U1) samples
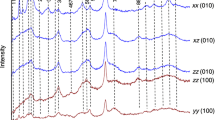
The main goal of the present work was the investigation of the hydroxyl sites and possible ion substitutions around them for the different tourmaline species, in addition to their dependence on the ion substitutions in the X, Y and Z sites. In view of that, a detailed Raman spectroscopy study of the O–H stretching bands around 3,500 cm−1 was performed. Figure 3 shows the Raman spectra in the high-frequency range, associated with OH-stretching mode, obtained for the green uvite, magnesio-foitite and pink elbaite samples, with the incident polarization parallel and perpendicular to the c-axis, zz and xz polarizations, respectively. In the spectra in Fig. 3, the OH-stretching features are only present for parallel polarizations along the c-axis (zz configuration). Thus, the OH-stretching mode must be associated with a totally symmetric A, irreducible representation. This reveals a preferential orientation of the hydroxyl ions in the W[O(1)] site along the c-axis. The hydroxyl ions must occupy the W[O(1)] and V[O(3)] sites of the general tourmaline structure. The W site is in the center of the SiO4 ring and belongs to the C 3v point group. The other hydroxyl is in the V site and its vibration is also associated with a totally symmetric, irreducible representation. In the case of O(3) sites, the hydroxyl must be oriented along the O(3)–O(5) direction, in order to be invariant under the symmetry operations of C 3v group. The same polarization behavior of the hydroxyl modes on the Raman spectra is observed for all uvite, fluor-uvite, magnesio-foitite, dravite, and elbaite samples.

Polarized Raman spectra in the region of the OH-stretching mode of a uvite (U1), b magnesio-foitite (Mg-F) and c elbaite (E2) samples
The lineshapes of the OH Raman stretching bands for the different tourmaline groups are clearly very different from each other, as shown in Fig. 4. For all samples, the OH bands can be decomposed into Gaussian lines with equal linewidths for all tourmaline species, but with different numbers of features and spectral frequencies. In principle, one would expect only two different frequencies for the OH vibrations, because in the tourmaline structure there are two different OH ions, occupying the W or V sites. However, due to the large number of possible cationic substitutions in the Y and Z sites, coordinated to the W and V sites, the OH-stretching frequencies may have slightly different values if the hydroxyl can be surrounded by different cations. This explains the fact that more than two Gaussian features are observed in the spectra. Different features are observed for the different tourmaline species. The frequency of the hydroxyl stretching vibration on the O(1) site is strongly influenced by the charge of the cation in the X site, and it is also coordinated by the three Y sites. Similar arguments for the incorporation of F in the W site were made in the paper of Henry and Dutrow (2011). On the other hand, the OH on the O(3) site is coordinated by one Y site and two Z sites, and its frequency depends on the charge of the cations on these sites. Indeed, the frequency of the OH vibration follows an inverse relation with the sum of the charge of cations coordinated to the hydroxyl site (Martínez-Alonso et al. 2002).

Gaussian fit of the OH-stretching Raman mode of the eight tourmaline samples: a uvite (U1), b uvite (U2), c fluor-uvite (F-U1), d fluor-uvite (F-U2), e magnesio-foitite (Mg-F), f dravite (D), g elbaite (E1) and h elbaite (E2)
From the data of Table 2, the probabilities of simultaneous occupation of the sites Y and 2Z coordinating the V site were obtained by the expression \(P\left( \% \right) = \left( {\frac{{x_{Y} }}{3} \cdot \frac{{x_{Z1} }}{6} \cdot \frac{{x_{Z2} }}{6}} \right) \times 100\), where x Y , x Z1 and x Z2 are the atomic occupation of the Y and the two Zs coordinating the V site. The results are presented in Table 3. The calculated probabilities were compared with the integrated area of the Gaussian lines obtained from the fitting of the OH-stretching modes in Fig. 4. Thus, the assignment of the features in the OH Raman spectrum was obtained based on the relation between intensity and wavenumber, respectively, with probability of occupation and the cationic charge.
For example, in the case of uvites, the Y site can be occupied mostly by Mg, and the Z site mostly by Al. Thus, the highest probability of occupation is YMgZAlZAl, and it must be associated with the main features in the uvite spectra. In the uvite U1, the Z site can also be occupied by Mg. Thus, two other possibilities of occupation that can be associated to features in the spectrum are YMgZAlZMg and YMgZMgZMg, both located at lower wavenumbers related to the first one. All the other ion combinations happen with very low probability and cannot be associated with the features observed in the spectra. The same kind of analysis was performed for all the samples.
