
Abstract
Radiotherapy (RT) utilizes the DNA-damaging properties of ionizing radiation to control tumor growth and ultimately kill tumor cells. By modifying the tumor cell phenotype and the tumor microenvironment, it may also modulate the immune system. However, out-of-field reactions of RT mostly assume further immune activation. Here, the sequence of the applications of RT and immunotherapy is crucial, just as the dose and fractionation may be. Lower single doses may impact on tumor vascularization and immune cell infiltration in particular, while higher doses may impact on intratumoral induction and production of type I interferons. The induction of immunogenic cancer cell death seems in turn to be a common mechanism for most RT schemes. Dendritic cells (DCs) are activated by the released danger signals and by taking up tumor peptides derived from irradiated cells. DCs subsequently activate T cells, a process that has to be tightly controlled to ensure tolerance. Inhibitory pathways known as immune checkpoints exist for this purpose and are exploited by tumors to inhibit immune responses. Cytotoxic T lymphocyte antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1) on T cells are two major checkpoints. The biological concepts behind the findings that RT in combination with anti-CTLA-4 and/or anti-PD-L1 blockade stimulates CD8+ T cell-mediated anti-tumor immunity are reviewed in detail. On this basis, we suggest clinically significant combinations and sequences of RT and immune checkpoint inhibition. We conclude that RT and immune therapies complement one another.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neoplastic diseases are very complex and tumors are far more than just an accumulation of tumor cells. Non-cancerous cells such as fibroblasts, cells that comprise the blood vessels and immune cells are highly interconnected with the tumor cells and form the cancer microenvironment. The tumors are characterized by sustained proliferation, avoidance of growth regulation, cell death resistance, genomic instability and mutations, modified energy supply, angiogenesis, invasion and metastases, and immortalization, as well as inflammation and active immunosuppression [1]. Tumor cells and immune cells are in a dynamic process and the immune system therefore not only protects against cancer development but also shapes and impacts on the phenotype of emerging tumors [2]. The immunoediting concept of Schreiber and colleagues that comprises the elimination, equilibrium and escape phase [3] opened the minds of oncologists of different specializations to the prospect that modulation of the immune system can strongly contribute to cancer therapy success.
Immunogenicity of chemotherapy and radiotherapy
But how do classical treatment modalities such as chemotherapy and radiotherapy that are commonly known to induce immunosuppression fit into this concept?
For chemotherapy, it has become clear that distinct chemotherapeutic agents such as oxaliplatin and anthracyclines induce immunogenic cell death which is characterized by the exposure of endoplasmic reticulum-derived proteins such as calreticulin (CRT) and by the release of immune-activating danger signals such as high-mobility group box 1 (HMGB1) protein, heat shock protein 70 (Hsp70) and adenosine triphosphate (ATP) [4, 5]. Such immunogenic drugs are currently being tested in clinical trials [6].
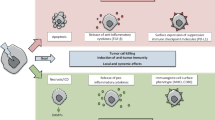
Radiotherapy uses high energy radiation to locally treat cancer. It primarily aims to induce DNA damage and ER stress via reactive oxygen species (ROS). These targeted effects result in cell cycle arrest of the tumor cells that try to repair the induced damage and concomitantly in reduction in the clonogenic potential. The latter is termed “clonogenic survival” by radiobiologists. It describes the capability of a single cell to form colonies again, but does not state anything about cell death. Cells whose cell cycle is arrested, as also occurs in senescence, should be regarded as alive, and the expression “replicative cell death” which alludes to the loss of the clonogenic capacity should be avoided [7]. Besides the targeted effects of ionizing radiation (X-ray) used in RT, non-targeted, systemic effects also exist [8]. In response to radiation, an increased expression of MHC-I and MHC-II molecules, CD80, adhesion molecules, stress ligands, Hsp70 and death receptors on tumor cell surfaces can be observed [9]. Furthermore, immune-activating chemokines, cytokines, exosomes and danger signals are released [10]. To summarize, this may result in activation of dendritic cells (DCs) that then initiate innate and adaptive immune responses [11] (Fig. 1).

