
Abstract
Histone deacetylase (HDAC) inhibitors impair tumor cell proliferation and alter gene expression. However, the impact of these changes on anti-tumor immunity is poorly understood. Here, we showed that the class I HDAC inhibitor, entinostat (ENT), promoted the expression of immune-modulatory molecules, including MHCII, costimulatory ligands, and chemokines on murine breast tumor cells in vitro and in vivo. ENT also impaired tumor growth in vivo—an effect that was dependent on both CD8+ T cells and IFNγ. Moreover, ENT promoted intratumoral T-cell clonal expansion and enhanced their functional activity. Importantly, ENT sensitized normally unresponsive tumors to the effects of PD1 blockade, predominantly through increases in T-cell proliferation. Our findings suggest that class I HDAC inhibitors impair tumor growth by enhancing the proliferative and functional capacity of CD8+ T cells and by sensitizing tumor cells to T-cell recognition.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is one of the leading causes of cancer death in women and claims more than 41,000 lives each year in the United States [1]. Triple-negative breast cancer (TNBC), which lacks the estrogen and progesterone receptors as well as the receptor tyrosine kinase, HER2, is a particularly aggressive form of the disease that often develops resistance to conventional chemotherapy [2]. Interestingly, patients whose TNBC tumors express major histocompatibility complex class II (MHCII) proteins have more tumor-infiltrating lymphocytes (TILs) and experience prolonged survival [3]. The expression of MHCII on murine breast tumor cells stimulates tumor-infiltrating CD4+ T cells enhances the local inflammatory response and augments the recruitment, expansion, and function of tumor-specific CD8+ T cells [4,5,6], thereby facilitating tumor rejection. Thus, finding ways to promote the expression of MHCII on MHCII-non-expressing tumor cells should be clinically advantageous.
Changes in the genetic and epigenetic regulation of gene expression are common characteristics of malignant cells [7, 8]. For example, tumor cells often have decreased histone acetylation [9], which changes gene expression by altering nucleosome structure and DNA accessibility [10]. Importantly, histone acetylation is dynamically regulated by histone acetyl transferases (HATs) and histone deacetylases (HDACs), the latter of which is a family of eighteen enzymes categorized based on sequence homology to yeast enzymes [11]. Given that malignant cells often exhibit a perturbed balance of HAT and HDAC expression [12,13,14], a variety of HDAC inhibitors are being investigated as anti-cancer agents [15]. Indeed, HDAC inhibitors generally induce cell cycle arrest, apoptosis, and differentiation of breast cancer cells [16, 17].
In addition to their effects on tumor cell proliferation and differentiation, HDAC inhibitors can modulate the immune response. For example, HDAC inhibitors can induce the expression of MHCI and MHCII proteins as well as costimulatory molecules like CD80, CD86, and CD40 on tumor cells [18, 19]. Conversely, they also promote the expression of inhibitory ligands like PD-L1 [20, 21], suggesting that they may have paradoxical effects on anti-tumor immunity. HDAC inhibitors also impair immune suppressive cell types, including myeloid-derived suppressor cells and Tregs [22,23,24,25,26], thereby increasing productive immunity. However, a direct connection between HDAC inhibition, altered gene expression profiles, and tumor-specific T-cell responses has not been established.
Here, we showed that the class I HDAC inhibitor, entinostat (ENT), impaired tumor cell proliferation and promoted the expression of MHCII and PD-L1 on murine breast tumors in vitro. Tumors in ENT-treated mice also grew more slowly and expressed higher levels of MHCII and PD-L1; however, the in vivo effects of ENT were completely dependent on both CD8+ T cells and IFNγ. Importantly, ENT promoted the proliferation and enhanced the effector activities of tumor-infiltrating CD8+ T cells. Interestingly, ENT sensitized tumor cells to the effects of IFNγ and, more importantly, sensitized tumors to the effects of PD1 blockade, primarily by further enhancing T-cell proliferation. Our findings suggest that class I HDAC inhibitors impair tumor growth by enhancing the functional and proliferative capacities of CD8+ T cells and by sensitizing tumor cells to T-cell recognition.
Materials and methods
Cell culture
TS/A cells were cultured in DMEM/high glucose supplemented with 10% fetal bovine serum (both from Hyclone Laboratories, Inc.). 4T1 cells were cultured in RPMI-1640 (Lonza) supplemented with 10% FBS (Hyclone Laboratories, Inc.). Cells were grown to 80% confluency, dissociated with 0.05% trypsin, 0.53 mM EDTA (Corning), washed, and plated in fresh media.
Chemicals and reagents
Panobinostat (LBH589) and entinostat (MS275) were obtained from LC laboratories. Murine recombinant IFNγ was purchased from EMD Millipore.
RNA isolation, real-time PCR, NanoString, and TCR repertoire analysis
Total RNA was purified from excised tumors with the RNeasy Mini kit (Qiagen) following the manufacturer’s instructions and quantified by optical density at 260 nm using the DeNovix DS-11 spectrophotometer (DeNovix Inc.). RNA quality was assessed with a Bioanalyzer 2100 (Agilent) and final concentration was calculated with Qubit (Life Technologies). For RT-PCR, ~ 500 ng of RNA was reverse transcribed into cDNA using SuperScript VILO Master Mix (Thermo Fisher) according to the manufacturer’s instructions. RT-PCR was performed with the resulting cDNA using Power SYBR green PCR master mix (Life technology) on an ABI 7900 (Applied Biosystems). GAPDH, HPRT, PGK1, SDHA, β-actin, and 18S rRNA were used as controls. Relative mRNA was quantified using the comparative Ct method (ΔΔ Ct method). Primers include: CIITA-F: 5′-TGCGTGTGATGGATGTCCAG-3′, CIITA-R: 5′-CCAAAGGGGATAGTGGGTGTC-3′; CD74-F: 5′-CCGCCTAGACAAGCTGATT-3′, CD74-R: 5′-ACAGGTTTGGCAGATTTCGGA-3′; IAα-F: 5′-GACCACGTAGGCACCTATGG-3′, IAα-R: 5′- CTACAGCTATGTTTTGCAGTCCA-3′; IEβ-F: 5′-GCGGAGAGTTGAGCCTACG-3′, IEβ-R: 5′-CCAGGAGGTTGTGGTGTTCC-3′; GAPDH-F: 5′-GGAGCCAAAAGGGTCATCATCTC-3′, and GAPDH-R: 5′-GAGGGGCCATCCACAGTCTTCT-3′. Nanostring analysis as well as TCR repertoire sequencing and analysis were performed as previously described [4].
