
Abstract
The thermoelastic parameters of the CAS phase (CaAl4Si2O11) were examined by in situ high-pressure (up to 23.7 GPa) and high-temperature (up to 2,100 K) synchrotron X-ray diffraction, using a Kawai-type multi-anvil press. P–V data at room temperature fitted to a third-order Birch–Murnaghan equation of state (BM EOS) yielded: V 0,300 = 324.2 ± 0.2 Å3 and K 0,300 = 164 ± 6 GPa for K′ 0,300 = 6.2 ± 0.8. With K′ 0,300 fixed to 4.0, we obtained: V 0,300 = 324.0 ± 0.1 Å3 and K 0,300 = 180 ± 1 GPa. Fitting our P–V–T data with a modified high-temperature BM EOS, we obtained: V 0,300 = 324.2 ± 0.1 Å3, K 0,300 = 171 ± 5 GPa, K′ 0,300 = 5.1 ± 0.6 (∂K 0,T /∂T) P = −0.023 ± 0.006 GPa K−1, and α0,T = 3.09 ± 0.25 × 10−5 K−1. Using the equation of state parameters of the CAS phase determined in the present study, we calculated a density profile of a hypothetical continental crust that would contain ~10 vol% of CaAl4Si2O11. Because of the higher density compared with the coexisting minerals, the CAS phase is expected to be a plunging agent for continental crust subducted in the transition zone. On the other hand, because of the lower density compared with lower mantle minerals, the CAS phase is expected to remain buoyant in the lowermost part of the transition zone.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The CAS phase is a Ca-rich aluminosilicate that was first observed with the composition Ca0.8Al3.6Si2.4O11 (with a small amount of Na2O and K2O) in the decomposition product of sediments and continental crust at P, T conditions of the mantle transition zone (Irifune et al. 1994). Later, experiments also found CAS as a liquidus phase in melting experiments on mid-ocean ridge basalt (MORB) compositions at 26–27 GPa and ~2,500 K (Hirose and Fei 2002; Wang and Takahashi 1999). According to the previous studies, the CAS phase could represent up to 10 vol% of the subducted continental crust or 30 vol% of the solid fraction of partially molten MORBs in the deep mantle.
The Ca-rich end-member of CAS was first observed with composition CaAl4Si2O11 in the high P, T assemblage of CaAl2Si2O8 anorthite at 14 GPa and ~1,800 K (Gautron et al. 1996). The calcium end-member is also reported as an accessory phase in solid–solid reactions of formation of actinide-bearing Ca-perovskites at 18 GPa and 2,000 K (Gautron et al. 2006; Gréaux et al. 2009). A natural occurrence of the Na-rich CAS phase with the composition Ca0.29Na0.83Al2.91Si2.95O11 has been discovered in heavily shocked Martian meteorites (Beck et al. 2004). Thus, the CAS phase might be an important constituent mineral of sediments and basalts subducted into the deep mantle, and therefore, the description of its thermoelastic behavior is important to infer the appropriate mineralogical model of basaltic and continental crust subducted at the depths of the mantle’s transition zone.
The CAS phase with the composition CaAl4Si2O11 is known to have a “barium ferrite”-type hexagonal structure with the P6 3 /3mmc space group (Gautron et al. 1999). The structure consists of one dodecahedral site occupied by Ca, and two octahedral (M1, M2) and one trigonal bipyramidal (T) sites that are occupied by Al and Si. A recent determination of Si–Al distribution in CaAl4Si2O11 phase synthesized at 20 GPa and 1,700–1,900 K showed a disordered Si–Al distribution in the M1 and T sites, whereas M2 site is fully occupied by Al (Xue et al. 2009).
Only a few studies have been devoted to the CAS phase at high P and T. Only recently, the high P, T phase relations (up to 23 GPa and 2,100 K) and the stability limit of CaAl4Si2O11 were investigated by Ishibashi et al. (2008), Zhai and Ito (2008), and Akaogi et al. (2009), showing that the CAS phase becomes stable at about 12–14 GPa and ~1,600 K. The dissociation of the CAS phase to an assemblage of CaSiO3 perovskite, Al2O3 corundum, and SiO2 stishovite was reported above 30 GPa and 2,000 K by Ishibashi et al. (2008) who used a laser-heated diamond anvil cell. Ono et al. (2005) reported the room temperature compressibility of the CAS phase up to 44 GPa, using a laser-heated diamond anvil cell.