Contribution of local and systemic effects of radiotherapy to tumor cell killing. Radiotherapy-induced increased generation of reactive oxygen species (ROS) induces DNA damage that is immediately sensed and, if possible, repaired (DNA damage response). Cells are arrested in the cell cycle and no longer proliferate. Senescent cells may result or the cells are driven toward death during the course of mitosis (mitotic catastrophe). Unrepaired DNA damage finally results in cell death. Additionally, systemic responses can be initiated arising from ROS and endoplasmic reticulum (ER) stress by modification of the tumor cell surface after exposure to ionizing radiation and by release of danger signals, inflammatory cytokines, exosomes and (modified) tumor antigens. Dendritic cells (DCs) mature are activated and initiate innate and adaptive immune responses. Ags antigens, CRT calreticulin, iDC immature DC, mDC mature DC
Systemic immune responses induced by radiation with further immune stimulation
However, the clinical routine shows that patients who receive RT very rarely develop regression of metastases outside of the irradiation field. This suggests that out-of-field reactions of RT need further immune modulation. This has been proven by preclinical models, such as the syngeneic mammary carcinoma 67NR mouse model. Regression of the non-irradiated ectopic tumor, outside of the irradiation field of the other tumor on the opposite flank, only occurred when additional immune activation was initiated. In this case, the number of DCs was enhanced with the growth factor Fms-related tyrosine kinase 3 ligand (Flt3-L). The resulting systemic immune reaction was tumor specific and dependent on T cells [12]. We found that local anti-tumor immune reactions that are induced by multimodal treatment of the tumor with RT, CT, hyperthermia and additional immune stimulation with the pan-caspase inhibitor zVAD-fmk are also mediated by T cells and dependent on the danger signals HMGB1 and nucleotides like ATP [13]. We are currently analyzing how zVAD-fmk impacts on systemic immune responses against tumor masses outside the irradiation field, since it fosters the induction of necroptosis by blocking apoptosis. Necroptosis is a programmed form of necrosis [14], considered to be highly immunogenic and therefore ideal for triggering systemic immune responses.
Nevertheless, radiation by itself can also be a beneficial trigger to recruit and activate immune cells. In the rat insulin promoter (RIP)1-Tag5 (RT5) mouse model of spontaneous tumors, the capability of tumor antigen-reactive T cells to infiltrate malignant tissue is lost during tumor progression. This results in tumor outgrowth. A single irradiation with 2 Gy restored and even increased the capability of tumor-specific CD4+ and CD8+ T cells to migrate into the tumor [15]. Radiation triggers the normalization of the vasculature by reprogramming M2 macrophages into M1 like macrophages that display inducible nitric oxide synthase (iNOS) activity. Combination of radiation and NO-dependent normalization of the tumor vasculature in combination with adoptive T cell transfer therefore results in efficient tumor cell killing and control. Systemic application of pro-inflammatory reagents such as cytosine–guanine-rich (CpG) motifs render tumors permissive for immune infiltration [16] and RT could be an inducer of such immune stimulators [17].
Importance of the timing of combination of RT with immune stimulation
To summarize, combination of RT with additional immunotherapy is a promising approach for inducing specific anti-tumor immune responses. One must always keep in mind that the sequence of the applications is crucial. As outlined above, first, the immune cell recruitment into the tumor by RT has to be enabled, and then additional immunotherapy should be performed. Witek et al. [18] demonstrated that radiation serves as an adjuvant and preparatory step for tumor vaccination. Tumor-specific T cell responses were amplified in mice with tumors irradiated before or after vaccination with an adenoviral-based vaccine against the colorectal cancer antigen guanylyl cyclase C. Here, fractionated radiation-induced similar effects compared to single high-dose irradiation with 8 Gy.
