
Abstract
Regulatory T cells (Tregs) mediate immunosuppressive signals that can contribute to the progression of head and neck squamous cell carcinoma (HNSCC). Interleukin-33 (IL-33) is defined as an ‘alarmin’, an endogenous factor that is expressed during tissue and cell damage, which has been shown to promote Treg proliferation in non-lymphoid organs. However, the interaction between IL-33 and Tregs in the HNSCC tumor microenvironment remains uncertain. In this study, we examined IL-33+ and Foxp3+ cells by immunohistochemistry in 68 laryngeal squamous cell cancer patients, followed by functional analysis of IL-33 in Tregs. In addition, the suppressive function of Tregs was assessed by cell proliferation assays. The level of stromal IL-33 was significantly upregulated in advanced versus early stage HNSCC patients and positively correlated with Foxp3+ Treg infiltration as well as a poor prognosis. ST2 is regarded as the only receptor of IL-33. Infiltrated ST2-expressing Tregs were responsive to IL-33, and the percentage of Tregs was increased upon IL-33 stimulation. Functional investigation demonstrated that IL-33 increased the proportion of Foxp3+GATA3+ Tregs and improved the suppressive functions of Tregs by inducing IL-10 and TGF-β1 as well as decreasing the proliferation of responder T cells. Blockade of ST2 abrogated the immunosuppression caused by IL-33. Our data demonstrate that stromal IL-33 both expands the Treg population and enhances their functions in the tumor microenvironment. Furthermore, stromal IL-33 has prognostic value for tumor progression. Thus, stromal IL-33 is a potential target for future HNSCC immunotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common malignancy worldwide [1]. Immunotherapies, such as immune checkpoint-targeting and anticancer vaccines, have shown promising outcomes for HNSCC patients [2,3,4]. Nevertheless, cancers are still able to escape the immune system by suppressing the local immune response, leading to limited effects on tumor regression [5].
The immune system, although complex, is believed to play an important role in the malignant progression of HNSCC [6, 7]. Various immune cell types, including T cells, B cells, dendritic cells, natural killer cells, and macrophages, undergo intricate regulation for surveillance of cancer in our body. Among these cell types, tumor-infiltrating CD4+ regulatory T cells (Tregs) play a crucial role in suppressing local antitumor immune responses and accelerating tumor progression [8]. We recently reported increased numbers of tumor-infiltrating Tregs in HNSCC patients, and that blockade of Treg accumulation in tumors enhances antitumor immune responses [9, 10]. However, factors that facilitate Treg accumulation and their functions at the tumor site remain unknown.
Interleukin (IL)-33, as a member of the IL-1 family, acts as an alarmin signal that is released upon tissue damage to alert immune cells expressing the ST2 receptor (IL-1RL1) [11]. Alarmin signaling molecules, such as IL-33, are implicated in tumor-associated inflammation in the tumor microenvironment (TME) and appear to be a major mechanism of tumor immune tolerance. These signaling molecules may contribute to metastasis and account for the potential failure of current immunotherapies [12]. The major targets of IL-33 in vivo are tissue-resident immune cells including Tregs [13]. GATA3, a Th2 marker [14, 15], is highly expressed in ST2+ Tregs and has been shown to regulate Foxp3 expression through activation [16]. A recent study showed that IL-33 promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs) [17] and Foxp3+ Tregs [18]. However, little information is currently available regarding the role of IL-33 in Tregs of HNSCC patients.
We have previously demonstrated that depletion of Treg accumulation in the TME is an effective therapeutic strategy for HNSCC [9, 10]. In this study, we evaluated the correlation between IL-33 and Tregs in the HNSCC microenvironment. Our data revealed that stromal IL-33 was a negative prognostic factor that promoted the immune-suppressive function of Tregs, and blockade of ST2 reduced the ability of Tregs to suppress the immune response. Moreover, GATA3 may be involved in the promotion of the Treg-mediated suppression. Our results identified stromal IL-33 as a critical factor that contributes to the enhanced suppressive functions of Tregs within the TME, which can be potentially targeted to reverse Treg-mediated tumor immune escape.
Materials and methods
Donor samples
Ten pairs of laryngeal tumor and adjacent tissues were obtained after surgical resection. Peripheral blood mononuclear cells (PBMCs) were obtained from 20 healthy donors and 15 laryngeal squamous cell carcinoma (LSCC) patients who had not received any previous oncological treatment.