Here, we report the pressure–volume–temperature (P–V–T) relations of the CAS phase based on multi-anvil press experiments coupled with energy-dispersive X-ray diffraction (EDXRD) using synchrotron radiation at beamline BL04B1 (SPring-8, Japan). The use of a large-volume apparatus allowed a precise control of the experimental conditions over a large range of pressure and temperature (Rubie 1998). Energy-dispersive powder diffraction pattern profiles were measured in situ at high pressure and at high temperature. Our results are compared with previous data and discussed with respect to a model of continental crust subducted to the P, T conditions of the mantle transition zone.
Experimental methods
A glass of CaAl2Si2O8 anorthite composition was first prepared by mechanical mixing of commercial oxides in molar proportions, CaCO3 Al2O3 2SiO2, and placed for 24 h at 1,173 K in order to remove any carbonates. The mixture was then melted at ~1,700 K (Osborn and Muan 1960), which after quenching produced a homogeneous glass with the following composition (in wt%): CaO, 19.60; Al2O3, 35.87; SiO2, 45.04 (i.e. Ca0.96Al1.93Si2.06O8). The starting material for high-pressure experiments was prepared by mixing the anorthite glass with Al2O3 in the molar proportions: 1CaAl2Si2O8 1Al2O3.
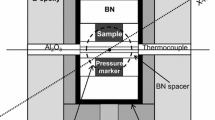
In situ high-pressure and high-temperature experiments were carried out using a Kawai-type multi-anvil press apparatus, SPEED-1500, located at beamline BL04B1 at the synchrotron radiation facility, SPring-8 (Hyogo Pref., Japan). High pressure was generated within the so-called 8/3 assemblies, which consist of a Co-doped MgO octahedron pressure medium, with a side length of 8.0 mm, and tungsten carbide anvils with a 3.0-mm truncation edge length, supported by pyrophilite gaskets. A cylindrical TiB2-doped BN heater that is transparent to X-rays was equipped with LaCrO3 thermal insulators and molybdenum electrodes, in order to generate high temperature during the HP experiments (see details in Fig. 1). Temperature was measured by a W97Re3-W75Re25 thermocouple, which was prevented from touching the furnace by Al2O3 tubes on both sides. The thermocouple was maintained between the two capsules and protected by MgO powder that also served as soft pressure-transmitting medium inside of the furnace. Capsules consisted of cylindrical graphite with gold foils at each end. Powder of the starting material was mixed with gold powder (~10/1 wt). Pressure was calibrated using the equation of state of gold proposed by Tsuchiya (2003).

Schematic drawing of the experimental cell assembly. Dashed lines represent the path for the thermocouple through the pressure medium and furnace
Four experiments were performed at high pressure and high temperature, in the P, T range of 0.0–23.7 GPa and 300–2,100 K (see Fig. 2; Table 1). The general procedure for each heating cycle was to compress the sample and then heat up to a target temperature. Data were collected at high temperature by intervals of 200 K, in the P, T stability field of the CAS phase, following previous studies (see Fig. 2 and references therein). The samples were then quenched to room temperature and heated a second time up to 700 K while data were collected in steps of 200 K. The aim of this procedure was to avoid the decomposition of the CAS phase at intermediate temperatures (~800–1,400 K) to the phase assemblage made of grossular garnet+ stishovite+ corundum (Akaogi et al. 2009; Zhai and Ito 2008). The next heating cycle started after the sample was quenched to room temperature, and the press load increased in order to reach higher pressure.