Mice, tumor administration, and treatment regimens
BALB/c or BALB/c.scid mice were injected with 1 × 105 TS/A or 4T1 cells into the mammary pad on day 0. The length and width of tumors were measured by caliper and tumor volume calculated by 0.4 × length × width2. Tumor-bearing mice were treated daily with 20 mg/kg ENT (unless otherwise indicated) dissolved in 15% DMSO, 20% Kolliphor EL, 65% PBS or with the same volume of vehicle alone via intraperitoneal injection beginning on day 4 after tumor cell injection and continuing for 2 weeks unless otherwise stated. The chosen dose was based on that used in the previous studies [22, 23].
Depleting antibodies against CD4 (GK1.5), CD8 (2.43), CD25 (PC61.5.3), CD19 (1D3), B220 (RA3.3A1/6.1), anti-rat kappa (Mar 18.5), and isotype control (2A3) were obtained from BioXCell. For CD4, CD8, and CD25 depletion, 200 μg antibody was administered intraperitoneally on days 3, 1, and weekly intervals thereafter. For B-cell depletion, 200 μg of anti-CD19 and anti-B220 was administered on day 10 and the anti-rat-kappa given on day 8. 200 μg anti-CD19 was then given on day 5, day 3, day 1, and weekly intervals thereafter. For combination experiments, 200 μg anti-PD1 (BioXCell, clone RMP1-14) or isotype antibody (BioXCell, clone 2A3) was administered intraperitoneally on days 10, 13, and 16.
Tumor dissociation, T-cell restimulation, and flow cytometry
Excised tumors were diced and digested with collagenase (c7657, Sigma) and DNase (d5025, Sigma) for 35 min at 37 °C as described [4]. Cell suspensions were filtered and subsequently used for flow cytometry or T-cell restimulation as described [4]. A list of antibodies, conjugates, and dilutions is provided in (Supplemental Table 1). Flow cytometry samples were analyzed on a BDFACS Canto II (BD Biosciences) and data were analyzed using FlowJo version 9.9.
Statistical analysis
Determination of statistical significance between group means at each time point was done using multiple unpaired independent t tests, correcting for multiple comparisons using the Holm–Sidak method, and assuming alpha of 0.05. For determination of significance between more than two group means, one-way ANOVA was used with Tukey’s method for multiple comparisons. With the exception of repertoire data, analysis was done in GraphPad Prism version 7.0a. The TCR high-throughput sequencing data were analyzed in R environment using the tcR package [27] and common R routines. TCR repertoire diversity was assessed using Inverse Simpson index and directly observed richness (number of unique clonotypes in a sample) available in the tcR package. Comparative analysis of TCR repertoire richness was performed after normalization of the HTS depth. The total data sets were downsampled to 145,000 randomly chosen sequencing reads using bootstrapping with 100 iterations. For the comparative analysis of TCR repertoire diversity indices, a downsampling of repertoire sizes to that of the smallest repertoire was performed using bootstrapping of 1047 clones with 1000 iterations. A median of each simulated richness and diversity distribution was used as estimated richness or diversity, accordingly [28]. The treatment groups were compared using the non-parametric Kruskal–Wallis test.
Results
HDAC inhibition promotes the expression of immune-modulatory molecules by tumor cells
Given that MHCII expression on TNBC cells is associated with increased TILs and improved clinical outcomes [3, 4] and that HDAC inhibitors can promote MHCII expression on some tumor cells [18, 29, 30], we tested whether HDAC inhibitors could promote MHCII expression in breast cancer cells. We initially cultured the murine mammary carcinoma cell lines, 4T1 and TS/A, with panobinostat (PAN) or entinostat (ENT), and measured the expression of the class II transcriptional activator, CIITA, and the MHCII chaperone protein, CD74, by real-time PCR. We found that PAN and ENT increased CIITA and CD74 mRNA in both cell lines (Fig. 1a, b).

HDAC inhibition induces the expression MHCII-related genes in tumor cells. Real-time PCR of RNA from 4T1 cells (a) or TS/A cells (b) cultured with vehicle, 5 μM ENT, or 10 nM PAN for 24 h or 72 h. Graphs show mean ± SD of three samples/group. c–e Representative histograms of MHCII (c), CD74 (d), and PD-L1 (e) on 4T1 and TS/A cells cultured with vehicle, PAN, ENT or IFNγ for 72 h. The mean fluorescent intensity (MFI) is shown in the graphs as the mean ± SD of two samples/group/dose
We next assessed the effect of HDAC inhibition on cell-surface expression of MHCII and CD74 by flow cytometry. As a positive control, we also treated cells with IFNγ, which induces expression of MHCII pathway components by activating the pIV promoter of CIITA [31]. We found that both inhibitors increased surface expression of MHCII and CD74 on 4T1 and TS/A cells in a dose-dependent fashion, but that 4T1 cells were more responsive to IFNγ than TS/A cells (Fig. 1c, d). Consistent with previous results [20], we also found that HDAC inhibitors as well as IFNγ increased PD-L1 expression on both 4T1 and TS/A cells (Fig. 1e). Again, 4T1 cells were more responsive than TS/A cells.