Impact of the radiation dose and fractionation on immune stimulation
The activation of human monocyte-derived DCs was also similar when coming into contact with norm- or hypofractionated irradiated human colorectal cancer cells [19]. Nevertheless, more data are urgently needed in order to draw conclusions on whether continued fractionated application of 2 Gy that is used in classical tumor therapy is as immunogenic as fewer applications with higher single doses or a very high single dose (radiosurgery). Irradiation of intracranial GL261-luc glioblastoma mouse tumors with 10 Gy with a dose rate of 1.9 Gy/min resulted in tumor growth retardation that was significantly improved through combination with immunotherapy using anti-programmed cell death protein 1 (PD-1) monoclonal antibody (see also below). The local RT with 10 Gy enhanced the pro-inflammatory profile of GL261 gliomas and paved the way for an increased influx of CD8+ effector T cells when combined with anti-PD-1 immunotherapy [20]. However, in a TSA mouse breast carcinoma model, fractionated irradiation with single doses of 8 or 6 Gy, in particular, in combination with an antibody against the immune checkpoint protein cytotoxic T lymphocyte antigen 4 (CTLA-4) resulted in significant enhanced tumor growth retardation in the tumor outside of the irradiation field [21]. Nevertheless, single doses of 6 or 8 Gy are still high doses. Of note is that a single high dose of 20 Gy was as effective as 3 × 8 Gy or 5 × 6 Gy in retarding growth of the irradiated tumor. This indicates that local and systemic responses always have to be considered when RT is part of multimodal tumor therapies. Furthermore, it is most likely that promotion of immune responses can be induced by many single doses and fractionations of X-ray. The mechanistic basis, as the CD8+ T cell-mediated tumor cell killing, might be common, but further mechanisms might differ in part. Lower single doses impact on tumor vascularization and consecutive infiltration of immune cells in particular [22], while higher doses impact on intratumoral induction and production of type I interferon (IFN) with consecutive triggering of innate and adaptive immune mechanisms [23]. The induction of immunogenic cell death by the many doses of radiation and fractionation schemes that are applied in tumor therapy seems in turn to be a common mechanism [24].
In addition, one has to keep in mind that low and intermediate doses of ionizing radiation (≤1.0 Gy) have opposed immunological effects. Here, inflammation is attenuated and the viability of many cells is not strongly influenced [25]. This might, however, also be beneficial for anti-tumor immunity, since chronic inflammation promotes tumor outgrowth [26]. A deeper understanding of the interconnection of inflammation, cancer and DNA damage responses should lead to further improvements in radio- and immunotherapy used to treat cancer in the future.
Immune checkpoint inhibition in combination with RT
As outlined above, DCs play a central role in starting innate and adaptive immune responses by being activated by damage-associated molecular patterns (DAMPs) and by taking up tumor peptides derived from irradiated cells. Subsequently, DCs have to deliver diverse signals to T cells to activate them and ensure their survival, and to initiate their differentiation. As a safety system of the immune system, T cell activation has to be highly controlled to ensure tolerance and to avoid severe autoimmune reactions. For this purpose, in addition to multiple co-stimulatory signals, inhibitory pathways exist that are known as immune checkpoints.
It has become clear that tumors exploit immune checkpoint pathways as a mechanism of inhibiting immune responses, particularly against tumor-specific cytotoxic T cells [27]. Activated T cells are inhibited via the binding of CD80 or CD86 on DCs to CTLA-4 on T cells. CTLA-4 (CD152) was discovered in the late 1980s by French researchers who discovered that it avoids overreactions of the immune system [28]. It regulates early T cell activation. The antibody ipilimumab against CTLA-4 was approved by the US Food and Drug Administration (FDA) in 2011, and together with the checkpoint inhibitors anti-PD-1/anti-PD-L1 (approved in 2014), a breakthrough in melanoma therapy was made [29]. PD-1 was discovered in the early 1990s by a Japanese biologist who found it as a molecule expressed on dying T cells [28]. It was therefore named programmed cell death protein 1 (CD279). It limits activity of T cells in peripheral tissues at the time of inflammatory response and therefore later than CTLA-4. Hence, PD-1 is the major immune resistance mechanism in the tumor microenvironment. Figure 2 depicts the immunobiological concept of why RT in combination with anti-PD-L1 blockade stimulates CD8+ T cell-mediated anti-tumor immunity. Currently, in 2015, the largest group of the over 250 trials registered at clinicaltrials.gov with immunotherapies is of checkpoint inhibitors [30]. However, one has to acknowledge that although the responses in some patients are impressive, they are often of limited duration and not present in the majority of patients. This implies that combination of checkpoint inhibitors and classical therapies such as RT have to be reinforced.