Pressure–temperature conditions during in situ measurements at high P, T. Each symbol represents a P, T condition where XRD pattern of the CAS phase was collected (yellow S2210, red S2402, green S2458, blue S2460). Solid and dashed lines indicate the transition boundaries of CaAl4Si2O11 determined, respectively, by Akaogi et al. (2009), and Zhai and Ito (2008). Dotted line represents the high-pressure stability limit of the CAS phase as reported by Ishibashi et al. (2008). Gr Ca3Al2Si3O12 grossular garnet, Ky Al2SiO5 kyanite, Cor Al2O3 corundum, St SiO2 stishovite, CAS CaAl4Si2O11 CAS phase, CPv Al-bearing CaSiO3 perovskite
Energy-dispersive X-ray diffraction (EDXRD) data at HP-HT were collected at the beamline BL04B1 at SPring-8 (project 2009A1300). Ge-solid detector was calibrated using characteristic fluorescence lines of metal standards (Cu, Mo, Ag, Ta, Pt, Au, and Pb). In all the measurements, we used a 2-theta angle of 6°, which was calibrated prior to experiments from the diffraction lines of Au. Collection time for each XRD pattern was about 300 s. Unit-cell parameters of recovered samples were also examined using a micro-focused X-ray diffractometer, Bruker M21X-SRA (Model No 3100-VR), equipped with a Cu rotating target, at the Geodynamics Research Center (Ehime University). The X-ray diffractometer was operated at 40 kV and 200 mA, and the profiles were collected for a 2-theta range of 20°–90°, for approximately 30 min for each pattern. Analysis of the X-ray diffraction patterns was carried out by means of the Le Bail multiphase profile-fitting technique implemented in the EXPGUI/GSAS software package (Le Bail et al. 1988; Larson and Von Dreele 2000; Toby 2001). Figure 3 shows two representative diffraction patterns collected in situ at high P, T conditions (Fig. 3a) and after quenching at room P, T conditions (Fig. 3b), respectively. The diffraction patterns are well explained by diffraction peaks of the CAS phase, stishovite, and gold. In all our diffraction profiles, we observed small peaks of stishovite coexisting with the CAS phase at high P, T. The relative peak intensities of both phases indicate that the CAS is the major phase in all samples and that stishovite can be considered as an accessory phase in the present high P, T experiments. The lattice parameters of the CAS phase measured in the quench samples are in good agreement with the literature (see Table 2 and references therein).

Two examples of diffraction patterns collected in the same sample a in situ at high pressure and high temperature (24 GPa, 1,700 K) and b after quenching to room P, T conditions (0 GPa, 300 K). CAS CAS phase, St stishovite, G gold diffraction lines, F gold fluorescence lines, *unidentified peaks
The quenched samples were examined using a JEOL JSM-5610LV scanning electron microscope (SEM) equipped with an energy-dispersive X-ray spectrometer (EDXS). The average chemical composition of the CAS phase in all the run products was Ca0.99Al3.94Si2.04O11, which is consistent with that reported by previous studies (Gautron et al. 1996; Ono et al. 2005; Zhai and Ito 2008).
Results and discussion
Pressure–volume data at room temperature
P–V data between 0.0 and 18.6 GPa, weighted by the uncertainties in pressure and volume, were fitted with a third-order Birch–Murnaghan equation of state (BM3 EOS), with the general form as follows:
where K 0,300, K′ 0,300, and V 0,300 are the isothermal bulk modulus, its pressure derivative, and the unit-cell volume at room P, T conditions, respectively. Figure 4 shows the refined isothermal compression curve at room temperature. Fitting the Eq. 1 with K 0,300, K′ 0,300 and V 0,300 as free parameters, we obtain a bulk modulus of K 0,300 = 164 ± 6 GPa, with its first pressure derivative K′ 0,300 = 6.2 ± 0.8 and V 0,300 = 324.2 ± 0.2 Å3 (see Table 3). When K′ 0,300 is fixed to 4.0, the fitting gives K 0,300 = 180 ± 1 GPa and V 0,300 = 324.0 ± 0.1 Å3. The refined room P volume is similar to that of Ono et al. (2005) (V 0 = 324.7 ± 0.8 Å3) and also to XRD measurements from the current and various studies on samples quenched from high P and T (see Table 2 and references therein). However, the unit-cell volumes measured at high pressure and the bulk modulus values derived from the BM3 EOS show a significant disagreement with those of Ono et al. (2005), who reported a refined K′ 0,300 of 4.7 ± 0.7 for K 0,300 equal to 229 ± 9 GPa.