Immunohistochemistry (IHC)
IL-33 and Foxp3 expression in HNSCC tissue was evaluated by immunohistochemistry. The main clinical and pathological characteristics of 68 patients are presented in Supplementary Table 1. Clinical staging and the anatomical site of tumors were assessed according to the 6th edition of the Union for International Cancer Control (UICC 2008) TNM classification of malignant tumors. After antigen retrieval by microwaving, IHC staining was performed on 4 µm-thick, paraffin-embedded serial sections of tissue samples using 10 µg/ml human anti-IL-33 (AF3625, R&D Systems) and human anti-Foxp3 (ab20034, Abcam) antibodies, according to the manufacturer’s instructions. Each primary antibody-probed section was incubated with the corresponding secondary antibody, followed by a horseradish peroxidase-labeled streptavidin reaction (Anti-goat: PV-9003, Zhongshanjinqiao; anti-mouse: K5007, Agilent) manually. Immune complexes were detected using 3–3′-diamino-benzidine tetra-hydrochloride (DAB) chromogen mixed with 2 ml DAB substrate, and the sections were counterstained with hematoxylin. The specificities of the staining reactions were confirmed using nonimmune serum instead of the primary antibody.
Evaluation of IHC staining
Digital images of the sections were obtained at 10 × and 40 × magnifications by light microscopy (Axio Scan Z1, Carl Zeiss). IL-33 and Foxp3 staining results were classified by the number of immune-reactive cells (highlighted by nuclear staining). Five areas at 40 × magnification were counted by two scientists, and the numbers within tumor and stromal regions were counted separately. Patients were divided into two groups according to IL-33+ cell numbers for survival analysis. Patients with cell numbers greater than the median were defined as ‘high’ and those were smaller cell numbers were defined as ‘low’[19].
Tissue-infiltrating lymphocyte isolation
Tissue-infiltrating lymphocytes (TILs) were obtained from dissected tumors or adjacent tissues using a digested mixture, as described elsewhere [20]. Dissected tissues were minced into small pieces and immersed in digestion mixture (Collagenase IV, C5138; DNase, D5025; Hyaluronidase, H6254; all from Sigma–Aldrich). The samples were incubated on a rotating platform at 37 °C for 20 min, and the resulting cell suspensions were filtered through 70-µm cell strainers (431751, Corning) and isolated by lymphocyte separation medium (0850494, MP Biomedical). Separated mononuclear cells were resuspended in RPMI 1640 cell culture medium for further analysis.
LSCC cell lines and preparation of tumor culture supernatants (TSNs)
Human LSCC cell line SNU899 was kindly provided by Professor Ja-Lok Ku (Seoul National University College of Medicine). TSNs were obtained as described previously [20]. In brief, TSN was prepared by cultivating 5 × 106 tumor cells in 10 mL complete RPMI 1640 medium in 100 mm dishes for 24 h. The medium was then changed to complete medium supplemented with 10% human AB serum instead of fetal bovine serum. After 2 days, the supernatant was centrifuged and stored at − 80 °C.
Preparation of DCs
DC preparation were performed as described previously [21]. PBMCs were isolated from healthy donors using density gradient centrifugation. CD14+ cells were isolated from PBMCs using CD14 microbeads (130-050-201, Miltenyi Biotec) and cultured at a density of 1 × 105 cells/well in flat-bottomed 24-well plates in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% penicillin and streptomycin, 50 ng/ml granulocyte macrophage-colony stimulating factor (300-03, PeproTech), and 20 ng/ml recombinant human IL-4 (200-04, PeproTech) for 6 days. Then, 1 µg/ml lipopolysaccharide was added to induce maturation of DCs. DC phenotypes were determined by flow cytometry. The maturation of DCs was determined by expression of DC maturation marker CD83, an MHC-II molecule (HLA-DR), and co-stimulatory molecules (CD80, CD86, and CD40), as described in our previous study [21].
To prepare SNU899-stimulated DCs, TSN from the SNU899 cell line was added at day 6 for 24 h. Both control and SNU899-stimulated DCs were harvested at day 7 for co-culture.
T cell subpopulation isolation and cell culture
CD4+ T cells were purified from healthy donor PBMCs using CD4 microbeads (130-045-101, Miltenyi Biotec). Cells were seeded at 2 × 105 cells per well in flat-bottomed 24-well plates in RPMI 1640 medium supplemented with 1% penicillin and streptomycin, and 10% fetal bovine serum. For Treg cell generation, SNU899 TSN-stimulated DCs were added to CD4+ T cells at a ratio of 1:10 and co-cultured for 5 days with 2 µg/ml anti-CD3 (555336, BD Bioscience) with or without 1 ng/ml recombinant human IL-33 (3625-IL-010, R&D System).
For neutralization experiments, a human anti-ST2 mAb (3 µg/ml, AF523, R&D Systems) was added at the start of the co-culture. Cells were harvested at the indicated times and analyzed by flow cytometry.