Using NanoString analysis of RNA extracted from cells treated in vitro, we observed increased MHCI expression in both cell lines in response to ENT, PAN and IFNγ (Supplemental Table 2). However, we also found that both inhibitors increased expression of the costimulatory molecules, 41BB, CD40, and ICOSL, the inhibitory ligand, PD-L1, and the T-cell chemoattractant, CXCL10 (Supplemental Table 2). Moreover, both inhibitors modestly increased expression of the transcription factor and histone acetyltransferase NCOA1, whereas ENT increased expression of STAT1 more so than PAN. Not surprisingly, IFNγ dramatically increased expression of STAT1, PD-L1, and CXCL10 in both cell lines (Supplemental Table 2). Collectively, these results demonstrate that HDAC inhibitors influence the expression of a variety of immune-modulatory genes in breast cancer cells.
HDAC inhibitors reduce proliferation of breast tumor cells
HDAC inhibition often impairs tumor cell proliferation [16, 17]. To assess the impact of HDAC inhibitors on the proliferation of TS/A and 4T1 cells, we cultured them with ENT, PAN, or IFNγ, and measured cellular DNA content via propidium iodide staining. We found that PAN and ENT, but not IFNγ, reduced the frequency of 4T1 cells in S phase and increased the frequency of cells in G0/G1 phase (Supplemental Fig. 1a, c). We observed similar changes in TS/A cells (Supplemental Fig. 1b, d). In addition, we observed that both PAN and ENT reduced cell recovery in cultures of 4T1 and TS/A at 48 h and 72 h (Supplemental Fig. 1e, f). Thus, HDAC inhibition reduces the proliferation of 4T1 and TS/A breast cancer cells in vitro.
HDAC inhibition increases MHCII expression and reduces tumor burden in mice
To test whether HDAC inhibition increased MHCII expression and reduced tumor growth in vivo, we implanted TS/A tumor cells into the mammary fat pads of BALB/c mice and, beginning on day 4, treated them daily with PAN or ENT. We found that on day 14, only about 1% of the tumor cells from control mice expressed MHCII, but that treatment with PAN or ENT increased the frequency of MHCII-expressing tumor cells to about 7% (Fig. 2a). Given that PAN-treated mice experienced notable weight loss, suggesting toxicity, we limited further experiments to ENT.

ENT induces expression of MHCII and PD-L1 and impairs tumor growth. a–f BALB/c mice were injected with TS/A cells and treated daily with the indicated inhibitors beginning on day 4. a Tumors were dissociated on day 14 and the frequency of MHCII+ cells CD45neg gate was determined by flow cytometry. Graph shows individual data as well as mean ± SEM of 3–4 samples/group. b RNA was extracted from whole tumors on day 14 and gene expression was determined by RT-PCR and normalized to vehicle. Graph shows individual data as well as mean ± SEM of 3–4 samples/group and is representative of three independent experiments. Flow plots show MHCII (c) and PD-L1 (d) expression on live, CD45neg cells from dissociated tumors on days 10 and 17. The mean ± SD is indicated in each plot with five mice/group/timepoint. e Tumor growth in mice treated with vehicle (19 mice), 20 mg/kg ENT (nine mice), or 25 mg/kg ENT (19 mice) daily for 2 weeks starting on day 4. f Tumor mass on day 17. Graphs show mean ± SEM. *p < 0.05, **p < 0.005, #p < 0.0005. Data are representative of three independent experiments
We next determined the expression of CIITA, CD74, and MHCII (IAα and IEβ1) mRNAs in whole tumors using RT-PCR, and found that each was similarly increased by treatment with ENT (Fig. 2b). We also assessed tumor expression of other molecules that are critical for immune-mediated recognition. We found that ENT increased expression of MHCI and Fas on TS/A cells in a dose-dependent fashion (Supplemental Fig. 2a). Treatment of TS/A tumor-bearing mice with ENT also increased the surface expression of MHCI and FAS on CD45neg cells (Supplemental Fig. 2b).
To determine whether MHCII expression on tumor cells was dependent on either the dose or duration of treatment, we treated TS/A tumor-bearing mice with 20, 25, or 30 mg/kg/day ENT or vehicle control beginning on day 4 and measured MHCII expression on tumor cells by flow cytometry on days 10 and 17. We found that the frequency of MHCII-expressing tumor cells increased with in a dose- and time-dependent fashion (Fig. 2c), with around 30% of tumor cells expressing MHCII at the highest dose on day 17. We also observed a time-dependent increase in PD-L1 expression on tumor cells following ENT treatment (Fig. 2d). Additionally, we found that ENT significantly impaired tumor growth in terms of size (Fig. 2e) and excised tumor mass on day 17 (Fig. 2f).
To determine how ENT affected gene expression within the tumor microenvironment, we extracted RNA from treated and control tumors on day 14 and analyzed mRNA expression using a custom NanoString probe set. We found that the gene expression profiles of untreated tumors clustered separately from those of treated tumors (Fig. 3a), with substantial increases in the expression of T-cell-related genes, such as granzyme B (GZMB), perforin (PRF-1), interferon-γ (IFNG), and l-selectin (SELL), in treated samples. Surprisingly, we only observed modest increases in MHCII-related genes, including I-E, I-A, and CD74, in ENT-treated samples (Fig. 3a). However, we did observe increased expression of the inhibitory ligands PD-L1 and PD-L2, as well as the chemokines, CXCL13 and CXCL10 (Fig. 3a).