P–V data for the CAS phase at room temperature and the isothermal curve at 300 K obtained by a third-order Birch–Murnaghan equation of state fit (V 0,300 = 324.2 ± 0.2 Å3 and K 0,300 = 164 ± 6 GPa for K′ 0,300 = 6.2 ± 0.8). Error bars are of the same size of the symbols. P–V data (black dots) and isothermal curve (dashed line) at 300 K obtained by Ono et al. (2005) are also shown for comparison (V 0,300 = 324.7 ± 0.8 Å3 and K 0,300 = 229 ± 9 GPa for K′ 0,300 = 4.7 ± 0.7)

Unit-cell volume of the CAS phase as a function of pressure and temperature. Error bars are of the same size of the symbols. The solid and dashed lines represent the HT-Birch–Murnaghan equation of state fit at 300, 500, 700, 1,500, 1,700, 1,900, and 2,100 K, respectively, with the following parameters: V 0,300 = 324.2 ± 0.1 Å3, K 0,300 = 171 ± 5 GPa, K′ 0,300 = 5.1 ± 0.6, α0,300 = 3.09 ± 0.25 × 10−5 K−1, and (∂K 0,T /∂T) P = −0.023 ± 0.006 GPa K−1 (RMS misfit = 0.218 GPa)
Ono et al. (2005) calculated the compressibility of the CAS phase based on 7 data-points collected during one run up to 44 GPa. In the present study, 38 data-points were collected inside and outside the stability field of the CAS phase over 4 different runs at high P, T (see Fig. 2). Based on our room temperature data, the CAS phase would be ~25% more compressible than that reported by Ono et al. (2005). One of the possibilities to explain such an inconsistency could be the different P-calibrant. Ono et al. (2005) used platinum and its EOS derived by Holmes et al. (1989), whereas we used gold and its EOS proposed by Tsuchiya (2003). Another possibility is the differential stress in the diamond anvil cell, which could have a significant effect on the compressibility measurements of the CAS phase. However, Ono et al. (2005) claimed that their experimental conditions were near hydrostatic conditions up to 44 GPa after annealing their sample at about ~2,000 K. It is then difficult to discuss the different results obtained in this study and those of Ono et al. (2005), and therefore, alternative methods (i.e. Brillouin scattering and high-pressure interferometry) are needed to resolve the current discrepancies.
P–V–T data and thermoelastic parameters
The pressure–volume–temperature relations were described with a modified HT-Birch–Murnaghan equation of state, with the following form:
In Eq. 2, we assumed that the bulk modulus P-derivative (K′ 0,T ) is constant with temperature, and therefore K′ 0,T = K′ 0,300. The thermal dependence of the unit-cell volume (at ambient pressure) is given by the thermal expansion coefficient α 0,T (Eq. 3), with the empirical assumption: α0,T = a 0 + b 0 T. The temperature dependence of the bulk modulus is expressed by a linear function of the temperature, assuming the T-derivative (∂K 0,T /∂T) P to be constant (Eq. 4).
Our P–V–T data were fit using the Eq. 2, refining all 6 parameters V 0,300, K 0,300, K′ 0,300 (∂K 0,T /∂T) P , a 0 and b 0 simultaneously. Figure 5 shows the isothermal compression curves at different temperatures, and Table 4 lists the thermoelastic parameters and the root-mean-square (RMS) misfit in pressure. The values of K 0,300 = 171 ± 5 GPa and K′ 0,300 = 5.1 ± 0.6 differ slightly from those determined from the fit at room temperature. The large uncertainties of b 0 (Eq. 3) is due to the lack of data-points in the range 700–1,700 K. Unfortunately, this issue cannot be solved experimentally due to the relatively narrow P, T stability field of the CAS phase.
Variations of a- and c-axis against P and T
Figure 6 shows the compression curves of the lattice parameters at different temperatures. The “linearized” HT-Birch–Murnaghan equation of state was used to fit the data, assuming that K′ 0,T is constant with temperature, as described in the previous section. All the thermoelastic parameters were not resolvable simultaneously, so we fixed K′ 0,300 = 5.1, which corresponds to the value obtained for the HT-BM EOS fit of the unit-cell volume. Table 5 summarizes the compressibility and thermal expansivity of the a- and c-axis. To avoid confusion, the thermal expansion was rewritten as s0,T = x 0 + y 0 T.