For cell suppression assays, CD4+CD25+CD127low Tregs were sorted using a Regulatory T Cell Isolation Kit (130-094-775, Miltenyi Biotec) from co-cultures containing SNU899-stimulated DCs, as described above. Autologous CD4+CD25− T cells (responder cells) sorted from the same co-culture were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE, 65-0850-84, eBioscience) and seeded together with the sorted Tregs in round-bottomed 96-well plates coated with 10 µg/ml anti-CD3 in complete medium containing 2 µg/ml soluble anti-CD28 (555725, BD Bioscience) with or without 40 ng/ml rIL-33. The ratios of Tregs and responder cells were 1:2 or 1:4. Proliferation of responder cells was measured by flow cytometry after 3 days.
Flow cytometric analysis
To determine the expression of ST2, the following antibodies were used: anti-CD3-PE-Cy7 (25-0036-42, eBioscience), anti-CD4-PerCP-Cy 5.5 (45-0048-42, eBioscience), anti-Foxp3-PE (12-4777-42, eBioscience), and anti-ST2-APC (FAB5231A, R&D Systems).
To determine the Treg proportion and GATA3 expression, the following antibodies were used: anti-CD3- PE-Cy7, anti-CD4-PerCP-Cy 5.5, anti-Foxp3-PE, and anti-GATA3-Alexa Fluor 488 (560163, BD Biosciences).
All flow cytometry antibodies were acquired from Invitrogen unless specified otherwise. Experiments were performed according to the manufacturers’ protocols. Cells were acquired with a Cytoflex S (Beckman Coulter) and analysis was performed using Flowjo VX software (Flowjo LLC).
Cytokine detection
The concentrations of IL-10 and TGF-β1 in cell culture supernatants were determined using ELISA kits (IL-10: ELH-IL10; TGF-β1: ELH-TGFb1, both from RayBiotech), according to the manufacturer’s protocols. The sensitivity limits of the ELISA kits ranged from 1 to 150 pg/mL for IL-10 and from 80 pg/mL to 60 ng/mL for TGF-β1. Mean values are presented and the inter- and intra-assay variations were less than 15%.
Statistical analysis
Statistical analyses were performed using SPSS software version 22.0 (IBM Inc.). The Kaplan–Meier method was used to evaluate survival curves and differences between curves were analyzed by the log-rank test. The relationship between IL-33 and Foxp3 expression was evaluated using Spearman’s correlation test. The Student’s t test and ANOVA were used to analyze data from IHC, flow cytometry, ELISAs, and the cell proliferation assay, and Bonferroni adjustment for multiple comparisons was used for between-group comparisons. Data are expressed as means and the standard error of mean (SEM). P < 0.05 was considered as significant.
Results
Stromal IL-33+ cells in HNSCC are associated with poor patient survival
To determine the clinical relevance of IL-33 in HNSCC, we first evaluated the prognostic value of IL-33 expression in 68 HNSCC patient specimens by IHC. Patients were divided into high (33/68) or low (35/68) IL-33+ expression groups as described in the “Materials and methods”.
The number of stroma-infiltrated IL-33+ cells in HNSCC tumors showed a positive correlation with the clinical stage (Fig. 1a) of the primary tumor. By further analyzing the associations between IL-33+ cell number and clinicopathological features of the cohort (Table 1), we found a significantly higher number of stromal IL-33+ cells in tumors with advanced clinical stages (III and IV) or T stage than in those with early clinical stages (I and II) or T stage tumors (P < 0.001, Fig. 1b, c). In contrast, the number of IL-33+ cells among tumor epithelial cells was negatively correlated with T staging and clinical staging (Supplementary Fig. 1). Subsequent Kaplan–Meier survival analysis showed that HNSCC patients with more stromal IL-33+ cells had worse overall survival (OS) and progress-free survival (PFS) than those with less IL-33+ cells in their tumors (Fig. 1d, e).