Based on the analysis described above and on the cationic occupation on X, Y and Z sites, an assignment is made for the different frequencies observed for the OH-stretching vibrations. For uvites, the main feature in the spectra (~3,592 cm−1) must be associated with the occupation of the Y site by Mg2+ and the two Z sites by Al3+ (YMgZAlZAl). The higher frequency feature in both spectra, 3,645 cm−1 for dravite and 3,660 cm−1 for elbaite, are associated with hydroxyl located at the O(1) site. This frequency is mainly dependent on the occupation of the X site, which is mostly occupied by Ca2+, in the case of uvite, and by Na+ in the case of dravite and elbaite. The higher cationic charge of Ca2+ is responsible for the lower frequency of this vibrational mode in uvite to 3,635 cm−1. The difference in the frequencies for dravite/Mg-foitite and elbaite is associated with the occupation of the Y sites, since the O(1) is coordinated by three Y sites. These sites in elbaite can be occupied by Li+ and Al3+, and by Mg2+ in the case of dravite/Mg-foitite. The additional high-frequency feature at 3,774 cm,−1 observed in the spectra of dravite/Mg-foitite, may be associated with an unoccupied X site.
The O(3) sites are coordinated by one Y and two Z sites. In the case of the uvite, these sites can be occupied by Mg2+ and Al3+ or Mg2+, respectively. Thus, there are three different possibilities of coordination of the O(3) sites in the uvite: ZAlZAlYMg, ZAlZMgYMg and ZMgZMgYMg. These three possibilities of occupation are responsible for the three frequencies ν2, ν3 and ν4 (see Table 2), related to the vibrations of the OH located on the O(3) sites. The observed band positions in all spectra and coordination assignments are given in Table 4. In the case of dravites and magnesio-foitite, the Y and Z sites are mostly occupied by Mg2+ and Al3+, respectively. However, the different features observed in the spectra may be associated with another cationic substitution such as Fe3+, also present in these samples. Finally, in the elbaites, the Y and Z sites can be occupied by Li+ or Al3+ and Al3+, respectively, originating two possible arrangements around the O(3) site: ZAlZAlYLi and ZAlZAlYAl. These two possible arrangements are responsible for the two strong features observed in the OH vibration spectra at 3,478 and 3,593 cm−1. Such observed spectra and the dependence of the OH-stretching frequencies on the short-range arrangements around W and V for elbaites are in agreement with recently reported results for this type of tourmaline (Skoggby et al. 2012). Similar assignments have also been observed for elbaite–schorl series obtained by infrared spectroscopy (Castañeda et al. 2000). Additional weak features observed in the elbaite spectra in the present case may be associated with the presence of Fe2+ substitutions, especially in the case of the green elbaite (see Table 2). All the observed band positions and possible coordination assignments for the eight samples studied here are summarized in Table 4.
Note that the OH band in dravite/magnesio-foitite extends from ~3,480 to ~3,630 cm−1, while in uvite it extends from ~3,520 to ~3,630 cm−1. In addition, the OH band in dravite/magnesio-foitite is broadened and more Gaussian lines are necessary to fit the experimental spectra. This can be understood by considering that for these species, the W site is typically occupied only by OH, and according to our chemical analysis there is no F in these samples. The presence or absence of F can be observed on the OH Raman spectra by the feature identified as ν5 or ν6 in Table 4, which is associated with the OH in the W site that can be occupied by F−. This feature is very strong in the case of dravite and magnesio-foitite samples where there is no F, its intensity is reduced in uvites and elbaites, and this feature is not present in the spectra of the fluor-uvites, where higher amounts of F− ions substituting OH− are present. The intensities of ν6 and ν7 features are also reduced or even negligible in uvites and fluor-uvites due to the OH/F substitutions. A similar result was observed for OH/F substitution in topaz minerals (Pinheiro et al. 2002), where the intensity of the OH-stretching Raman mode could be associated with the extension of F/OH substitutions.
Comparing the spectra of the different uvite samples with each other, it is observed that spectra of samples that present pleochroic effect (samples F-U1 and F-U2) are slightly narrower than those of samples U1 and U2 where that effect is not observed, and can be fitted by using only three Gaussian lines. This result suggests a lower degree of chemical cationic disorder in these F-uvite samples, which may be responsible for their strong pleochroism. However, further studies regarding the physical mechanism responsible for the pleochroism are still necessary.
Infrared spectroscopy measurements
In good agreement with the Raman results, the profiles of the infrared absorption bands of the hydroxyl groups also show characteristic features for each particular group of tourmalines. Figure 5 shows the infrared absorption spectra, in the OH vibrational range, for the uvite, fluor-uvite, magnesio-foitite, dravite and elbaite samples studied here. The similarity between the lineshapes of FTIR and Raman OH bands is clearly seen by comparing Figs. 4 and 5. As in the case of Raman bands, the infrared OH bands of the dravite and magnesio-foitite samples are broadened in comparison with other tourmalines, and requires at least one additional Gaussian feature for the spectral fitting. Moreover, the integrated areas of the OH bands for fluor-uvites are lower because of the higher amount of F in these samples.

Infrared absorption spectra in the OH-stretching frequency range of the different uvite, fluor-uvite, magnesio-foitite, dravite and elbaite samples
Conclusions
Polarized Raman spectroscopy allowed us to identify the symmetry of the OH vibrational-stretching modes in five different species of tourmaline: uvite, fluor-uvite, magnesio-foitite, dravite and elbaite, and revealed that these vibrational modes are oriented along the crystal c-axis. The profiles of the OH features for the investigated tourmaline series are very different from each other because of the many possibilities of cationic substitutions in Y and Z sites. Moreover, we observed a higher integrated area for the infrared OH-stretching modes of dravite and magnesio-foitite, suggesting a higher amount of OH in these types of tourmaline, and, consequently a lower OH/F substitution in them as well. This result is consistent with the broader Raman and IR bands for this material. Finally, narrower OH bands are observed for pleochroic uvites, which are classified as fluor-uvites, when compared with other uvite samples. This result suggests that the pleochroism observed in this type of tourmaline is somehow related to a lower degree of chemical disorder in its structure.