Variation of the normalized lattice parameters a/a 0 (circle symbols) and c/c 0 (square symbols) as a function of pressure and temperature. Error bars are of the same size of the symbols. The solid and dashed lines represent the linearized HT-Birch–Murnaghan equation of state fit at 300, 500, 700, 1,500, 1,700, 1,900, and 2,100 K, respectively
The calculated unit-cell parameters at zero pressure and room temperature are in good agreement with the XRD data obtained from the quenched samples and with the data reported in the literature (see Table 2 and references therein). The values of the “linearized” bulk moduli at room temperature suggest that the c-axis is remarkably more compressible than the a-axis. These results agree well with the previous experimental observations at high pressure and room temperature (e.g. Ono et al. 2005). Figure 7 shows the variation against temperature of the isothermal bulk modulus, K 0,T (Fig. 7a), the thermal expansion, α0,T (Fig. 7b), and the thermal pressure estimated by P Th = α 0,T × K 0,T × T (Fig. 7c), for the a- and c-axis. When temperature increases, our data show that the a-axis softens faster than the c-axis (Fig. 7a). However, even at high temperature, the c-axis remains more compressible than the a-axis. The c-axis has a larger thermal expansivity than the a-axis, but this difference decreases considerably at high temperature (Fig. 7b). The calculated thermal pressures are higher for the a-axis at any temperature. These results suggest that the stiffness of the a-axis is significantly larger than the c-axis both in compression and in thermal expansion.

a Isothermal bulk modulus K 0,T = K 0,300 + (∂K 0,T /∂T) P × (T–300), b thermal expansion α 0,T = x 0 + y 0 × T, and c thermal pressure P Th = α 0,T × K 0,T × T against temperature for the lattice parameters of the CAS phase. Solid and dashed lines represent the a-axis and the c-axis, respectively
The different behaviors of the unit-cell axes under high P, T could be explained on the basis of the crystal structure of the CAS phase, and in particular by the linkage of (Si,Al)O6 octahedra. The spinel-like sheet of M1 octahedra forms, along the a-axis, a column of edge-sharing octahedra that are expected to be stiff and therefore less flexible at high P, T than the corner-sharing network of M1 and M2 octahedra along the c-axis. Also, the CAS phase displays two half-occupied tetragonal sites, which form a trigonal bipyramid (T-site) oriented parallel to the c-axis. Displacement of (Si, Al)-atoms in the mirror plan of the T sites are also expected to favor the compressibility along the c-axis. Note that such a mechanism would trigger the transition of Si and/or Al to a fivefold coordination. Silicon coordinated by five oxygen atoms is believed to play an important role in the transport properties of minerals through the formation of oxygen vacancies (e.g. Angel et al. 1996).
Implications for subducted continental crust
We calculated density of a model of hypothetical subducted continental crust containing 10 vol% CaAl4Si2O11, using the CAS phase’s EOS parameters obtained in the present study and those of other constituent minerals proposed by previous studies. Former studies have shown that the proportion of minerals in subducted slabs might significantly vary depending on temperature, pressure, and bulk composition (Irifune et al. 1994; Ono et al. 2001; Rapp et al. 2008). Yet, it is difficult to provide quantitative analyses on subducted slabs as their compositions and temperature might significantly vary from one region to another. Irifune and coauthors calculated that under conditions corresponding to the mantle transition zone, the continental crust is mainly composed of K-hollandite, majorite garnet, stishovite, and some amount of CAS phase (~10 vol%).
We first calculated densities of a hypothetical continental crust (HCC), which consists of stishovite, majorite, and K-hollandite with 1:1:1 ratio in volume (i.e. continental crust minus CAS composition). At P > 20 GPa, CaSiO3 perovskite is gradually exsolved from the majorite garnet (Saikia et al. 2008) and would constitute ~6 vol% of the continental crust (Irifune et al. 1994). Therefore, at P > 20 GPa, the HCC model becomes stishovite, majorite, K-hollandite, and Ca-perovskite with the ratio 1:0.8:1:0.2 in volume. We calculated densities of a model of continental crust (CC) based on the combination of the HCC model with constituent minerals of the CaAl4Si2O11 system with the ratio 0.9:0.1 in volume (e.g. Irifune et al. 1994). In this model, we assumed that the formation and proportions of CAS phase and Ca-perovskite are unrelated, and therefore, those two Ca-rich silicates could coexist in a large range of P and T as proposed by a recent study (Gréaux et al. 2011b).
Previous studies showed that the phase relation of the CaAl4Si2O11 system present three domains: (1) grossular garnet, kyanite, and corundum below ~14 GPa; (2) grossular garnet, stishovite, and corundum above 14 GPa and below 1,400 K; and (3) the CAS phase at above 14 GPa and 1,400 K. Thus, the triple point of the CaAl4Si2O11 system is located at 14.7 GPa and ~1,550 K (Akaogi et al. 2009; Zhai and Ito 2008). Here, we adopted the phase boundaries calculated by Akaogi et al. (2009) using thermodynamics data (see Fig. 8b). In their calculations, they also proposed that the CAS phase dissociates to a mixture of Al-bearing Ca-perovskite, stishovite, and corundum at ~25 GPa and T > 1,500 K, which is consistent with the previous experimental determination of Ishibashi et al. (2008).