Stromal IL-33+ cells are associated with poor prognoses of HNSCC. a Representative images of IL-33+ cells (red arrows) in the tumor stroma from clinical stage I to IV. b, c IL-33 expression was higher in advanced T stages (T3 and 4) and advanced clinical stages (III and IV) than early T stages (T1 and 2) and early clinical stages (I and II). The Student’s t test was used for data analysis (b: 5.04 ± 0.44 vs. 9.19 ± 0.86; c: 4.78 ± 0.42 vs. 9.36 ± 0.78). d, e Accumulation of stromal IL-33+ cells predicted poor survival of patients with HNSCC. Kaplan–Meier curves for overall survival (d) and progress-free survival (e) indicated poor survival of patients with high expression of IL-33 (OS: HR 3.87, 95% CI 1.49–10.06; PFS: HR 2.45, 95% CI 1.04–5.78). Data are the mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001
IL-33+ cell accumulation is positively correlated with Treg infiltration in HNSCC tissues
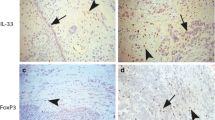
The abovementioned data indicated a correlation between stromal IL-33 and poor prognosis, but the target of IL-33 remains uncertain. Previous studies have shown that IL-33 accelerates tumor progression by increasing the accumulation of immunosuppressive cells such as MDSCs and Tregs [17, 18]. Considering these data and our results showing that Treg infiltration into HNSCC was negatively correlated with prognosis, we speculated that stromal IL-33 might affect intratumoral accumulation of Tregs in HNSCC. To test this hypothesis, we examined the prevalence of tumor-infiltrating Tregs and IL-33+ cells in serial sections by IHC of HNSCC tumors. Tregs were mainly found to infiltrate the tumor stroma (Fig. 2a). We calculated the number of IL-33+ cells and Foxp3+ Tregs in the same area of serial sections (Fig. 2b, c), and then analyzed their correlation. The level of Treg accumulation was higher in the high IL-33 group compared with the low IL-33 group (P < 0.001, Fig. 2d). Moreover, Spearman’s correlation was used to assess the relationship between Tregs and IL-33+ cells. A strong positive correlation was found between these two cell types in tumor stroma (r2 = 0.31, P < 0.001, Fig. 2e).

Correlation between IL-33 and Tregs in HNSCC patients. a Representative images of Foxp3 staining (green arrows) in tumor and stroma. b, c Representative images of Foxp3 (b, green arrows) and IL-33 (c, red arrows) staining in stroma of the same tissue area. d Treg infiltration in low IL-33-expressing patients was lower than in that in high IL-33-expressing patients. The Student’s t test was used for data analysis (2.88 ± 0.77 and 7.7 ± 1.11). e Correlation between IL-33+ cells and Treg infiltration within tumors (r2 = 0.31). Data are the mean ± SEM. ***P < 0.001
ST2 expression in Tregs from HNSCC tissue is higher than that in Tregs from peripheral blood
Our results indicated that stromal IL-33 was positively correlated with Tregs in the HNSCC TME. To further investigate the biological interactions between IL-33 and Foxp3+ Tregs, we analyzed the expression of ST2 in CD4+Foxp3+ Tregs among both healthy donor and patient PBMCs as well as TILs from dissected tumor tissues (Fig. 3b). Quantitative analyses showed that the proportion of ST2+ Tregs among total Tregs was significantly higher in TILs compared with those among PBMCs from healthy control or HNSCC patients and no significant difference was found between healthy donor and patient PBMCs (Fig. 3c). To further determine whether the TME induced an increase of ST2+ Tregs, we cultured CD4+ T cells from healthy donors with control DCs or SNU899-stimulated DCs at a 10:1 ratio, as described in the “Materials and methods” [21]. As expected, co-culture with SNU899-stimulated DCs increased the proportion of ST2+ Tregs among PBMCs compared with control DCs (Fig. 3d, e). Moreover, IL-33 was not detected in SNU899 TSN by an ELISA (data not shown). These observations indicated that the microenvironment of LSCC increased the proportion of ST2-expressing Tregs in response to IL-33.

ST2-expressing Tregs are enriched in the HNSCC TME. a Gating strategy for CD3+CD4+ cells. b Proportions of ST2-expressing Tregs among healthy donor PBMCs, (PBMC-HD), PBMCs from HNSCC patients (PBMC-HNSCC), and TILs. c Quantitative analysis of the percentage of ST2+Foxp3+ Tregs among total Tregs. d Proportion of ST2-expressing Tregs in co-cultures (DCs and CD4+ T cells) containing control DCs (control DC) or SNU899-stimulated DCs (SNU899 DC). Data were analyzed by one-way ANOVA. e Quantitative analysis of the percentage of ST2+Foxp3+ Tregs among total Tregs in co-cultures of control DCs or SNU899-stimulated DCs. The Student’s t test was used for data analysis. Data are the mean ± SEM and representative of three independent experiments. n.s. not significant, ***P < 0.001
IL-33 expands the Treg population, induces suppressive cytokine production, and enhances Treg-mediated suppression of proliferation in vitro
To further investigate the effect of IL-33 on Tregs, recombinant human IL-33 was added to co-cultures of SNU899-stimulated DCs and healthy donor CD4+ T cells at a 1:10 ratio, as described in the Materials and Methods. More Foxp3+ Tregs were derived from the additional IL-33 group than the control group. Moreover, an anti-ST2 mAb abrogated the effect of IL-33 (Fig. 4a). To investigate the overall effect of IL-33 on the suppressive functions of Tregs, we examined the levels of IL-10 and TGF-β1 in co-cultures by ELISAs. IL-10 and TGF-β1 levels were only correspondingly increased in conditioned supernatants with additional IL-33, whereas blockade of ST2 weakened the production of these cytokines (Fig. 4b). Taken together, these results indicated that IL-33 increased Treg proportions and induced the production of immunosuppressive cytokines.