Immune activation in ENT-treated tumors. a Heat map of gene expression (Nanostring counts) in RNA extracted on day 14 from tumors treated with vehicle, 20 mg/kg ENT, or 30 mg/kg ENT starting on day 4. b–e Tumor-bearing mice were treated with vehicle or 20 mg/kg ENT and tumors were dissociated on days 10 and 14. b Representative flow plots of CD25 and FoxP3 expression after gating on live, CD3+CD4+ cells. c MFI of FOXP3 expression in CD3+CD4+CD25+FOXP3+ cells. d Number of Tregs normalized to tumor mass. e CD8/Treg ratio. Graphs show individual data as well as mean ± SEM of 4–5 samples/group
Consistent with the previous studies [25], ENT reduced FoxP3 mRNA expression (Fig. 3a). To test whether the reduction in FoxP3 mRNA expression affected the numbers or phenotype of FoxP3-expressing Tregs, we enumerated CD4+CD25+FoxP3+ Tregs by flow cytometry. Indeed, we found that ENT decreased the frequency of Tregs in ENT-treated tumors on day 10 (Fig. 3b), and also decreased the per-cell expression of FoxP3 (Fig. 3c). Moreover, the total number of Tregs in ENT-treated tumors was reduced relative to controls (Fig. 3d), an observation that correlated with an increase in the CD8:Treg ratio (Fig. 3e).
Anti-tumor activity of ENT requires T-cell-mediated adaptive immunity
To test whether adaptive immunity was required for the anti-tumor effects of ENT [32], we injected TS/A cells into BALB/c.scid mice, which lack T and B cells, and treated tumor-bearing mice with ENT beginning on day 4. We found that in the absence of T and B cells, treatment with ENT at either 20 or 25 mg/kg/day had only a modest impact on tumor growth (Fig. 4a) and failed to upregulate MHCII expression (Fig. 4b).

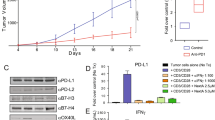
Anti-tumor activity of ENT requires adaptive immunity. a, b Tumor-bearing BALB/c.scid mice were treated with ENT or vehicle daily starting on day 4. a Tumor growth. Graph shows mean ± SEM of ten mice/group; *p < 0.05. b Representative flow plots showing MHCII expression on live, CD45neg cells on day 20. The mean ± SD is indicated. c, d Tumor-bearing BALB/c mice were treated with combinations of 20 mg/kg ENT and blocking antibodies. c Tumor growth. Graph shows mean ± SD of five mice/group; **p < 0.005. d Representative flow plots of showing MHCII expression on live, CD45neg cells on day 17. The mean ± SD is indicated. e TS/A cells were cultured with combinations of 2.5 μM ENT and 100 U/mL IFNγ for 24 h or 72 h, and the expression of MHCII, CD74, and PD-L1 was measured by flow cytometry. Graphs show mean ± SEM of two replicates/group. Data are representative of two independent experiments. f The indicated depleting antibodies were administered to ENT-treated, tumor-bearing mice and tumor growth was monitored. Graph shows mean ± SEM of five mice/group. #p < 0.0005
Given the ability of IFNγ to trigger MHCII and PD-L1 expression, we next used an IFNγ-blocking antibody to test whether reductions in tumor growth and increases in MHCII and PD-L1 expression in ENT-treated mice were dependent on IFNγ. We found that ENT impaired tumor growth control mice, but failed to slow tumor growth in mice given IFNγ-blocking antibody (Fig. 4c). Moreover, ENT promoted MHCII expression on tumor cells in control mice, but not in those treated with IFNγ-blocking antibody (Fig. 4d). Similarly, PD-L1 was only upregulated in control mice and not in mice treated with IFNγ blocking antibody (Fig. 4d). These data indicate that the anti-tumor effects of ENT in vivo are dependent on IFNγ and likely on T cells.
Given that ENT alone could promote MHCII expression on TS/A cells, but IFNγ alone poorly triggered MHCII expression (Fig. 1c), even though it clearly impacted gene expression (Supplemental Table 2), we next tested the possibility that ENT and IFNγ could cooperatively increase MHCII expression by culturing TS/A cells with ENT and IFNγ alone or together and measuring the cell-surface expression of MHCII, CD74, and PD-L1. We found that although IFNγ by itself had almost no effect on MHCII or CD74 expression, and modestly increased PD-L1, it acted in concert with ENT to more-than-additively enhance the expression of these proteins (Fig. 4e).
We next tested which adaptive immune cell population(s) was required for the anti-tumor effect of ENT by administering depleting antibodies against CD4+ cells, CD8+ cells, both CD4+ and CD8+ cells, Tregs, or B cells to mice prior to injection of tumor cells and at weekly intervals thereafter. Our depletion strategy was effective, as the relevant populations were eliminated in both the tumor and lymph node (Supplemental Fig. 3). Importantly, we found that depletion of CD4+ T cells or B cells modestly increased tumor growth in ENT-treated mice, whereas depletion of CD8+ T cells or both CD4+ and CD8+ T cells dramatically increased tumor growth in ENT-treated mice (Fig. 4f). Conversely, co-treatment with anti-CD25 (to inhibit Treg activity) led to tumor rejection in 4/5 of ENT-treated mice (Fig. 4f). Moreover, these mice were subsequently able to reject TS/A tumors, but not 4T1 tumors, injected on the opposite side 60 days later with no additional treatment (not shown), suggesting that the combination of ENT and anti-CD25 promoted a functional memory response. Interestingly, treatment with ENT alone actually led to significant reduction of B cells in tumors and draining lymph nodes (Supplemental Fig. 4a, b). Treatment with ENT also depleted Natural Killer (NK) cells in both tumors and draining lymph nodes (Supplemental Fig. 4a, b). Together, these findings demonstrate that IFNγ and CD8 T cells are integral to ENT-mediated tumor control.