Immune-modulating irradiation as a basis for combination of radiotherapy with checkpoint inhibitors. Immune-stimulating irradiation results in the release of damage-associated molecular patterns (DAMPs) and (modified) tumor antigens. Dendritic cells (DCs) are recruited and activated and take up the antigens (Ags). DCs then migrate to lymph nodes and cross-present the tumor Ags to T cells. The priming of T cells is highly controlled to avoid autoimmune reactions and to maintain self-tolerance. There are various ligand–receptor interactions between antigen-presenting cells such as DCs and T cells that regulate the T cell response to Ag. The up-regulation of the co-stimulatory molecules CD80 and CD86 on DCs in response to radiation-induced DAMPs delivers co-stimulatory signals to T cells. However, mostly pairs of co-stimulatory–inhibitory receptors bind the same ligand(s) (e.g., CD28 and cytotoxic T lymphocyte antigen 4 (CTLA-4) both bind to CD80/CD86). Of note is that the inhibitory receptor is commonly up-regulated later on, namely after T cell activation. A longer-lasting T cell activation can therefore be achieved when antibodies designed to relieve immunosuppression such as anti-CTLA-4 are administered in addition to RT. Besides these immune-activating effects of RT, potentially deleterious effects exist. Tumor cells can up-regulate programmed cell death protein 1 ligand (PD-L1) in response to radiation. PD-L1 interaction with PD-1 on T cells then shuts down the cytotoxic T cell response within the tumor. This is another reason for combining RT with Abs that inhibit immune checkpoint proteins or the interaction with their respective receptors. Since anti-CTLA-4 and anti-PD-L1 use non-redundant immune mechanisms to enhance anti-tumor immunity and act at different time points during establishment of anti-tumor immune reactions, dual immune checkpoint inhibition in combination with RT might be the therapy of choice for the future
Single immune-modulatory agents have been shown to enhance the anti-tumor effects of RT, even in poorly immunogenic tumors. In the metastatic mouse mammary carcinoma 4T1 model, for example, only combination of RT with a single dose of 12 Gy with anti-CTLA-4 mAb 9H10 extends the survival of mice in a CD8+ T cell-dependent manner [31]. Nevertheless, tumor-induced immunosuppression often restricts the success of radio-immunotherapies. Therefore, combination of antibodies (Abs) designed to stimulate immunity (e.g., anti-CD137 or anti-CD40) or relieve immunosuppression (anti-PD-1) with RT might overcome this suppression. Mice bearing established orthotopic AT-3 mammary tumors were all cured when anti-CD137 and anti-PD-1 monoclonal Abs were combined with single (12 Gy) or fractionated (4 × 4 Gy or 4 × 5 Gy) RT [32]. Furthermore, combination of two checkpoint inhibitors that target immune checkpoints both at early (anti-CTLA-4) and later (anti-PD-1) time points of T cell activation in concerted action with RT could be another solution. Addition of PD-L1 blockade reinvigorates exhausted T cells and improves the response to radiation plus anti-CTLA4 Ab treatment. While anti-CTLA-4 treatment predominantly decreased regulatory T cells, anti-PD-L1 treatment increased the frequency of tumor-infiltrating CD8+ T cells [33]. Kim et al. [34] recently demonstrated in C57BL/6 mice that were implanted with the mouse glioma cell line GL261 transfected with luciferase that combination of anti-TIM-3 with anti-PD-1 and stereotactic radiosurgery was synergistic and conferred a significant survival benefit.
Importance of the timing of RT with immune checkpoint inhibition
A big challenge is to identify not only the most beneficial combination of certain radiation schedules with immunotherapies but also the chronological sequence. Immune cells are recruited into the tumor after irradiation. Since RT is mostly delivered in fractions, re-irradiation of the tumor might kill the infiltrating immune cells that are already fighting the tumor or are activated for migration into the next lymph node to prime cytotoxic T cells. Our own work demonstrates that when starting 2 days after the last irradiation with 2 × 5 Gy, immune cells migrate into CT-26 colorectal tumors and rest there only for a few days (Frey et al. [35] manuscript in preparation). Additional immune activation by immunotherapy should be administered in this time frame and RT should be paused. This suggests that immunotherapies should be applied in the middle to the end of the working week to exploit the radiation pause at the week ends.
