
Abstract
Groundwater quality in the Santo Domingo Irrigation District area in Baja California Sur, Mexico, indicates the presence of various salinization processes, (1) the geological matter of marine origin comprising the aquifer material suffers diagenetic effects due to its interaction with groundwater of low salinity, (2) the effects of intensive agriculture practices produce effluents that infiltrate to the saturated zone, and (3) the extraction of groundwater causes modifications in the natural flow system induces lateral flow of seawater from the coast line. However, groundwater management has been carried out with the belief that the latter is the main source of salinization. This has resulted in a policy of installing wells increasingly far from the coast, which is not solving the problem. Irrigation-return and seawater that remains in the geological units have been identified as major sources of salinization. Controls should be imposed when installing wells in contact with clayey units that form the base of the aquifer. Extracted groundwater consists of a mixture of (1) groundwater of relatively low salinity that circulates in the aquifer and (2) an extreme member with salinity different to seawater contained mainly in formations that have low permeability, which limits the aquifer underneath. The geochemistry of carbonates and cation-exchange reactions (both direct and reverse) control the concentration of Ca, Mg, Na, and HCO3, as well as pH values. The concentrations of dissolved trace elements (F, Li, Ba, Sr) suggest that the extreme saline member is different from the average seawater composition. A distinction between the salinization caused by farming practices and that blamed on seawater is defined by the use of NO3.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In Mexico, as in other countries, uncontrolled groundwater extraction in coastal aquifers produces environmental impacts such as (1) a decline in water levels, which can reaching below average sea level; this causes an inflow of groundwater from the coast line to uplands detected as an increase in salinity above background values, and (2) coastal ecosystem impacts related to changes in salinity. Salinization is one of the most widespread forms of water contamination in the world (Richter and Kreitler 1993).
Natural topographic conditions in unconfined upland coastal aquifers generate driving forces where groundwater flows to the sea. In past geological time, such flow has been flushing trapped saline water stored in coastal aquifers. Flushing of saline water depends on different factors, mainly (1) the amount and surface distribution of natural upland recharge, (2) hydraulic head, and (3) hydraulic conductivity of aquifer units. Incomplete flushing generates freshwater lenses floating over saline water, a typical example in Mexico was studied in the Yucatan Peninsula, where groundwater extraction has modified the freshwater/seawater interface producing mixing and salinization of fresh water (Graniel and others 1999).
Under natural steady-state conditions in a coastal aquifer, low salinity groundwater flow discharges into the sea restricting the landward invasion of seawater. The position of the freshwater/saltwater interface depends on the difference in densities and hydraulic heads in both liquids (Custodio and Llamas 1983). It is well known that development of groundwater resources in unconfined coastal aquifers modifies the natural discharge conditions of freshwater into the sea. Extraction lowers the water table and disturbs the dynamic balance between fresh and salt water, resulting in a reversal of groundwater flow and the possibility of inland lateral movement of seawater. This phenomenon has been described in several parts of the world, for example Schmorak (1967) reported it in Israel and Richter and Kreitler (1993) depicted illustrative examples from the United States of America. Seawater intrusion in coastal aquifers has been evaluated with different methodologies. Analytic (Glover 1959; Bear and Dagan 1962; Sakr 1999) and numerical (Rivera and others 1990; Calvache and Pulido-Bosch 1994, 1997; Zhou and others 2000; Sadeg and Karahanolu 2001) approaches have attempted to ascertain the position of the freshwater/saltwater interface, and to propose a prediction of changes in water levels and salinity resulting from different extraction schemes. The understanding of processes and chemical reactions throughout seawater intrusion and re-freshening in coastal aquifers has been investigated by several authors, for instance, Giménez and Morell (1997), using hydrogeochemical analyses, studied a coastal aquifer affected by salinization in Spain; Petalas and Diamantis (1999) identified different sources of saline water with hydrochemical techniques in a coastal aquifer of northeastern Greece; and Stuyfzand (1999) showed chemical changes related to seawater intrusion and re-freshening in Dutch coastal dunes. A modeling approach of the multicomponent ion-exchange process in a coastal aquifer of Argentina was presented by Martínez and Bocanegra (2002). Lambrakis and Kallergis (2001), with a similar methodology, defined an estimate of re-freshening time under natural recharge conditions for three coastal aquifers in Greece.