Effects of IL-33 on Tregs in vitro. a CD4+T cells from healthy donors were co-cultured with SNU899-stimulated DCs under the following treatments: control medium, IL-33, and IL-33 with anti-ST2 for 3 days. Representative images of Foxp3+ Treg proportions under different treatments (left) and the percentage of Tregs among CD4+ T cells (right) are shown. b Levels of Treg-secreting cytokines TGF-β1 and IL-10 in conditioned supernatant were detected by ELISAs. c CD4+CD25+CD127low Tregs and CD4+CD25− T responder cells were isolated by magnetic cell separation from CD4+ T cells collected in co-culture with SNU899-stimulated DCs. Tregs were seeded together with responder cells labeled with CFSE at ratios of 1:2 and 1:4 in the presence of anti-CD3 and anti-CD28 antibodies. Proliferation of responder cells was examined by flow cytometry after 72 h. One representative image is shown. d Proliferation rates of responder cells under different treatment conditions. Data are the mean ± SEM and representative of three independent experiments. Data were analyzed by one-way ANOVA. **P < 0.01, ***P < 0.001
The suppressive function is a characteristic of Tregs. To verify that IL-33 modulated the functions of Tregs, especially Treg-mediated suppression, we isolated CD4+CD25+CD127low Tregs and CD4+CD25− responder cells from CD4+ T cells co-cultured with SNU899-stimulated DCs because of the low yield of Tregs from TILs. When stimulated with anti-CD3/anti-CD28 without Tregs, responder T cells proliferated and IL-33 only mildly promoted proliferation. Nevertheless, additional Tregs significantly decreased the proliferation of responder T cells, while the presence of IL-33 further enhanced the immunosuppressive function of Tregs (Fig. 4c). Consistent with the above observations, blockade of ST2 attenuated this suppressive effect. Quantitative analyses showed that IL-33 mildly increased the percentage of proliferating T-responder cells in the co-culture system, while significantly decreasing the percentage of proliferating T-responder cells in the presence of Tregs (Fig. 4d), suggesting that Tregs were more responsive to IL-33 than responder T cells. In addition, the effect of rIL-33 was dose dependent (Supplementary Fig. 2). These results suggested IL-33 upregulated the suppressive function of Foxp3+ Tregs in vitro.
GATA3 is involved in the IL-33/ST2 signaling pathway acting on Foxp3+ Tregs
GATA3 is a known transcription factor that regulates ST2 expression in Th2 cells. Recent studies have revealed that the Foxp3 promoter contains GATA3-binding sites, and that GATA3 is important for Treg functions [16, 22, 23]. Because of the above observations, we postulated that IL-33 promoted Foxp3+ Treg proliferation through GATA3 activation. To test this hypothesis, we examined expression of GATA3 in Tregs from co-cultures if SNU899-stimulated DCs and healthy donor CD4+ T cells under different interventions. As shown in Fig. 5a, IL-33 increased the percentage of Foxp3+GATA3+ Tregs in co-cultures, while an anti-ST2 mAb abrogated this effect. Further quantitative analysis showed that Foxp3+GATA3+ Tregs were significantly increased by stimulation with IL-33 and downregulated by the anti-ST2 mAb (Fig. 5b). Taken together, these data indicate that IL-33/ST2 signaling facilitates Foxp3+ Tregs, and GATA3 may be involved in this pathway.

IL-33 regulates Foxp3 through promotion of GATA3 expression. a CD4+ T cells cultured with SNU899-stimulated DCs under different treatment conditions were collected and stained for Foxp3 and GATA3. The percentage of Foxp3+GATA3+ Tregs was then examined by flow cytometry with gating on CD4+Foxp3+ Tregs. Representative scattergrams are shown. b Quantitative analysis of the percentage of Foxp3+GATA3+ double-positive Tregs. Data were analyzed by one-way ANOVA. Data are the mean ± SEM and representative of three independent experiments. **P < 0.01
Discussion
Tregs have been suggested to contribute to tumor progression by suppressing local antitumor immunity in the TME [8, 24]. Our previous studies demonstrated the association between Tregs and HNSCC progression [9, 25, 26]. However, many aspects of the highly complex process of Treg activation and regulation upon recruitment in the TME remain incompletely understood.