Adaptive immune resistance mechanisms further determine the design of multimodal therapies. When a weak endogenous immune response is present, no up-regulation of PD-L1 ligand on tumor cells is likely. In this case, therapy with anti-PD-L1/anti-PD-1 Abs is not effective. Inducers of anti-tumor immunity such as RT also have some deleterious effects and can induce expression of PD-L1 ligand on tumor cells and tumor-associated antigen-presenting cells (APCs). However, here consecutive therapy with anti-PD-L1/anti-PD-1 Abs becomes beneficial and finally results in induction of anti-tumor immunity [27]. Three days after irradiation with 12 Gy of a spontaneous mammary tumor (TUBO) in BALB/c mice, an increase in the expression of PD-L1 was observed on DCs and on tumor cells. Combined treatment of the tumor with RT plus anti-PD-L1 was most effective and dependent on CD8+ T cells that induce apoptosis of myeloid-derived suppressor cells (MDSCs) through tumor necrosis factor (TNF) alpha [36]. Furthermore, interferon gamma produced by CD8+ T cells is responsible for mediating PD-L1 up-regulation on tumor cells after delivery of fractionated RT with 5 × 2 Gy, as demonstrated with the CT-26 colorectal tumor model. Of special note is that again the dosing schedule is critical to the outcome with RT: The survival of the mice was significantly improved only when anti-PD-L1 Ab was given concomitantly, namely at the beginning or the end of the fractionated irradiation. Delivery of anti-PD-1 Ab one week after the last irradiation did not improve the sole effects of RT on the tumor [37].
Clinical evidence of beneficial interactions of RT with immunotherapy
Although there is strong evidence from preclinical work that RT and immunotherapy fit together, clinical reports detailing the interaction of RT with immunotherapies are limited, but on the way (summarized in [38]). Immunological effects of RT alone are mostly described in patients with melanoma. Furthermore, DCs loaded with antigens from irradiated autologous proliferating tumor cells are superior to vaccination with antigens only. For the combination of IL-2 with RT, higher single doses seem to be of advantage. Abscopal, namely immune-mediated systemic, anti-tumor effects of RT are predominantly observed after combination with anti-CTLA-4 Ab and anti-PD-L1 Ab. Again, tumor regressions are only observed in a subset of patients. Metastatic melanoma patients with tumors showing high PD-L1 expression did not respond to RT plus anti-CTLA4. This suggests that radiation, anti-CTLA4 and anti-PD-L1 have distinct effects on the TCR repertoire, regulatory T cells and T cell exhaustion. Their combination promotes response and immunity through distinct mechanisms, as outlined above [33]. A further breakthrough is the study of Golden et al. [39] demonstrating that combination of radio(chemo)therapy with granulocyte-macrophage colony-stimulating factor (GM-CSF) generates systemic anti-tumor responses not only in melanoma, but also in other solid tumor entities, such as non-small cell lung carcinoma (NSCLC) and breast cancer. Immunotherapy works best for tumors with high somatic mutation prevalence. Melanoma, lung cancer and colorectal cancer have this prevalence [40]. Nevertheless, it clearly also works in breast cancer and glioma, which display lower prevalence. Further stress stimuli such as RT and CT might enhance the mutation rates and thereby support additional immunotherapy.
Tremelimumab and ipilimumab are two fully humanized anti-CTLA-4 antibodies that have advanced to testing in clinical trials. Again, in the beginning most studies focused on melanoma [41], but there are currently several ongoing trials on combination of RT and anti-CTLA-4 antibody therapy for other tumor entities, such as unresectable pancreatic cancer (NCT02311361), metastatic castration-resistant prostate cancer (NCT00861614), metastatic NSCLC (NCT02221739), advanced cervical cancer (NCT01711515), and metastatic cancers of the liver and lungs (NCT02239900). Single-agent trials have already been initiated with the anti-PD-1 antibodies nivolumab, pembrolizumab and pidilizumab, and current efforts focus on trials of combination treatments together with efforts to identify predictive and prognostic biomarkers [42].
Conclusions and challenges
Many preclinical and more and more clinical data prove that RT and immunotherapies fit together. Concurrent application of immune checkpoint inhibitors and RT makes sense with the current knowledge of the biological modes of action. However, more clinical studies are urgently needed in order to gain deeper knowledge of side effects, such as severe inflammation and strong autoimmune reactions. No conclusive preclinical data on the most beneficial dose and fractionation of RT are available so far. From the immunological point of view, hypofractionated RT might be ahead, since in the longer radiation pauses the immune system has time to act and react. Combined therapies work best for tumors with mutational processes such as melanoma, lung cancer, bladder cancer, esophageal cancer, colorectal cancer, cervical cancer, and head and neck cancer, but others should not remain unconsidered. Furthermore, also malignancies of hematopoietic origin have to be followed up since they are also capable of co-opting their local environment in order to escape immune attack. In selected subtypes of Hodgkin lymphoma (nodular sclerosing Hodgkin lymphoma), for example, PD-1 ligands are over-expressed due to a genetic amplification of the loci encoding them [43]. They should therefore be ideal candidates for combined treatment with RT plus immune checkpoint inhibitors. Since RCT is usually performed in clinics for most tumor entities, chemotherapeutic agents that induce immunogenic cancer cell death, such as anthracyclines and oxaliplatin, should be considered.