In coastal areas, the chemical composition of extracted water from wells is often a non-conservative mixture of freshwater from an aquifer unit and saltwater from the sea. However, other anthropogenic and natural sources can also contribute to salinization of groundwater in coastal aquifers. When development for agriculture or public water-supply are based on water extracted from a coastal aquifer in a semiarid area, other sources of salinity can contribute to salinization of extracted groundwater. These sources could be salts concentrated in the unsaturated zone, dissolution of salts in the saturated zone, as well as effluents related to anthropogenic activities. Other examples of groundwater salinization connected with seawater intrusion and contamination processes related to dumping of urban solid waste on the Italian coast have been described, by Chiocchini and others (1997), as an environmental concern that compromises domestic use. In northwestern Mexico, seawater intrusion and nitrate contamination, linked with irrigation that uses raw sewage (Steinich and others 1998), has been detected as a main environmental problem affecting groundwater resources. Groundwater salinization in agricultural zones may also be produced by irrigation-return effluents. Evapotranspiration, dissolution of minerals, and fertilizers in the unsaturated zone increase irrigation-return flow salinity above the initial water conditions: their infiltration thereby increases groundwater salinity.
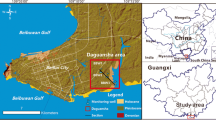
Arid climatic conditions and intensive agricultural development of particular coastal zones in northwestern Mexico encourage salinization of soil and groundwater. The Santo Domingo Irrigation District (SDID), located close to the Pacific coast of Baja California Peninsula (Fig. 1), is the largest agricultural center in that part of Mexico. The farming area has increased from 260×106 m2 in 1956 to 648×106 m2 in 1990. The number of active irrigation wells has increased from 543 in 1970, to 631 in 1977, to ~700 wells in the most recent survey (1985). Also, there are 30 wells for potable water use and 69 for cattle use. Natural recharge estimates made with numerical modeling are around 180×106 m3/year, an amount substantially lower than the average annual extraction rate for the 1957–1996 period, estimated at about 300–350×106 m3. Observed average drawdown for the period of 1957–1996 was 23 m (0.6 m/year), the average water-level elevation in 1957 was estimated to be 13 m above sea level (a.s.l.), in 1980 it was 0 m a.s.l., and in 1996, the mean water level elevation was 10 m below sea level (b.s.l.; DESISA 1997).

Location of study area and geological cross sections, showing the Santo Domingo Irrigation District
Salinity is the total amount of inorganic solid material dissolved in any natural water; it is normally expressed in terms of total dissolved solids (TDS). Over the years, salinization of extracted groundwater in several wells of the SDID has been detected. Figure 2 shows the salinization trends of extracted water from selected wells: initial TDS concentrations were quite high for some wells, but an increase is observed in all of them. This salinity increase reduces crop yield and irrigation water must be applied in excess quantities to provide considerable leaching of salts in the soil. Without presenting sound hydrogeological or hydrogeochemical evidence, early studies proposed that extracted groundwater salinization was associated with a conventional seawater intrusion phenomenon developed in the region (ACSA 1969; TMI 1979). Therefore, the implemented aquifer management strategy to reduce salinization was to abandon wells with high salinity and drill new ones in areas further away from the coast. This enabled good quality water to be obtained for only a short period of time because these new wells subsequently showed salinity increases. This is shown in Fig. 3, where the percentage of wells with extracted saline water in 1968 (before reallocation of most of the high salinity wells) and 1995 is represented. It is clear that, in the long term, a substantial improvement in water quality was not achieved with the implemented management strategy. Such results suggest that a conceptual hydrogeological model that is based only on conventional seawater intrusion may not be describing the prevailing phenomena, and imply that additional potential salinity sources need to be understood within the study area.