a Calculated density profiles of a hypothetical continental crust + 10 vol% CaAl4Si2O11 at pressure conditions of the mantle transition zone, for the geotherms of an “average” mantle (Brown and Shankland 1981; red solid line), a hot subduction (Thompson 1992; purple solid line) and a cold subduction (Thompson 1992; blue solid line). The densities of the CAS phase are shown (dashed line) for the mantle geotherm. The black heavy solid line represents a 1-dimensional density profile that is plotted after the Preliminary Reference Earth Model (PREM, Dziewonski and Anderson 1981). b Pressure–temperature–composition relation along the geotherms shown in Fig. 7a. The phase relations (black solid lines) are from Akaogi et al. (2009). Gr Ca3Al2Si3O12 grossular garnet, Ky Al2SiO5 kyanite, Cor Al2O3 corundum, St SiO2 stishovite, CAS CaAl4Si2O11 CAS phase, CPv CaSiO3 perovskite
Three density profiles are proposed here, which differ by their temperature profiles and mineral proportions. We first assumed that minerals are thermally equilibrated with the surrounding mantle and used the geotherm proposed by Brown and Shankland (1981). Then, we used two geotherms of cold and hot subducted slabs, respectively, which are based on the temperature profiles proposed by Thompson (1992). For comparison, we plotted the densities of the CAS phase along the mantle geotherm as well as a seismological 1D density profile (PREM, Dziewonski and Anderson 1981).
Table 6 summarizes the thermoelastic parameters used in our calculation. In the present models, we assumed pure end-member compositions for all the phases and used the thermal EOS parameters determined in the present study for the CAS phase and those obtained in previous studies for the grossular garnet (Gréaux et al. 2011a), kyanite (Fei 1995; Yang et al. 1997), stishovite (Nishihara et al. 2005), corundum (Hama and Suito 2001), K-hollandite (Nishiyama et al. 2005), and Ca-perovskite (Wang et al. 1996). It is known that the composition of majorite garnet changes with pressure (Irifune et al. 1994); indeed, we chose and employed the EOS parameters of majorite garnet, proposed by Wang et al. (1998).
According to the phase boundaries proposed by Akaogi et al. (2009), the CAS phase should not crystallize along the cold subduction geotherm; therefore, the larger densities of this model are essentially due to the presence of stishovite and K-hollandite in the phase assemblages. The density profile following the hot subduction geotherm is generally similar to the cold subduction profile in the upper-to-middle part of the transition zone and the lower mantle, the differences being ascribed to the higher temperatures in the hot slab. However, we observe a marked decrease in the density at the depths of the middle-to-lowermost part of the transition zone, which is due to the crystallization of the CAS phase at ~20 GPa and 1,500 K (Fig. 8b). One can note that, in natural systems, corundum often dissociates in surrounding minerals, and therefore, the densities showed here might be a little lower at P, T conditions where the CAS phase does not crystallize. The density profile following a normal mantle geotherm shows the smallest densities. The CAS phase is expected to form at ~14 GPa and 1,700 K and would be stable from the uppermost transition zone to the shallower lower mantle. In this region, the calculated densities for the CAS phase are higher than the surrounding mantle minerals. In contrast, the CAS phase is less dense than the surrounding minerals in the uppermost lower mantle. The densities of the CAS phase in the transition zone are ρCAS = 4.02–4.20 g cm−3, whereas the surrounding mantle is ρPREM = 3.72–4.00 g cm−3. Above 660 km depth, the CAS phase is 0.18 g cm−3 less dense than lower mantle minerals.
Conclusions
Using a Kawai-type multi-anvil press coupled with synchrotron X-ray diffraction, we carried out P–V–T measurements on the CAS phase with the composition CaAl4Si2O11, at pressures between 0 and 24 GPa and temperatures up to 2,100 K. This is the first report of a complete thermal equation of state for the CAS phase.