Of further note is that the time frame of response differs between classical therapies and immunotherapies. The response of CT and small molecule inhibitors is within weeks, but the response of immune checkpoint inhibitors is within months. Sometimes initial increase in metastatic lesions due to infiltrating immune cells even occurs. This provides a case for re-evaluation of response criteria such as time to progression, to name just one [27]. Since multiple and diverse anti-tumor and immune escape mechanisms are triggered by distinct checkpoint inhibitors and radiation schemes, clinical trials should always be accompanied by translational research projects to define prognostic and predictive immunobiomarkers [44]. Furthermore, since damage induced by ionizing radiation strongly impacts on the tumor microenvironment, but also on the environment of healthy tissue, multiple targets for radiosensitization and radioprotection arise. Combinations of RT with immunomodulation, vascular normalization and antifibrotic therapy have to be carefully designed and evaluated in the near future [45]. Nevertheless, it has already become clear that RT complements immune checkpoint therapies and thereby acts locally and systemically to combat cancer [46, 47].
Abbreviations
- Ab:
-
Antibody
- Ag:
-
Antigen
- APC:
-
Antigen-presenting cell
- ATP:
-
Adenosine triphosphate
- CpG:
-
Cytosine–guanine-rich motifs
- CRT:
-
Calreticulin
- CT:
-
Chemotherapy
- CTLA-4:
-
Cytotoxic T lymphocyte antigen 4
- DAMP:
-
Damage-associated molecular pattern
- DC:
-
Dendritic cell
- DNA:
-
Deoxyribonucleic acid
- ER:
-
Endoplasmic reticulum
- FDA:
-
US Food and Drug Administration
- Flt3-L:
-
Fms-related tyrosine kinase 3 ligand
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- HMGB1:
-
High-mobility group box 1
- Hsp70:
-
Heat shock protein 70
- ICAM-1:
-
Intercellular adhesion molecule-1
- IFN:
-
Interferon
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- MDSC:
-
Myeloid-derived suppressor cell
- MHC:
-
Major histocompatibility complex
- NSCLC:
-
Non-small cell lung cancer
- PD-1:
-
Programmed cell death protein 1
- ROS:
-
Reactive oxygen species
- RT:
-
Radiotherapy
- RT5:
-
Rat insulin promoter (RIP)1-Tag5 tumor mouse model
- TNF:
-
Tumor necrosis factor
- X-ray:
-
Ionizing radiation
- zVAD-fmk:
-
Z-Val-Ala-DL-Asp-FMK
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. doi:10.1016/j.cell.2011.02.013
Mittal D, Gubin MM, Schreiber RD, Smyth MJ (2014) New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol 27:16–25. doi:10.1016/j.coi.2014.01.004
Dunn GP, Old LJ, Schreiber RD (2004) The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21:137–148. doi:10.1016/j.immuni.2004.07.017
Kepp O, Senovilla L, Vitale I et al (2014) Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 3:e955691. doi:10.4161/21624011.2014.955691
Apetoh L, Ghiringhelli F, Tesniere A et al (2007) Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13:1050–1059. doi:10.1038/nm1622
Vacchelli E, Aranda F, Eggermont A, Galon J, Sautes-Fridman C, Cremer I, Zitvogel L, Kroemer G, Galluzzi L (2014) Trial watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology 3:e27878. doi:10.4161/onci.27878
Kroemer G, El-Deiry WS, Golstein P et al (2005) Classification of cell death: recommendations of the nomenclature committee on cell death. Cell Death Differ 12(Suppl 2):1463–1467. doi:10.1038/sj.cdd.4401724
Salomaa SI, Wright EG, Hildebrandt G, Kadhim MA, Little MP, Prise KM, Belyakov OV (2010) Editorial. Non-DNA targeted effects. Mutat Res 687:1–2. doi:10.1016/j.mrfmmm.2010.01.018
Hodge JW, Ardiani A, Farsaci B, Kwilas AR, Gameiro SR (2012) The tipping point for combination therapy: cancer vaccines with radiation, chemotherapy, or targeted small molecule inhibitors. Semin Oncol 39:323–339. doi:10.1053/j.seminoncol.2012.02.006
Frey B, Rubner Y, Kulzer L, Werthmoller N, Weiss EM, Fietkau R, Gaipl US (2014) Antitumor immune responses induced by ionizing irradiation and further immune stimulation. Cancer Immunol Immunother 63:29–36. doi:10.1007/s00262-013-1474-y