Salinity evolution trend in extracted water from selected wells. The location of the wells is presented in Fig. 7

Water salinity distribution for 1968 and 1995 in SDID wells. N represents the amount of sampled wells
Potential sources of groundwater salinization in agricultural zones in a coastal aquifer display specific physical and chemical characteristics, from which each can be individually identified through different hydrogeochemical techniques. In coastal regions, an understanding of the origin and nature of groundwater quality problems is needed as a starting point for mitigation: it is necessary to differentiate sources and establish their relative importance in the salinization process. This information is needed to define an optimal management strategy to control salinization of water and soil. Therefore, the general objective of this paper is to evaluate salinization sources in a coastal aquifer in an arid region in northwestern Mexico that has significant agricultural development. The SDID is used as a reference because it is regarded as a typical agricultural center supported by coastal aquifers that exist in the Baja California Peninsula and northwestern Mexico. Specific objectives include (1) definition of the mechanisms that determine the increase of salinity in the groundwater and (2) identification of the processes and chemical reactions involved. This is to be achieved using chemical inorganic indicators (major, minor and trace elements) within the context of groundwater flow systems.
Study area
The area of interest is in the state of Baja California Sur, and comprises a surface extent of about 1,900 km2 (Fig. 1). It consists of a broad plain with an altitude of less than 100 m a.s.l., and is included in the Coastal Plain Physiographic Province. It is bounded to the east by the mountain belt of the Sierra la Giganta and to the west by the Pacific Ocean. The SDID forms part of the drainage basins of the Bramonas and Santo Domingo streams, both with intermittent runoff discharging in an east–west direction to the Pacific Ocean. The region experiences an arid climate, with summer rains and extreme temperature variations. Annual rainfall is approximately 140 mm in the plain, but increases towards the mountains in the east. There is a distinct dry period in mid-summer, but winter precipitation is about 30% of the total. Records indicate an average annual temperature of 24 °C, with maximum values of 40 °C.
Geology
The geology of the study area comprises a thick sequence of sediments and volcanic rocks in two regional geological provinces: (1) Vizcaino-Purisima Basin, Purisima Sub-basin and (2) la Giganta Volcanic Belt. Basement rocks of the Purisima Sub-basin include Triassic sedimentary rocks, an ophiolithic sequence, and partially serpentized ultramafic rocks (Mina 1957). Lithological units comprising the Purisima Sub-basin within the study area include Paleocene lutites (Santo Domingo Formation) and Paleocene–Eocene sandstone and lutite sequences (Tepetate Formation). From a structural point of view, the Purisima Sub-basin forms part of a syncline approximately 600 km long, occupied by Upper Cretaceous to Lower Tertiary sediments (Cserna de 1989).
La Giganta Volcanic Belt forms a broad region (500 km long by 30–50 km wide) and consists of volcanic material (pyroclasts, lava flows, and breccias) and continental sand (Comondu Formation) with a composite thickness of around 1,200 m (Morán-Zenteno 1994). This volcanic sequence overlies a Mesozoic batholith consisting of felsic and metamorphic rocks. The Salada Formation discordantly overlies Santo Domingo, Tepetate, and Comondu Formations; it is considered to be mainly of Pliocene–Pleistocene age and comprises sediments deposited in a marine environment. It has widespread outcrops in areas adjacent to the western edge of Sierra la Giganta and in the plain of the Santo Domingo stream. The Salada Formation consists predominantly of light brown fine to medium sand, medium to poorly consolidated, with variable proportions of silt and clay. The sand particles are composed of quartz, feldspar and other igneous materials. The layers are 0.30 to 0.40 m thick and, in some places, the sand layers are interlayered with poorly consolidated conglomerates. Abundant secondary carbonate has been reported in some places in the form of calcrete layers and as a cementing material. Maximum reported thickness in the north zone of the area is ~185 m (ACSA 1969). Figure 4 shows the distribution and geometry of the Salada Formation discordantly underlying the Quaternary granular materials of diverse origin (fluvial, eolian, and alluvial) and overlying clayey formations.

General longitudinal a and transversal b geological cross sections through the SDID showing distribution of Salada Formation. Location of cross sections is shown in Fig. 1
Hydrogeology
An interpretation of surface geological information, an analysis of drill cuttings from wells, and geophysical (vertical electric soundings) data were used to define an unconfined granular aquifer constituted mainly by the Salada Formation and in some locations by Quaternary sediments (ACSA 1969; TMI 1979; DESISA 1997). Its lower vertical boundary consists of clayey formations for which a low hydraulic conductivity is inferred. The south, east, and north boundaries of the aquifer are those of outcropping volcanic material (Comondu Formation) that composes the Sierra La Giganta. Geophysical and geological evidence suggest that the amount of silt and clay in the Salada Formation increases towards the west, from where a reduction in its hydraulic conductivity could be assumed. This granular aquifer used to have a maximum saturated thickness of ~140 m; now, over the years, it has decreased to about 100 m. The aquifer is heterogeneous in regard to the spatial distribution of hydraulic conductivity. An interpretation of pumping tests with a numerical radial-flow model, indicate values of 5–60 m/day. The high values are found in the central-east and north; low values correspond to western (outside the SDID limits) and southern regions (DESISA 1997).