The present results suggest that the CAS phase would have intermediate densities compared with other coexisting minerals stable at the P,T conditions of the transition zone. Therefore, an increasing proportion of the CAS phase with increasing temperature (T > 1,500 K) would contribute to plunge the slab deeper into the transition zone. For equal proportions, the plunging efficiency of the CAS phase would be comparable with K-hollandite but lower than stishovite and Ca-perovskite. In natural systems, it is known that the CAS phase incorporates some amount of Na2O and K2O (Beck et al. 2004; Irifune et al. 1994), but to this day, no study reports the effect of those elements on its thermoelastic properties. Considering the low amount of Na2O and K2O in natural mantle, it is indeed expected that this effect could be negligible.
The densities calculated for a continental crust containing 10 vol% CaAl4Si2O11 showed that reactions of formation and dissociation of the CAS phase could explain multiple seismic reflections in regions of hot slabs subducted to the upper-to-middle part of the MTZ and uppermost lower mantle, respectively. However, the density crossover near the 660 km depth discontinuity might hinder the subduction of the continental crust into the lower mantle. Therefore, the CAS phase would act as an agent of buoyancy within hot slabs in the lowermost part of the transition zone. Thus, the CAS phase could play an important role in hosting and recycling Ca and Al in stagnant slabs and/or upwelling plumes in the deeper parts of the mantle transition zone.
References
Akaogi M, Haraguchi M, Yaguchi M, Kojitani H (2009) High-pressure phase relations and thermodynamic properties of CaAl4Si2O11 CAS phase. Phys Earth Planet Inter 173:1–6
Angel RJ, Ross NL, Seifert F, Fliervoet TF (1996) Structural characterization of pentacoordinate silicon in a calcium silicate. Nature 384:441–444
Beck P, Gillet P, Gautron L, Daniel I, El Goresy A (2004) A new natural high-pressure (Na, Ca)-hexaluminosilicate (CaxNa1–x)Al3+xSi3-xO11 in shocked Martian meteorites. Earth Planet Sci Lett 219:1–12
Brown JM, Shankland TJ (1981) Thermodynamic parameters in the Earth as determined from seismic profiles. Geophys J R Astr Soc 66:579–596
Dziewonski AM, Anderson DL (1981) Preliminary reference Earth model. Phys Earth Planet Inter 25:297–356
Fei Y (1995) Thermal expansion. In: Ahrens TJ (ed) Mineral physics and crystallography, A handbook of physical constants. AGU Reference Shelf 2: 29–44
Gautron L, Kesson SE, Hibberson WO (1996) Phase relations for CaAl2Si2O8 (anorthite composition) in the system CaO-Al2O3-SiO2 at 14 GPa. Phys Earth Planet Inter 97:71–81
Gautron L, Angel RJ, Miletich R (1999) Structural characterisation of the high-pressure phase CaAl4Si2O11. Phys Chem Miner 27:47–51
Gautron L, Gréaux S, Andrault D, Bolfan-Casanova N, Guignot N, Bouhifd MA (2006) Uranium in the Earth’s lower mantle. Geophys Res Lett 33. doi:10.1029/2006GL027508
Gréaux S, Gautron L, Andrault D, Bolfan-Casanova N, Guignot N, Bouhifd MA (2009) Experimental high pressure and high temperature study of the incorporation of uranium in Al-rich CaSiO3 perovskite. Phys Earth Planet Inter 174:254–263
Gréaux S, Kono Y, Nishiyama N, Kunimoto T, Wada K, Irifune T (2011a) P-V-T equation of state of Ca3Al2Si3O12 grossular garnet. Phys Chem Miner 38:85–94
Gréaux S, Nishiyama N, Kono Y, Gautron L, Ohfuji H, Kunimoto T, Menguy N, Irifune T (2011b) Phase transformations of Ca3Al2Si3O12 grossular garnet to the depths of the Earth’s mantle transition zone. Phys Earth Planet Inter. doi:10.1016/j.pepi.2011.02.001
Hama J, Suito K (2001) Thermoelastic model of minerals: application to Al2O3. Phys Chem Miner 28:258–267
Hirose K, Fei Y (2002) Subsolidus and melting phase relations of basaltic composition in the uppermost lower mantle. Geochim Cosmochim Acta 66:2099–2108
Holmes NC, Moriarty JA, Gathers GR, Nellis WJ (1989) The equation of state of platinum to 660 GPa (6.6 Mbar). J Appl Phys 66:2962–2967