ENT enhances the function and clonal expansion of TILs
Given the requirement for T cells and IFNγ, we next enumerated cytokine-producing, tumor-infiltrating CD4+ and CD8+ T cells on days 10 and 17 in ENT-treated and control mice. Surprisingly, we found that fewer CD4+ T cells infiltrated ENT-treated tumors than control tumors (Fig. 5a), even though ENT-treated tumors expressed MHCII. Despite the differences in total infiltration, we observed similar production of the effector cytokines, TNFα and IFNγ (Fig. 5b, c). In contrast, we found that slightly more CD8+ T cells infiltrated ENT-treated tumors than control tumors (Fig. 5d) and that they produced strikingly more granzyme B and IFNγ (Fig. 5e, f).

ENT promotes T-cell effector function and expansion. a–f Tumors from ENT-treated and control mice were dissociated on days 10 and 17 and infiltrating CD4+ T cells (a–c) and CD8+ T cells (d–f) were evaluated by flow cytometry. a Number of CD4+ T cells normalized to tumor mass. b Representative flow plots of CD4+ T cells expressing IFNγ and TNFα. c Number of CD4+ T cells producing IFNγ and TNFα normalized to tumor mass. d Number of tumor-infiltrating CD8+ T cells normalized to tumor mass. e Representative flow plots of CD8+ T cells expressing IFNγ and TNFα. f Number of CD8+ T cells producing IFNγ and granzyme B (GZB) normalized to tumor mass. Graphs in a, c, d, and f show mean ± SEM of 14 mice/group/timepoint; **p < 0.005, #p < 0.0005. Data are representative of two independent experiments. g–i TCRβ repertoire on day 14. g Tree plots showing relative TCRβ frequencies. h Unique sequences in each sample. i Inverse Simpson index quantifying the repertoire diversity of each sample. Box and whiskers plots show median and min and max values of 4–5 mice/group. Non-parametric statistical difference assessed using Kruskal–Wallis test (h, i) and expressed as §§p < 0.005, §§§p < 0.0005
An increase in highly functional CD8+ T cells could be reflective of increased proliferation of individual clones or greater recruitment of newly stimulated clones. To distinguish these possibilities, we evaluated the TCRβ repertoire in ENT-treated and control tumors, and found that ENT promoted dramatic expansions of T-cell clones in each tumor (Fig. 5g). We also found fewer unique TCR sequences in ENT-treated tumors (Fig. 5h), leading to about a one-third reduction in TCRβ diversity (Fig. 5i). Thus, ENT treatment significantly impacts the T-cell repertoire, promoting the clonal expansion of T cells that express cytolytic molecules.
ENT sensitizes tumors to the effects of PD1 blockade
Recent data link an IFNγ-induced gene expression signature with the response of patients to PD1 blockade [33]. Although our microarray did not capture every gene in this signature, many of the same genes were also upregulated by ENT (Fig. 6a). As a result, we next tested whether ENT could sensitize established tumors to PD1 blockade. We found that TS/A tumors were refractory to anti-PD1 monotherapy, as there was no change in tumor growth relative to controls (Fig. 6b). However, treatment with ENT dramatically enhanced the effects of anti-PD1, such that 80% of mice had rejected the tumor by day 28 (Fig. 6b). Moreover, these same mice subsequently rejected secondary TS/A tumors without additional treatment (not shown).

ENT sensitizes tumors to PD1 blockade. a Gene set enrichment analysis comparing genes differentially expressed genes in ENT-treated and control tumors and a list of IFNγ-inducible genes that predict response to PD1 blockade; normalized enrichment score of 1.45 and p value of 0.033. b Tumor growth in mice treated with combinations of ENT and anti-PD1. Graph shows mean ± SEM of 5 mice/group. c, d Representative flow plots (c) and graphs (d) showing the frequency of restimulated CD8+ T cells that express IFNγ and TNFα. Graph shows mean ± SEM of 6–7 mice/group. Data are representative of two independent experiments. e, f Ki67 expression by CD8+ TILs (e) and CD4+ TILs (f) following treatment with ENT and anti-PD1. Representative histograms are color-matched to the graphs, which show the mean ± SEM of 6–7 mice/group. Data are representative of two independent experiments. g–i TCRβ repertoire on day 14; five mice/group. g Tree plots of relative TCRβ frequencies. h Unique sequences in each sample. i Inverse Simpson index quantifying the repertoire diversity of each sample. Box and whiskers plots show median and min and max values. Statistical difference is expressed as *p < 0.05, **p < 0.005, #p < 0.0005 for parametric analyses, using one-way ANOVA with Tukey’s method for multiple comparison (b, d–f). Non-parametric statistical difference assessed using Kruskal–Wallis test (h, i) and expressed as §p < 0.05
We reasoned that PD1 blockade might enhance T-cell effector functions and/or boost tumor-specific T-cell proliferation. To test these possibilities, we harvested tumors on day 17 and assessed T-cell effector function and proliferation. We found that infiltrating CD8+ T-cell function was moderately improved by PD1 blockade alone, but that the combination of PD1 blockade and ENT significantly increased T-cell effector function (Fig. 6c, d). Interestingly, PD1 blockade alone had a little impact on either CD4+ or CD8+ T-cell proliferation, whereas ENT alone modestly boosted CD8+ T-cell proliferation, but not CD4+ T-cell proliferation (Fig. 6e, f). However, the combination of ENT and anti-PD1 dramatically increased the proliferative capacity of tumor-infiltrating CD8+ T cells (Fig. 6e) as well as CD4+ T cells (Fig. 6f).
Given the increases in T-cell proliferation, we next tested the impact of combination therapy on the TCRβ repertoire. Although we found expanded clones in tumors from all treatment groups (Fig. 6g), the number of unique sequences was reduced in ENT-treated tumors with or without PD1 blockade (Fig. 6h). Similarly, the repertoire diversity was also reduced in ENT-treated tumors with or without PD1 blockade (Fig. 6i). These results suggest that ENT promotes the expansion of a few T-cell clones, but that anti-PD1 triggers an additional expansion of a larger cohort of infiltrating T cells. Collectively, these data demonstrate that ENT sensitizes non-responsive tumors to PD1 blockade by increasing T-cell quality and, more importantly, by enhancing the proliferative capacity of responding T cells.