The groundwater flow system of the study area is controlled by the uniform gentle topography of the Sierra La Giganta to sea level; it involves a regional flow system from the high topographic areas of the western edge of Sierra la Giganta towards the west (Pacific Ocean). Above this regional flow there are intermediate systems, which are connected with high localized recharge along the riverbeds. As no extensive surface manifestations of groundwater discharge were identified within the study area, the main pre-agricultural development discharge zone was along the coastline. When the operation of SDID started in the mid-1950s, general groundwater flow direction in the horizontal plane was from east to west. The first available potentiometric data (Fig. 5) show water level elevation lines for 1957 that probably represent a situation close to the original steady-state natural conditions prior to agricultural development. Water levels in wells were above sea level in the entire region; also, a groundwater divide is identified. Groundwater flow in the northern region was from east to west; however, in the southern part it was from east to southwest. The influence of natural recharge from Bramonas stream in the central part of SDID is clearly identified with the configuration of the equipotential lines. Some 40 years later (1996), the elevation of the static water level was 20 m a.s.l. towards the eastern region, but, within the SDID, the water level in wells was below mean sea level (Fig. 5). A drawdown-cone is defined by the equipotential line of 5 m b.s.l., including nearly 75% of the SDID (southern, central and northern regions); inside this region two local drawdown cones in the central portion were around 20–25 m b.s.l. A gradient reversal of 2.5×10-4 between the west of SDID and the coastline can be calculated using a linear water-table configuration. Wells near the seashore are scarce due to restrictions in the obtained water quality; however, water levels are reported to be around average sea level (DESISA 1997). A maximum drawdown of about 35 m for the 1957–1996 period is observed in the center of the SDID where a higher density of wells per square kilometer is present as compared with the southern and northern zones.

Water level lines in the horizontal plane, and flow lines for 1957 and 1995 (After DESISA 1997)
Methods
Available information regarding the chemistry of extracted groundwater in SDISD consists of annual records for several wells with routine water analyses, including the major elements (HCO3, SO4, Cl, Na, Ca, Mg), total dissolved solids (TDS), pH, and specific electrical conductivity (SEC) measurements. Reported data by the SDID laboratory are from 1968 to 1995. This information was used to establish the evolution of salinity trends for selected wells (Fig. 2), with a comparative examination of salinity distribution (Fig. 3) within the study area for 1968 and 1995 (Fig. 6).

Distribution of salinity in extracted water for 1968 and 1995. Points represent well location where salinity measurements were available. Positions of Santo Domingo and Bramonas ephemeral streams are also shown
For this study, a representative set of groundwater samples were obtained from 35 active wells selected on the basis of lithology and electric logs, screen length, and salinity distribution. Most selected wells are used for irrigation purposes (33) and are about 100–120 m deep. One well was used for drinking water supply (well 34, 70 m deep) and one for cattle use (well 35, 50 m deep). Wells selected to obtain water samples included the detected salinity range; all of them tap the Salada Formation. A pumping time of at least 4 h was required before the sample was collected. Extracted water from these wells represent a mixture of vertical water quality along the screen length (30–50 m for wells used for irrigation purposes). Field measurements of temperature, pH, Eh (Pt electrode) TDS, and SEC were made using an in-line flow-through isolation cell to ensure the exclusion of atmospheric gases and to enhance measurement stability. Calibration for pH measurement was made in every site using buffers at pH 7.0 and 4.0 and time was allowed for the water and electrode temperature to reach equilibration. Stabilization of readings for pH and redox potential was achieved in 10–20 min, and readings were taken after steady conditions were observed. One sample filtered (0.45 μm) in the field was taken at each site in an acid-washed, well-rinsed low density polyethylene bottle and acidified with 1% HNO3 to a pH of <2 to assure major cation (Na, K, Ca, Mg), trace metals (Li, Ba, Sr, Fe), and SiO2 stabilization. Another sample was taken unfiltered and no acid was added for HCO3, CO3, Cl, SO4, F, and B analyses. Samples for NO3 determination were filtered and acidified to pH 2 (H2SO4). Samples were stored at 4 °C for no more than 1 week prior to laboratory analyses, which was carried out in San Luis Potosi University.