Irifune T, Ringwood AE, Hibberson WO (1994) Subduction of continental crust and terrigenous and pelagic sediments: an experimental study. Earth Planet Sci Lett 126:351–368
Ishibashi K, Hirose K, Sata N, Ohishi Y (2008) Dissociation of CAS phase in the uppermost lower mantle. Phys Chem Miner 35:197–200
Larson AC, Von Dreele RB (2000) GSAS general structure analysis system. Operation manual. Los Alamos Nat Lab LAUR 86–748
Le Bail A, Duroy H, Fourquet JL (1988) Ab initio structure determination of LiSbWO6 by X-ray powder diffraction. Mater Res Bull 23:447–452
Nishihara Y, Nakayama K, Takahashi E, Iguchi T, Funakoshi KI (2005) P-V-T equation of state of stishovite to the mantle transition zone conditions. Phys Chem Miner 31:660–670
Nishiyama N, Rapp RP, Irifune T, Sanehira T, Yamazaki D, Funakoshi K (2005) Stability and P-V-T equation of state of KAlSi3O8-hollandite determined by in situ X-ray observations and implications for dynamics of subducted continental crust material. Phys Chem Miner 32:627–637
Ono S, Ito E, Katsura T (2001) Mineralogy of subducted basaltic crust (MORB) from 25 to 37 GPa, and chemical heterogeneity of the lower mantle. Earth Planet Sci Lett 190:57–63
Ono S, Iizuka T, Kikegawa T (2005) Compressibility of the calcium aluminosilicate, CAS, phase to 44 GPa. Phys Earth Planet Inter 150:331–338
Osborn EF, Muan A (1960) The system CaO-Al2O3-SiO2. Plate No. 1 in Phase equilibrium diagrams of oxide systems 219
Rapp RP, Irifune T, Shimizu N, Nishiyama N, Norman MD, Inoue T (2008) Subduction recycling of continental sediments and the origin of geochemically enriched reservoirs in the deep mantle. Earth Planet Sci Lett 271:14–23
Rubie DC (1998) Characterising the sample environment in multianvil high-pressure experiments. Phase Transit 68:431–451
Saikia A, Frost DJ, Rubie DC (2008) Splitting of the 520-kilometer seismic discontinuity and chemical heterogeneity in the mantle. Science 319:1515–1518
Thompson AB (1992) Water in the Earth’s upper mantle. Nature 358:295–302
Toby BH (2001) EXPGUI, a graphical user interface for GSAS. J Appl Crystallogr 34:210–213
Tsuchiya T (2003) First-principles prediction of the P-V-T equation of state of gold and the 660-km discontinuity in Earth’s mantle. J Geophys Res B 108:2462
Wang W, Takahashi E (1999) Subsolidus and melting experiments of a K-rich basaltic composition to 27 GPa: Implication for the behavior of potassium in the mantle. Am Miner 84:357–361
Wang Y, Weidner DJ, Guyot F (1996) Thermal equation of state of CaSiO3 perovskite. J Geophys Res B 101:661–672
Wang Y, Weidner DJ, Zhang J, Gwanmesia GD, Liebermann RC (1998) Thermal equation of state of garnets along the pyrope-majorite join. Phys Earth Planet Inter 105:59–71
Xue X, Zhai S, Kanzaki M (2009) Si-Al distribution in high-pressure CaAl4Si2O11 phase: A 29Si and 27Al NMR study. Am Miner 94:1739–1742
Yang H, Downs RT, Finger LW, Hazen RM, Prewitt CT (1997) Compressibility and crystal structure of kyanite, Al2SiO5, at high pressure. Am Miner 82:467–474
Zhai S, Ito E (2008) Phase relations of CaAl4Si2O11 at high-pressure and high-temperature with implications for subducted continental crust into the deep mantle. Phys Earth Planet Inter 167:161–167
Acknowledgments
The authors thank K. Funakoshi, Y. Higo, and T. Kunimoto for their helpful assistance in the experiments at BL04B1, at the synchrotron radiation facility, SPring-8 (Project 2010A1326). We are grateful for assistance of A. Yamada and H. Ohfuji in the preparation and analyses of starting materials. We acknowledge D. Gatta and M. Akaogi for valuable comments, which improved the manuscript, and C. McCammon for its editorial handling. This work was supported by the Global-COE program “Deep Earth Mineralogy”.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gréaux, S., Nishiyama, N., Kono, Y. et al. P–V–T equation of state of CaAl4Si2O11 CAS phase. Phys Chem Minerals 38, 581–590 (2011). https://doi.org/10.1007/s00269-011-0430-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-011-0430-7