Discussion
Our data show that class I HDAC inhibitors like ENT alter gene expression and impair tumor cell proliferation in vitro (Fig. 1 and Supplemental Fig. 1), but when used in vivo, require CD8+ T cells and IFNγ to mediate similar effects (Figs. 2, 3, 4). These data suggest that in vivo ENT has a significant impact on T cells, which target tumor cells via their effector functions, including the expression of IFNγ, TNF, and granzyme B (Figs. 3, 5, 6). Consistent with this idea, we find that CD8+ TILs from ENT-treated tumors are clonally expanded and express cytokines and other effector molecules for longer than their counterparts from control tumors (Fig. 5). These effects are even more pronounced in mice treated with the combination of ENT and anti-PD1 (Fig. 6). Consequently, ENT sensitizes PD1-unresponsive tumors to PD1 blockade, thereby promoting tumor rejection (Fig. 6). Taken together, these results demonstrate that class I HDAC inhibitors sensitize normally unresponsive tumors to the effects of PD1 blockade by altering gene expression and promoting intratumoral CD8+ T-cell responses.
In part, HDAC inhibitors like ENT act by altering the expression of tumor-specific genes, including those in the MHCII pathway. However, increased MHCII expression on tumor cells in ENT-treated mice is dependent on T cells and IFNγ. This result is not too surprising, as IFNγ is a known regulator of MHCII expression [31]. However, the anti-tumor effects of ENT are largely independent of CD4+ T cells, suggesting that the stimulation of CD4+ T cells by MHCII-expressing tumor cells is not a primary mechanism that mediates tumor cell killing. Instead, increased MHCII expression on tumor cells is most likely a consequence, rather than a cause, of robust anti-tumor immunity and probably reflects the ability of ENT to enhance the clonal expansion and effector activity of tumor-specific CD8+ T cells. A similar phenomenon likely occurs in patients, in which the expression of MHCII on solid tumors correlates with the presence of TILs and improved patient outcomes [3, 34, 35].
Interestingly, we find that tumor cells cultured with ENT are more sensitive to the effects of IFNγ, perhaps by opening chromatin structure around the genes encoding CIITA, MHCII, or PD-L1. Thus, a similar phenomenon may occur in vivo, in which ENT does not directly promote MHCII expression, but sensitizes tumor cells to the effects of IFNγ made by activated T cells. Regardless, the idea that ENT promotes MHCII expression via IFNγ-producing CD8+ T cells is consistent with the increased number of tumor-infiltrating, IFNγ-expressing CD8+ T cells.
Do CD4+ T cells have any role in this process? Our data suggest that their role is minor. For example, we find that the number of CD4+ T cells is actually decreased in the tumors of ENT-treated mice and that their cytokine-producing ability is unchanged. Moreover, depletion of CD4+ T cells from ENT-treated animals has a minimal effect on tumor growth, suggesting that, despite the additional expression of MHCII on tumor cells in ENT-treated mice, effector CD4+ T cells only modestly contribute to the enhanced anti-tumor immune response. Unlike CD4+ effector T cells, however, which should positively contribute to anti-tumor immunity, CD4+ Tregs often suppress anti-tumor immunity. In this regard, HDAC inhibitors like ENT prevent the deacetylation of STAT3, thereby compromising Treg activity by impairing FOXP3 expression [24,25,26]. Consistent with this idea, we find that FOXP3 expression in whole tumors is significantly reduced upon ENT treatment and that the number of FOXP3-expressing Tregs normalized to tumor mass is also substantially less. Moreover, the combination of ENT and anti-CD25 (an antibody blocks IL-2 signaling and impairs Tregs function [36]), dramatically impairs tumor growth and promotes durable tumor regression. ENT also impairs the suppressive activity of MDSCs [22, 23]. Although we did not examine MDSCs, the potential of ENT to suppress their function would also benefit the anti-tumor T-cell response. Thus, class I HDAC inhibitors seem to benefit anti-tumor immunity by promoting CD8+ T-cell activation and inhibiting the functions of suppressive cell types.
Although some reports suggest that HDAC inhibitors impair T-cell activation and effector functions [37,38,39], we observe increased expression of mRNAs associated with T-cell effector differentiation (IFNγ, STAT1, and IRF-1), cytotoxic functions (granzyme B and perforin), and recruitment (CXCL13 and CXCL10), in tumors from ENT-treated mice. Moreover, the number of tumor-infiltrating CD8+ T cells that co-express IFNγ and granzyme B is significantly increased in ENT-treated mice. Thus, ENT does not impair T-cell proliferation or effector activity at the time we are giving it. In this regard, HDAC inhibitors may impair the activation and proliferation of naïve T cells [39, 40], but also enhance the function and proliferation of previously activated effector T cells [15, 25, 41]. These apparently contradictory functions can be explained by the differences in the chromatin landscape in naïve and activated T cells [42]. Therefore, clinicians should carefully consider the timing of HDAC inhibitor administration relative to the timing of therapies that might trigger de novo immune responses or those intended to enhance potentially ongoing responses.
Despite its immune stimulatory effects, ENT also promotes the expression of the inhibitory ligands, PD-L1 and PD-L2. Like the expression of MHCII, the expression of PD-L1 on tumor cells is often dependent on IFNγ [43], but may be potentiated by ENT, perhaps by opening local chromatin structure surrounding the PD-L1 gene. In fact, many of the genes upregulated by ENT conform to the T cell-inflamed gene expression profile that predicts clinical responses to anti-PD-1 [33]. Although increases in PD-L1 expression are unlikely to benefit anti-tumor immunity, in this case, increased PD-L1 expression reflects local T-cell activation and IFNγ production. Unlike ENT, however, which promotes both T-cell proliferation and enhances T-cell effector function, PD1 blockade primarily stimulates T-cell proliferation. Repertoire analysis supports this conclusion and suggests that, whereas ENT promotes the hyperexpansion of a few T-cell clones, PD1 blockade more broadly enhances T-cell proliferation. Expansion of a greater diversity of tumor-reactive T cells likely prevents the development of antigen-escape mutants, thereby conferring a greater long-term benefit.