IL-33 is defined as an ‘alarmin’ and is implicated in tumor-associated inflammation and tumor progression through immune regulation [11, 18]. Some studies have shown that IL-33 is critical for HNSCC tumor invasion, and that it is associated with a poor prognosis [27, 28]. In addition, IL-33 has been shown to indirectly/directly promote Treg proliferation in several situations [16, 29, 30], and ST2 signaling is important for the functions of Treg [16]. To expand the understanding of the correlation between IL-33 and Tregs in the HNSCC TME, we detected the expression of IL-33 and Foxp3 in 68 newly presenting HNSCC patients that had not received any previous treatment for cancer. Our results revealed that the level of stromal IL-33 was significantly higher in advanced stage HNSCC than in the early stage and positively correlated with a poor prognosis and Foxp3+ cells. In contrast, IL-33 in tumor epithelial cells was significantly reduced in the advanced stages of HNSCC compared with early stages (Supplementary Fig. 1). This discrepancy indicates distinct biological functions between parenchymal and stromal IL-33. In normal tissues, IL-33 is mainly expressed in the nuclei of epithelial and endothelial cells, and is regarded as an ‘alarmin’ [31]. The lack of a ‘danger’ signal is a major mechanism of immune invasion in the TME [12]. Liu et al. found that IL-33 is lower in cancer cells than normal epithelium, especially in poorly differentiated cells [32]. Our results reflected these findings that IL-33 expression is lower in poorly differentiated cells than well or moderately differentiated cancer cells, and parenchymal IL-33 levels are lower in both the advanced T stage and clinical stages.
Stromal IL-33, which is derived from carcinoma-associated fibroblasts (CAFs) and bone marrow stromal tissues, is the main source of IL-33 in the TME [17, 27] and is important for the local immune status and tumor progression. In previous studies, Chen et al. found that CAFs in the TME promote tumor aggressiveness [27], and Xiao et al. showed that IL-33 expressed in the tumor stroma is crucial for accumulation of MDSCs [17]. These studies revealed the association between stromal IL-33 and tumor progression. Our results suggest that stromal IL-33, but not parenchymal IL-33, serves as a negative prognostic factor and is positively correlated with Foxp3+ Tregs.
Several studies have reported the effects of IL-33 on Tregs in various non-lymphoid organs. For example, IL-33 increases the suppressive function of ST2+ Tregs in the intestines and the expression of ST2 [16]. Visceral adipose tissue-resident Tregs expand and prevent inflammation after IL-33 stimulation [22]. IL-33 also participates directly in the intratumoral accumulation of Tregs in a breast cancer model [18]. Lung-resident Tregs are induced to Th2-like Tregs in the presence of IL-33 [33]. Thus, IL-33 plays a central role in modulating tissue-resident Tregs. Our finding that tumor-infiltrating Tregs expressed high levels of ST2, which was consistent with these previous observations, indicated that tissue-resident Tregs respond to IL-33 signals [16, 22, 23, 33]. We also found that IL-33 could induced naïve T cells to differentiate into ST2+Foxp3+ Tregs, and increased the proportion of Tregs and levels of cytokines such as IL-10 and TGF-β1 secreted by Tregs, promoting their suppressive function. Furthermore, the anti-ST2 antibody attenuated these effects, indicating that ST2 could be a potential therapeutic target. Taken together, IL-33 stimulation may facilitate Treg-mediated immune suppression in the TME.
GATA3 is a Th2 marker [14, 15] that is important for the expression of ST2 [34, 35]. Schiering’s group showed that the Foxp3 locus contains GATA3-binding sites in its promoter [16]. Hence, we propose the hypothesis that GATA3 is involved in the maintenance of Treg functions. Our data indicated that IL-33 increased the expression level of GATA3 in Tregs, and that blockade of ST2 impaired this effect, suggesting that GATA3 in Tregs promotes Treg-mediated suppression after IL-33 stimulation.
There are some limitations in this study. First, in vivo studies are needed to validate the effect of IL-33 on Tregs and the therapeutic effect of ST2 blockade. In addition, further studies are required to investigate the origin of IL-33 in the TME. Based on our results, we postulate that stromal IL-33 acts as a crucial factor in the expansion of Tregs by activation of ST2 and GATA3 in the HNSCC microenvironment to suppress antitumor immune responses. Thus, Tregs may serve as a potential target against HNSCC progression, which may offer a new strategy to improve the efficacy of immune therapy.