Gaipl US, Multhoff G, Scheithauer H, Lauber K, Hehlgans S, Frey B, Rodel F (2014) Kill and spread the word: stimulation of antitumor immune responses in the context of radiotherapy. Immunotherapy 6:597–610. doi:10.2217/imt.14.38
Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, Formenti SC (2004) Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys 58:862–870. doi:10.1016/j.ijrobp.2003.09.012
Werthmöller N, Frey B, Wunderlich R, Fietkau R, Gaipl US (2015) Modulation of radiochemoimmunotherapy-induced B16 melanoma cell death by the pan-caspase inhibitor zVAD-fmk induces anti-tumor immunity in a HMGB1-, nucleotide- and T-cell-dependent manner. Cell Death Dis 6:e1761. doi:10.1038/cddis.2015.129
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714. doi:10.1038/nrm2970
Klug F, Prakash H, Huber PE et al (2013) Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 24:589–602. doi:10.1016/j.ccr.2013.09.014
Garbi N, Arnold B, Gordon S, Hammerling GJ, Ganss R (2004) CpG motifs as proinflammatory factors render autochthonous tumors permissive for infiltration and destruction. J Immunol 172:5861–5869. doi:10.4049/jimmunol.172.10.5861
Frey B, Hehlgans S, Rödel F, Gaipl US (2015) Modulation of inflammation by low and high doses of ionizing radiation: implications for benign and malign diseases. Cancer Lett 368:230–237. doi:10.1016/j.canlet.2015.04.010
Witek M, Blomain ES, Magee MS, Xiang B, Waldman SA, Snook AE (2014) Tumor radiation therapy creates therapeutic vaccine responses to the colorectal cancer antigen GUCY2C. Int J Radiat Oncol Biol Phys 88:1188–1195. doi:10.1016/j.ijrobp.2013.12.043
Kulzer L, Rubner Y, Deloch L, Allgauer A, Frey B, Fietkau R, Dorrie J, Schaft N, Gaipl US (2014) Norm- and hypo-fractionated radiotherapy is capable of activating human dendritic cells. J Immunotoxicol 11:328–336. doi:10.3109/1547691x.2014.880533
Zeng J, See AP, Phallen J et al (2013) Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys 86:343–349. doi:10.1016/j.ijrobp.2012.12.025
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S (2009) Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res 15:5379–5388. doi:10.1158/1078-0432.ccr-09-0265
Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ (2002) Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res 62:1462–1470
Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, Fu YX, Auh SL (2011) The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res 71:2488–2496. doi:10.1158/0008-5472.can-10-2820
Rubner Y, Wunderlich R, Ruhle PF et al (2012) How does ionizing irradiation contribute to the induction of anti-tumor immunity? Front Oncol 2:75. doi:10.3389/fonc.2012.00075
Wunderlich R, Ernst A, Rodel F, Fietkau R, Ott O, Lauber K, Frey B, Gaipl US (2015) Low and moderate doses of ionizing radiation up to 2 Gy modulate transmigration and chemotaxis of activated macrophages, provoke an anti-inflammatory cytokine milieu, but do not impact upon viability and phagocytic function. Clin Exp Immunol 179:50–61. doi:10.1111/cei.12344
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140:883–899. doi:10.1016/j.cell.2010.01.025
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12:252–264. doi:10.1038/nrc3239
Couzin-Frankel J (2013) Breakthrough of the year 2013. Cancer Immunother Sci 342:1432–1433. doi:10.1126/science.342.6165.1432
Eggermont AM, Maio M, Robert C (2015) Immune checkpoint inhibitors in melanoma provide the cornerstones for curative therapies. Semin Oncol 42:429–435. doi:10.1053/j.seminoncol.2015.02.010
Pandha H, Pawelec G (2015) Immune checkpoint targeting as anti-cancer immunotherapy: promises, questions, challenges and the need for predictive biomarkers at ASCO 2015. Cancer Immunol Immunother 64:1071–1074. doi:10.1007/s00262-015-1748-7
Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, Formenti SC (2005) Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res 11:728–734