References
Alvarez MA, Coy-Yli R (1978) Raman-spectra of tourmaline. Spectrochim Acta A 34:899–908
Castañeda C, Oliveira EF, Gomes N, Soares ACP (2000) Infrared study of OH sites in tourmaline from elbaite–schorl series. Am Mineral 85:1503–1507
Dietrich RV (1985) The tourmaline group. Van Nostrand Reinhold Company, New York
Gasharova B, Mihailova B, Konstantinov L (1997) Raman spectra of various types of tourmaline. Eur J Mineral 9:935–940
Gonzalez-Carreno T, Fernandez M, Sanz J (1988) Infrared and electron microprobe analysis of tourmalines. Phys Chem Minerals 15:452–460
Griffith WP (1969) Raman spectroscopy of minerals. Nature 224:264–266
Hawthorne FC, Henry DJ (1999) Classification of the minerals of the tourmaline group. Eur J Mineral 11(2):201–215
Henry DJ, Dutrow BL (1996) Metamorphic tourmaline and its petrologic applications. In: Grew ES, Anovitz LM (eds) Boron: mineralogy, petrology and geochemistry. Mineralogical Society of America, Washington, pp 503–557
Henry DJ, Dutrow BL (2011) Incorporation of fluorine in tourmaline: external environmental vs. internal crystallographic influences. Can Mineral 49:41–56
Henry DJ, Novák M, Hawthorne FC, Ertl A, Dutrow BL, Uher P, Pezzota F (2011) Nomenclature of tourmaline-supergroup minerals. Am Mineral 96:895
Hoang LH, Hien NTM, Chen XB, Minh NV, Yang I-S (2011) Raman spectroscopic study of various types of tourmalines. J Raman Spectrosc 42:1442–1446
Martínez-Alonso S, Rustad JR, Goetz AFH (2002) Ab initio quantum mechanical modeling of infrared vibrational frequencies of the OH group in dioctahedral phyllosilicates. Part II: main physical factors governing the OH vibrations. Am Mineral 87:1224–1234
McKeown DA (2008) Raman spectroscopy, vibrational analysis, and heating of buerguerite tourmaline. Phys Chem Minerals 3:259–270
Mihailova B, Gasharova B, Konstantinov L (1996) Influence of non-tetrahedral cations on Si–O vibrations in complex silicates. J Raman Spectrosc 27:829–833
Oliveira EF, Castañeda C, Eeckhout SG, Gilmar MM, Kwitko RR, Grave ED, Botelho NF (2002) Infrared and Mössbauer study of Brazilian tourmalines from different geological environments. Am Mineral 87:1154–1163
Pinheiro MVB, Fantini C, Krambrock K, Persiano AIC, Dantas MSS, Pimenta MA (2002) OH/F substitution in topaz studied by Raman spectroscopy. Phys Rev B 65:104301
Prasad PSR, Sarma DS (2005) Study of structural disorder in natural tourmalines by infrared spectroscopy. Gondwana Res 8:265–270
Reddy BJ, Frost RL, Martens WN, Wain DL, Kloprogge T (2007) Spectroscopic characterization of Mn-rich tourmalines. Vib Spectrosc 44:42–49
Rosenberg PE, Foit FE (1979) Synthesis and characterization of alkali-free tourmaline. Am Min 64:180–186
Skoggby H, Bosi F, Lazor P (2012) Short-range order in tourmaline: a vibrational spectroscopic approach to elbaite. Phys Chem Minerals 39:811–816
Taran MN, Rossman GR (2002) High-temperature, high-pressure optical spectroscopy study of ferric iron-bearing tourmaline. Am Mineral 87:1148–1153
Zhang M, Scott JF (2007) Raman studies of oxide minerals: a retrospective on cristobalite phases. J Phys: Condens Matter 19:275201
Zhao C, Liao L, Xia Z, Sun X (2012) Temperature-dependent Raman and infrared spectroscopy study on iron-magnesium tourmalines with different Fe content. Vib Spectrosc 62:28–34
Acknowledgments
The authors acknowledge financial support from the Brazilian agencies CNPq and CAPES, the Minas Gerais state agencie FAPEMIG and VALE S.A. We are very grateful to L. Garcia from the Physics Department—UFMG—for the microprobe analyses.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fantini, C., Tavares, M.C., Krambrock, K. et al. Raman and infrared study of hydroxyl sites in natural uvite, fluor-uvite, magnesio-foitite, dravite and elbaite tourmalines. Phys Chem Minerals 41, 247–254 (2014). https://doi.org/10.1007/s00269-013-0642-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-013-0642-0