Abbreviations
- LSCC:
-
Laryngeal squamous cell carcinoma
- TSN:
-
Tumor supernatant
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA Cancer J Clin 65:87–108
Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, Worden F, Saba NF, Iglesias Docampo LC, Haddad R, Rordorf T, Kiyota N, Tahara M, Monga M, Lynch M, Geese WJ, Kopit J, Shaw JW, Gillison ML (2016) Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med 375:1856–1867
Harrington KJ, Ferris RL, Blumenschein G Jr, Fayette CAD, Licitra J, Kasper L, Even S, Vokes C, Worden EE, Saba F, Kiyota NF, Haddad N, Tahara R, Grunwald M, Shaw V, Monga JW, Lynch M, Taylor M, DeRosa F, Morrissey M, Cocks L, Gillison K, Guigay ML J (2017) Nivolumab versus standard, single-agent therapy of investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck (CheckMate 141): health-related quality-of-life results from a randomised, phase 3 trial. Lancet Oncol 18:1104–1115
Li Q, Prince ME, Moyer JS (2015) Immunotherapy for head and neck squamous cell carcinoma. Oral Oncol 51:299–304
Ferris RL (2015) Immunology and immunotherapy of head and neck cancer. J Clin Oncol 33::3293–3304
Houghton AN, Guevara-Patino JA (2004) Immune recognition of self in immunity against cancer. J Clin Invest 114:468–471
Mandal R, Senbabaoglu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N, Lee KW, Ganly I, Hakimi AA, Chan TA, Morris LG (2016) The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 1:e89829
Zou W (2006) Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 6::295–307
Sun W, Li WJ, Wei FQ, Wong TS, Lei WB, Zhu XL, Li J, Wen WP (2016) Blockade of MCP-1/CCR4 signaling-induced recruitment of activated regulatory cells evokes an antitumor immune response in head and neck squamous cell carcinoma. Oncotarget 7:37714–37727
Sun W, Wei FQ, Li WJ, Wei JW, Zhong H, Wen YH, Lei WB, Chen L, Li H, Lin HQ, Iqbal M, Wen WP (2017) A positive-feedback loop between tumour infiltrating activated Treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br J Cancer 117:1631–1643
Cayrol C, Girard JP (2014) IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol 31:31–37
Matzinger P (2002) The danger model: a renewed sense of self. Science 296:301–305
Peine M, Marek RM, Lohning M (2016) IL-33 in T cell differentiation, function, and immune homeostasis. Trends Immunol 37:321–333
Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, Paul W (2009) IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci USA 106:13463–13468
Maneechotesuwan K, Xin Y, Ito K, Jazrawi E, Lee KY, Usmani OS, Barnes PJ, Adcock IM (2007) Regulation of Th2 cytokine genes by p38 MAPK-mediated phosphorylation of GATA-3. J Immunol 178:2491–2498
Schiering C, Krausgruber T, Chomka A, Frohlich A, Adelmann K, Wohlfert EA, Pott J, Griseri T, Bollrath J, Hegazy AN, Harrison OJ, Owens BMJ, Lohning M, Belkaid Y, Fallon PG, Powrie F (2014) The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513:564–568
Xiao P, Wan X, Cui B, Liu Y, Qiu C, Rong J, Zheng M, Song Y, Chen L, He J, Tan Q, Wang X, Shao X, Liu Y, Cao X, Wang Q (2016) Interleukin 33 in tumor microenvironment is crucial for the accumulation and function of myeloid-derived suppressor cells. Oncoimmunology 5:e1063772
Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, Lukic ML (2014) Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer 134:1669–1682
Brunner SM, Rubner C, Kesselring R, Martin M, Griesshammer E, Ruemmele P, Stempfl T, Teufel A, Schlitt HJ, Fichtner-Feigl S (2015) Tumor-infiltrating, interleukin-33-producing effector-memory CD8(+) T cells in resected hepatocellular carcinoma prolong patient survival. Hepatology 61:1957–1967
Kuang DM, Peng C, Zhao Q, Wu Y, Chen MS, Zheng L (2010) Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology 51:154–164
Wei FQ, Sun W, Wong TS, Gao W, Wen YH, Wei JW, Wei Y, Wen WP (2016) Eliciting cytotoxic T lymphocytes against human laryngeal cancer-derived antigens: evaluation of dendritic cells pulsed with a heat-treated tumor lysate and other antigen-loading strategies for dendritic-cell-based vaccination. J Exp Clin Cancer Res 35:18
Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, Fagarasan S, Mielke LA, Afshar-Sterle S, Masters SL, Nakae S, Saito H, Wentworth JM, Li P, Liao W, Leonard WJ, Smyth GK, Shi W, Nutt SL, Koyasu S, Kallies A (2015) The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol 16:276–285
Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, Wagers A, Germain RN, Benoist C, Mathis D (2015) Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab 21:543–557
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10:942–949
Sun W, Li WJ, Wu CY, Zhong H, Wen WP (2014) CD45RA-Foxp3high but not CD45RA + Foxp3low suppressive T regulatory cells increased in the peripheral circulation of patients with head and neck squamous cell carcinoma and correlated with tumor progression. J Exp Clin Cancer Res 33:35
Sun W, Li WJ, Fu QL, Wu CY, Lin JZ, Zhu XL, Hou WJ, Wei Y, Wen YH, Wang YJ, Wen WP (2015) Functionally distinct subsets of CD4(+) regulatory T cells in patients with laryngeal squamous cell carcinoma are indicative of immune deregulation and disease progression. Oncol Rep 33:354–362
Chen SF, Nieh S, Jao SW, Wu MZ, Liu CL, Chang YC, Lin YS (2013) The paracrine effect of cancer-associated fibroblast-induced interleukin-33 regulates the invasiveness of head and neck squamous cell carcinoma. J Pathol 231:180–189
Ishikawa K, Yagi-Nakanishi S, Nakanishi Y, Kondo S, Tsuji A, Endo K, Wakisaka N, Murono S, Yoshizaki T (2014) Expression of interleukin-33 is correlated with poor prognosis of patients with squamous cell carcinoma of the tongue. Auris Nasus Larynx 41:552–557
Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, Turnquist HR (2014) IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2 + regulatory T cells. J Immunol 193:4010–4020
Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, Bluestone JA, Locksley RM (2015) Interleukin-33 and interferon-gamma counter-regulate group 2 innate lymphoid cell activation during immune perturbation. Immunity 43:161–174
Moussion C, Ortega N, Girard JP (2008) The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One 3:e3331
Liu J, Shen JX, Hu JL, Huang WH, Zhang GJ (2014) Significance of interleukin-33 and its related cytokines in patients with breast cancers. Front Immunol 5:141
Chen CC, Kobayashi T, Iijima K, Hsu FC, Kita H (2017) IL-33 dysregulates regulatory T cells and impairs established immunologic tolerance in the lungs. J Allergy Clin Immunol 140::1351–1363 e1357
Nawijn MC, Dingjan GM, Ferreira R, Lambrecht BN, Karis A, Grosveld F, Savelkoul H, Hendriks RW (2001) Enforced expression of GATA-3 in transgenic mice inhibits Th1 differentiation and induces the formation of a T1/ST2-expressing Th2-committed T cell compartment in vivo. J Immunol 167:724–732
Gachter T, Werenskiold AK, Klemenz R (1996) Transcription of the interleukin-1 receptor-related T1 gene is initiated at different promoters in mast cells and fibroblasts. J Biol Chem 271:124–129
Funding
Yi-hui Wen was supported by grants from the Natural Science Foundation of Guangdong Province, China (Nos. 2018A030313667, 2017A030310362, and 2015A030310236). Wei-ping Wen was supported by Guangzhou Science and Technology Programme (No. 201605030003) and the Sun Yat-Sen University 5010 Plan (2010004). Vivian Wai Yan Lui was supported by the General Research Fund from the Research Grant Council, Hong Kong (No. 1711484 and No. 17121616), Theme-based Research from the Research Grant Council, Hong Kong (T12-401/13-R), and a Direct Grant for Research (No. 2016.095), the Chinese University of Hong Kong. Wei Sun was supported by the Natural Science Foundation of China (No. 81602365) and Natural Science Foundation of Guangdong Province, China (No. 2016A030310153).
Author information
Authors and Affiliations
Contributions
YW, WS, and WW conceived and designed the experiments. HL and YW performed the experiments. HL and YZ evaluated immunohistochemistry. XW, LC, and WS made important contributions to collecting blood and tissue samples. HL, YW, and WW wrote the manuscript. VWYL commented on and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The study was approved by the Ethics Committee of the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China (Approval no. 2012-349). Informed consent was obtained from all participants prior to enrollment in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wen, Yh., Lin, Hq., Li, H. et al. Stromal interleukin-33 promotes regulatory T cell-mediated immunosuppression in head and neck squamous cell carcinoma and correlates with poor prognosis. Cancer Immunol Immunother 68, 221–232 (2019). https://doi.org/10.1007/s00262-018-2265-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-018-2265-2