Verbrugge I, Hagekyriakou J, Sharp LL et al (2012) Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res 72:3163–3174. doi:10.1158/0008-5472.can-12-0210
Twyman-Saint Victor C, Rech AJ, Maity A et al (2015) Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520:373–377. doi:10.1038/nature14292
Kim JE, Patel MA, Mangraviti A et al (2015) 143 the combination of anti-TIM-3 and anti-PD-1 checkpoint inhibitors with focused radiation resulted in a synergistic antitumor immune response in a preclinical glioma model. Neurosurgery 62(Suppl 1):212. doi:10.1227/01.neu.0000467105.60300.04
Frey B, Rubner Y, Wunderlich R, Weiss EM, Pockley AG, Fietkau R, Gaipl US (2012) Induction of abscopal anti-tumor immunity and immunogenic tumor cell death by ionizing irradiation—implications for cancer therapies. Curr Med Chem 19:1751–1764
Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX (2014) Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 124:687–695. doi:10.1172/jci67313
Dovedi SJ, Adlard AL, Lipowska-Bhalla G et al (2014) Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res 74:5458–5468. doi:10.1158/0008-5472.can-14-1258
Barker CA, Postow MA (2014) Combinations of radiation therapy and immunotherapy for melanoma: a review of clinical outcomes. Int J Radiat Oncol Biol Phys 88:986–997. doi:10.1016/j.ijrobp.2013.08.035
Golden EB, Chhabra A, Chachoua A et al (2015) Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol 16:795–803. doi:10.1016/s1470-2045(15)00054-6
Alexandrov LB, Nik-Zainal S, Wedge DC et al (2013) Signatures of mutational processes in human cancer. Nature 500:415–421. doi:10.1038/nature12477
Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD (2014) Immune modulation in cancer with antibodies. Annu Rev Med 65:185–202. doi:10.1146/annurev-med-092012-112807
Philips GK, Atkins M (2015) Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int Immunol 27:39–46. doi:10.1093/intimm/dxu095
Kline J, Bishop MR (2015) Update on checkpoint blockade therapy for lymphoma. J Immunother Cancer 3:33. doi:10.1186/s40425-015-0079-8
Specht HM, Ahrens N, Blankenstein C et al (2015) Heat shock protein 70 (Hsp70) peptide activated natural killer (NK) cells for the treatment of patients with non-small cell lung cancer (NSCLC) after radiochemotherapy (RCTx)—from preclinical studies to a clinical phase ii trial. Front Immunol 6:162. doi:10.3389/fimmu.2015.00162
Barker HE, Paget JT, Khan AA, Harrington KJ (2015) The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer 15:409–425. doi:10.1038/nrc3958
Ngiow SF, McArthur GA, Smyth MJ (2015) Radiotherapy complements immune checkpoint blockade. Cancer Cell 27:437–438. doi:10.1016/j.ccell.2015.03.015
Frey B, Gaipl US (2015) Radio-immunotherapy: the focused beam expands. Lancet Oncol 16:742–743. doi:10.1016/s1470-2045(15)00055-8
Acknowledgments
This work was partially funded by the German Federal Ministry of Education and Research (BMBF; m4 Cluster, 16EX1021R and GREWIS, 02NUK017G) and the European Commission (DoReMi, European Atomic Energy Community’s Seventh Framework Programme (FP7/2007-2011) under Grant Agreement No. 249689).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no competing interests.
Additional information
This paper is a Focussed Research Review based on a presentation given at the Fourth International Conference on Cancer Immunotherapy and Immunomonitoring (CITIM 2015), held in Ljubljana, Slovenia, 27th–30th April 2015. It is part of a series of Focussed Research Reviews and meeting report in Cancer Immunology, Immunotherapy.
Rights and permissions
About this article

Cite this article
Derer, A., Frey, B., Fietkau, R. et al. Immune-modulating properties of ionizing radiation: rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer Immunol Immunother 65, 779–786 (2016). https://doi.org/10.1007/s00262-015-1771-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-015-1771-8