In summary, our findings suggest a model wherein HDAC inhibitors like ENT enhance anti-tumor immunity by increasing CD8+ T-cell proliferation, cytokine expression, and functional activity, and by impairing Treg accumulation and FOXP3 expression within the TME. ENT also increases the sensitivity of tumor cells to IFNγ made by responding T cells, thereby enhancing the expression of MHC molecules as well as other immune-modulatory molecules on tumor cells. By changing gene expression in tumor cells, ENT also likely exposes new tumor antigens, thereby broadening the number of epitopes recognized by T cells. Importantly, the ENT-mediated changes in gene expression, in both tumor cells and infiltrating T cells, unleash the therapeutic potential of checkpoint inhibitors like anti-PD1. In other words, ENT turns “cold” tumors into “hot” ones, a process that should increase patient response rates to checkpoint blockade. Therefore, class I HDAC inhibitors like ENT are powerful immune activators that should be carefully integrated into combinatorial treatment strategies to maximize their anti-tumor effects.
Abbreviations
- ATCC:
-
American-Type Culture Collection
- CIITA:
-
Class II transcriptional activator
- ENT:
-
Entinostat
- GZB:
-
Granzyme B
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- MHCII:
-
MHC class II
- PAN:
-
Panobinostat
- TIL:
-
Tumor-infiltrating lymphocyte
- TNBC:
-
Triple negative breast cancer
References
Siegel RL, Miller KD, Jemal A (2018) Cancer statistics, 2018. CA Cancer J Clin 68:7–30
Gu G, Dustin D, Fuqua SAW (2016) Targeted therapy for breast cancer and molecular mechanisms of resistance to treatment. Curr Opin Pharmacol 31:97–103
Forero A, Li Y, Chen D, Grizzle WE, Updike KL, Merz ND, Downs-Kelly E, Burwell TC, Vaklavas C, Buchsbaum DJ, Myers RM, LoBuglio AF, Varley KE (2016) Expression of the MHC class II pathway in triple-negative breast cancer tumor cells is associated with a good prognosis and infiltrating lymphocytes. Cancer Immunol Res 4:390–399
McCaw TR, Li M, Starenki D, Cooper SJ, Liu M, Meza-Perez S, Arend RC, Buchsbaum DJ, Forero A, Randall TD (2018) The expression of MHC class II molecules on murine breast tumors delays T cell exhaustion, expands the T cell repertoire, and slows tumor growth. Cancer Immunol Immunother 68:175–188
Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS (2003) Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur J Immunol 33:1183–1192
Mortara L, Castellani P, Meazza R, Tosi G, De Lerma Barbaro A, Procopio FA, Comes A, Zardi L, Ferrini S, Accolla RS (2006) CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin Cancer Res 12:3435–3443
Berdasco M, Esteller M (2010) Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell 19:698–711
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M (2005) Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37:391–400
Bowman GD, Poirier MG (2015) Post-translational modifications of histones that influence nucleosome dynamics. Chem Rev 115:2274–2295
de Ruijter AJM, van Gennip AH, Caron HN, Kemp S, van Kuilenburg ABP (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370:737–749
Johnstone RW (2002) Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 1:287
Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M (2007) Expression profile of class I histone deacetylases in human cancer. Oncol Rep 18:769–774
Suzuki J, Chen YY, Scott GK, Devries S, Chin K, Benz CC, Waldman FM, Hwang ES (2009) Protein acetylation and histone deacetylase expression associated with malignant breast cancer progression. Clin Cancer Res 15:3163–3171
McCaw TR, Randall TD, Forero A, Buchsbaum DJ (2017) Modulation of antitumor immunity with histone deacetylase inhibitors. Immunotherapy 9:1359–1372
Munster PN, Troso-Sandoval T, Rosen N, Rifkind R, Marks PA, Richon VM (2001) The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Can Res 61:8492–8497
Vigushin DM, Ali S, Pace PE, Mirsaidi N, Ito K, Adcock I, Coombes RC (2001) Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin Cancer Res 7:971–976
Khan AN, Magner WJ, Tomasi TB (2007) An epigenetic vaccine model active in the prevention and treatment of melanoma. J Transl Med 5:64
Woods DM, Woan K, Cheng F, Wang H, Perez-Villarroel P, Lee C, Lienlaf M, Atadja P, Seto E, Weber J, Sotomayor EM, Villagra A (2013) The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res 23:341–348
Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J (2015) HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res 3:1375–1385
Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, Massengill C, Noyes DR, Martinez GV, Afzal R, Chen Z, Ren X, Antonia SJ, Haura EB, Ruffell B, Beg AA (2016) HDAC inhibitors enhance T Cell chemokine expression and augment response to PD-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res 22:4119–4132
Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, Diaz LA, Papadopoulos N, Kinzler KW, Vogelstein B, Zhou S (2014) Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci 111:11774–11779
Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, Chintala S, Ordentlich P, Kao C, Elzey B, Gabrilovich D, Pili R (2017) Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res 23:5187–5201
Pallandre J-R, Borg C, Rognan D, Boibessot T, Luzet V, Yesylevskyy S, Ramseyer C, Pudlo M (2015) Novel aminotetrazole derivatives as selective STAT3 non-peptide inhibitors. Eur J Med Chem 103:163–174
Shen L, Pili R (2012) Class I histone deacetylase inhibition is a novel mechanism to target regulatory T cells in immunotherapy. Oncoimmunology 1:948–950
Terranova-Barberio M, Thomas S, Munster PN (2016) Epigenetic modifiers in immunotherapy: a focus on checkpoint inhibitors. Immunotherapy 8:705–719