Alkalinity was measured using volumetric titration with bromocresol green-methyl red indicator. Major cations, as well as Si, Li, and Sr, were analyzed by ICP-AES using a Thermo Jarrell ASH IRIS/AP optical emission spectrometer (Table 1). Fluoride and Cl were determined by an ion-selective electrode. Sulfate was determined by a turbidimetric method using barium chloride. Nitrate concentrations and B were analyzed by automated colorimetry. Accuracy of ICP-AES analyses was controlled using laboratory standards. Duplicate analyses were performed for the anions and for B; reported values are an average. Major cations and anions were analyzed at independent laboratories within San Luis Potosi University. The calculated ionic balance is within 6% for 85% of the samples and less than 10% for the other 15% of samples.
Differences between potential salinization sources were investigated by analyses of major (HCO3, Cl, SO4, Na, Ca and Mg), minor (NO3, K), and trace elements (F, Li, Ba, Sr, Fe), as well as by colloids (SiO2) and specific geochemical relations (Na/Cl). The effect of water derived from irrigation-return was identified on the basis of the concentrations of SO4 and NO3 in conjunction with Cl and Na ions, which are generally concentrated in irrigation-return (Richter and Kreitler 1993).
Results
Salinity evolution 1968–1995
The distribution of TDS in extracted groundwater for 1968 and 1995 is presented in Fig. 6. Total dissolved solids values >1,500 mg/l for 1968 were identified mainly in the northern part of the SDID (vicinity of Santo Domingo stream) and in a central region between the ephemeral streams; both regions comprise about 15% of the irrigation district. Regions with TDS >1,500 mg/l by 1995 comprised 45% of the area, mainly in a zone between the streams. Another region south of Bramonas stream showed salinity values as high as 2,500 mg/l. The southern region lacks of water salinity information, but it is assumed to have salinity values >2,500 mg/l because, in 1995 irrigation was severely restrained in this part of the irrigation district. Including this region, a total of almost 70% of the irrigation district had groundwater with a salinity beyond a concentration of 1,500 mg/l. However, the salinity evolution in Fig. 6 suggests that, in some zones, the water salinity has not increased substantially. This effect can be observed in zones with preferential natural recharge. During exceptionally wet years, limited runoff is generated within the basin; infiltration of the overflow in Bramonas stream is considered to preserve groundwater quality in some parts of the SDID, especially the region east of the highway adjacent to both sides of the stream, which has been designed as a water reserve zone for potable water supply. The reallocation of irrigation wells is not permitted in this zone.
Groundwater groups and water mixing
The study area is within a coastal aquifer in a semiarid region where groundwater quality is influenced by various potential sources of salinity that determine the composition of water extracted from wells. Results of chemical analyses (Table 2) show large variations in chemical compositions and also indicate high salinity of some groundwater samples. Values of TDS are 612–3,430 mg/l (mean 1,300 mg/l) and SEC values are in the range of 878–4,910 μS/cm. Most samples are Na-Cl or Na-mixed-anion type, although the low salinity waters are Na-HCO3 or mixed-cation-HCO3 type. The high salinity waters are Na-Cl type (maximum Cl concentration 1,484 mg/l). Generally, groundwater temperature measured at well discharge is 29.7 °C, suggesting that the flow systems in the area circulate at a somewhat similar depth, of approximately 100 m. Chemical characterization of the study area involved the partitioning of water analyses into homogeneous groups using available graphical techniques, including a Stiff pattern diagram (Stiff 1951), an expanded Durov diagram, x–y plots, and mixing and dilution diagrams. For example, in Fig. 7, Stiff pattern diagrams and sampling locations are shown: the various shapes of diagrams indicate differences in extracted water chemistry. These illustrations and the expanded Durov diagram (Fig. 8) allowed the identification of five chemical groups. According to the results shown in Fig. 8, group 2 accounts for 35% of the samples, group 3 for 28%, group 4 for 17%, and groups 1 and 5 for 9% and 11%, respectively. Group 2 is found frequently in the northern part of the SDID, but Stiff pattern diagrams, shown in Fig. 7, lack an indication of a specific spatial distribution within the study area. However, according to the expanded Durov diagram, some processes can be delineated in specific paths.