Nazarov VI, Pogorelyy MV, Komech EA, Zvyagin IV, Bolotin DA, Shugay M, Chudakov DM, Lebedev YB, Mamedov IZ (2015) tcR: an R package for T cell receptor repertoire advanced data analysis. BMC Bioinform 16:175
Venturi V, Kedzierska K, Turner SJ, Doherty PC, Davenport MP (2007) Methods for comparing the diversity of samples of the T cell receptor repertoire. J Immunol Methods 321:182–195
Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F, Tomasi TB (2000) Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol 165:7017–7024
Zika E, Greer SF, Zhu XS, Ting JPY (2003) Histone deacetylase 1/mSin3A disrupts gamma interferon-induced CIITA function and major histocompatibility complex class II enhanceosome formation. Mol Cell Biol 23:3091–3102
Wright KL, Ting JP (2006) Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol 27:405–412
West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, Johnstone RW (2013) An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Can Res 73:7265–7276
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, Piha-Paul SA, Yearley J, Seiwert TY, Ribas A, McClanahan TK (2017) IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Investig 127:2930–2940
Callahan MJ, Nagymanyoki Z, Bonome T, Johnson ME, Litkouhi B, Sullivan EH, Hirsch MS, Matulonis UA, Liu J, Birrer MJ, Berkowitz RS, Mok SC (2008) Increased HLA-DMB expression in the tumor epithelium is associated with increased CTL infiltration and improved prognosis in advanced-stage serous ovarian cancer. Clin Cancer Res 14:7667–7673
Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, Feld E, Johnson AS, Greenplate AR, Sanders ME, Lovly CM, Frederick DT, Kelley MC, Richmond A, Irish JM, Shyr Y, Sullivan RJ, Puzanov I, Sosman JA, Balko JM (2016) Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun 7:10582
Kohm AP, McMahon JS, Podojil JR, Begolka WS, DeGutes M, Kasprowicz DJ, Ziegler SF, Miller SD (2006) Cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4 + CD25 + T regulatory cells. J Immunol 176:3301–3305
Brogdon JL, Xu Y, Szabo SJ, An S, Buxton F, Cohen D, Huang Q (2007) Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood 109:1123–1130
Long J, Chang L, Shen Y, Gao WH, Wu YN, Dou HB, Huang MM, Wang Y, Fang WY, Shan JH, Wang YY, Zhu J, Chen Z, Hu J (2015) Valproic acid ameliorates graft-versus-host disease by downregulating Th1 and Th17 Cells. J Immunol 195:1849–1857
Moreira JMA, Scheipers P, Sørensen P (2003) The histone deacetylase inhibitor trichostatin A modulates CD4 + T cell responses. BMC Cancer 3:1–18
Skov S, Rieneck K, Bovin LF, Skak K, Tomra S, Michelsen BK, Ødum N (2003) Histone deacetylase inhibitors: a new class of immunosuppressors targeting a novel signal pathway essential for CD154 expression. Immunobiology 101:1430–1438
Cao K, Wang G, Li W, Zhang L, Wang R, Huang Y, Du L, Jiang J, Wu C, He X, Roberts AI, Li F, Rabson AB, Wang Y, Shi Y (2015) Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene 34:5960–5970
Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, Hellmann MD, Wolchok JD, Leslie CS, Schietinger A (2017) Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545:452
Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, Chen L (2012) Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 4:127
Acknowledgements
The authors would like to thank Uma Mudunuru and Scott Simpler for animal husbandry, and Eddy Yang and Debbie Della Manna of the NanoString Laboratory and Sagar Hanamanthu of the Comprehensive Flow Cytometry Core for lending respective expertise.
Funding
This work was supported by the University of Alabama at Birmingham Comprehensive Cancer Center (P30 CA013148), National Institutes of Health grant CA216234, and by the Breast Cancer Research Foundation of Alabama.
Author information
Authors and Affiliations
Contributions
TRM designed, performed, and interpreted experiments and wrote the manuscript. ML designed, performed, and interpreted experiments. DS performed TCR repertoire sequencing and analysis. SJC performed TCR repertoire sequencing and analysis. ML designed, performed, and interpreted experiments. RCA designed and interpreted experiments and edited the manuscript. AF designed and interpreted experiments and edited the manuscript. DJB designed and interpreted experiments and edited the manuscript. TDR designed and interpreted experiments and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
All procedures involving animals were performed in accordance with the guidelines of the National Research Council (United States) Committee for the Update of the Guide for the Care and Use of Laboratory Animals and were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee (IACUC) in protocol 09854.
Animal source
BALB/c mice were purchased from Charles River Laboratories International, Inc. BALB/c.scid mice (CBySmn.CB17-PrkdcscidIJ) were purchased from The Jackson Laboratory.
Cell line authentication
TS/A murine mammary adenocarcinoma cells were provided by Dr. Roberto S. Accolla, Department of Clinical and Biological Sciences, University of Insubria, Italy. TS/A cells were obtained at passage 22 and passaged two times prior to freezing archival samples. 4T1 cells were purchased from the American Type Culture Collection (ATCC). Samples of 4T1 and TS/A cells were authenticated by IDEXX BioAnalytics: case #23272-2019. All cell lines tested negative for mycoplasma (and 13 other mouse pathogens) via PCR performed by Charles River Research Animal Diagnostic Services.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
McCaw, T.R., Li, M., Starenki, D. et al. Histone deacetylase inhibition promotes intratumoral CD8+ T-cell responses, sensitizing murine breast tumors to anti-PD1. Cancer Immunol Immunother 68, 2081–2094 (2019). https://doi.org/10.1007/s00262-019-02430-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-019-02430-9