Location of collected water samples and their Stiff Diagram pattern, identified homogeneous chemical groups are also shown

Expanded Durov diagram with detected process paths that affect water chemistry are also indicated. Numbers in parenthesis are used for field data identification
Selected statistics for identified homogeneous groups are presented in Table 3. The average values for each group show a continuous increase of TDS, Cl, SO4, K, Li, and Sr from groups 1–5. In contrast, other parameters, such as pH, HCO3, Na, Ca, Mg, and Ba, have a different behavior. In the case of pH, HCO3, and Na, calculated average values are higher for group 2 than for groups 1 and 3. Also, a distinction is made for Ca, Mg, and Ba because group 2 has lower concentration values than groups 1 and 3. A general description of the groups shows that group 1 represents a low salinity groundwater, whereas group 5 includes high salinity water. The progressive salinity increases from group 1 to 5 (Table 3), and the occurrence of seawater in the vicinity suggests a mixing model between two end members: a low salinity groundwater (group 1) and a high salinity water (seawater). As information regarding the composition of local seawater is unavailable, the mixing shown in Fig. 8 is considered in relation to average seawater (Lloyd and Heathcote 1985). A simple conservative mixing model between proposed end members could be represented by arrow B in Fig. 8. Most samples are not along this path; therefore, chemical reactions that modify the conservative mixing of some of the ions should prevail and have to be identified.
Cation-exchange reactions
The position of samples within the fields of the Durov diagram were used to identify processes and reaction paths such as mixing, cation exchange, and dissolution affecting groundwater composition. A detailed description of fields and their hydrogeochemical significance is presented by Lloyd and Heathcote (1985) and Petalas and Diamantis (1999). Field number 1 in Fig. 8 is usually the place for low-salinity recently infiltrated water in sand aquifers (HCO3-Ca type waters), CO2 addition from the root zone, and carbonate dissolution, which most of the time controls the reactions of water chemistry in this condition. Group 1 samples were taken in wells relatively close to Santo Domingo and Bramonas streams, where water-recharge conditions are implied. However, group 1 is located in fields 2 and 3, possibly due to relatively high values of Mg. Bicarbonate and Na are the prevailing ions in field 3, normally representing cation-exchanged waters; arrow A indicates the reaction path of group 1 samples affected by direct cation exchange, where Na existing in the clay fraction of the aquifer material was exchanged for Ca in groundwater (Appelo and Postma 1996). The mixing processes with the saline end member is nil in field 3; therefore, group 2 samples in this part of the diagram are considered to represent group 1 water modified by direct cation-exchange processes along the flow system. Other bivalent cations, such as Mg, Ba, and Sr, can also be exchanged with Na and K from the clay fraction of the aquifer material. Samples in field 5 are quite close to the conservative mixing-line (arrow B), but most samples in group 2 (50%) are in field 6, illustrating that both processes (mixing and direct cation exchange) are present. Assuming conservative mixing, an increase in the saline end-member proportion should produce water in field 9; however, most samples of groups 3, 4, and 5 are in field 8 because mixing and reverse cation-exchange reactions (arrow C) are affecting their composition. Reverse cation exchange consists of exchange Ca from the clay fraction in the aquifer material with Na from groundwater, resulting in an increase of Ca in groundwater and a reduction of dissolved Na. The effect of reverse cation exchange is less noticeable in two samples in field 9: they appear to represent conservative mixing between proposed end members.
A Cl-Na diagram (Fig. 9) illustrates identified reactions and processes. A principal feature for most group 2 samples is the enrichment in Na relative to Cl concentrations, considering the conservative mixing (between group 1 and average seawater) line. Chloride has a conservative behavior; deviations from conservative mixing that result from solubility or redox controls are not feasible. Consequently, the high Na concentrations in group 2 suggest a strong water–aquifer interaction, related to direct cation-exchange reactions between groundwater and the clay fraction of the aquifer material. Groups 3, 4, and 5 show Na deficiency with respect to the conservative mixing line due to reverse cation-exchange reactions. The Na/Cl (mg/l) ratio (Fig. 9) for group 2 samples shows consistently high values (zone B) above the corresponding seawater dilution line (C=0.53), whereas samples from groups 3, 4, and 5 lie below this line (zone A). Additionally, for Cl concentrations with values lower than about 230 mg/l, the Na/Cl ratio is consistently higher than the average seawater ratio. According to Nordstrom and others (1989), Na/Cl ratios above 0.5 are common in surface waters and low salinity groundwater, whereas those below that value correspond to natural waters with cations, such as Ca, which occurs in significant amounts. In the case of groups 3, 4, and 5, Ca and Mg increase (Table 2) to such an extent that their added value is bigger than Na content in most of the samples.


Variations of Cl concentrations as compared with Na, Na/Cl, NO3, Ca, Mg, Sr, Li, B, F, and Ba in groundwater
Agriculture impacts on groundwater quality
Initial and present agricultural management practices in the SDID have been a main concern of farmers and related authorities. Díaz-Jiménez (1981) indicates that only 14% of farmers have a high agricultural level of technological efficiency; recent estimates indicate that this fraction has not increased substantially (DESISA 1997). However, the level of specialization in cotton and grain production in relation to the national yield indicates that high crop harvests are usually obtained. In this regard, any typical land with available water and low technological efficiency requires the use of excess irrigation to enable leaching of salts from the soil. In addition, high amounts of fertilizers, herbicides, and pesticides are also needed to obtain an economic crop yield. An assessment of the irrigation-return flow in the SDID establishes a minimum volume of about 30–40×106 m3/year, equivalent to ≈12% of the estimated amount of extracted groundwater (DESISA 1997). Arid conditions create irrigation-return flow to become concentrated in some chemical constituents because of a number of processes such as evapotranspiration, mineral dissolution, and solutions of fertilizers and agricultural residues: net decreases in Ca, HCO3, and SO4 are common as a result of precipitation accompanied by a net gain of Na, K, Cl, and NO3 (Richter and Kreitler 1993). With a low technological efficiency, fertilizer is leached by excess water because crop recovery is as low as 50%, resulting in large amounts of N introduced into the groundwater system. In most cases, the natural baseline groundwater contains low N concentrations (≤10 mg/l); however, as irrigation-return flow almost always contains significant concentrations of N, this element could be used to distinguish salinization related to agricultural effluents from that produced by other sources. Natural N sources are included in soil organic matter, although, for the SDID, the principal identified sources comprise non-point sources such as the extensive application of different types of fertilizers (ammonium sulfate, monoammonium phosphate, urea, anhydrous ammonia) and manure to the soil. Under the oxidizing conditions prevailing in unsaturated and saturated zones, N is readily converted to NO3 throughout the nitrification process. NO3 is a highly soluble and mobile species that has the potential to leach down gradient along groundwater flow systems (Freeze and Cherry 1979). Other fertilizers used in the SDID, such as potassium nitrate, calcium nitrate, and ammonium nitrate, directly incorporate NO3 into irrigation-return flow.
Despite the fact that there are no field measurements of dissolved oxygen (DO), the redox potential (Eh values are positive and about 250–300 mV) suggests oxidizing conditions prevail within the aquifer. The very low FeT concentrations detected (Table 2) in extracted groundwater are in agreement with this condition. Under this aerobic condition, observed NO3 concentrations in groundwater are often high (up to 62 mg/l). Nitrate increases significantly with respect to Cl in some group 2 samples and in most of group 3–5 waters, as shown in Fig.9, indicating that both species are enriched in irrigation-return flow. The highest average NO3 concentrations were detected in groups 3–5 (Table 3) because these waters have the highest salinity. The influence of irrigation-return on the salinization processes is evident. The correlation between NO3 and Cl for some samples is strong: this effect is considered to be derived from the evapotranspiration process on fertilizers, although a component of mixing with a saline end member is expected to also be present. Nitrate values in the order of 8–10 mg/l (proposed natural baseline) are apparent from groundwater in which salinity is not significantly affected by irrigation-return and may reflect naturally low total N inputs from pre-agricultural development times.
A high NO3 concentration in groundwater is a cause for concern because a water reserve zone for potable water supply has been delineated within the agricultural area. Nitrate in drinking water has been linked to several human health hazards causing metahemoglobinemia, certain types of cancer, and negative reproductive outcomes (Rodvang and Simpkins 2001). About 89% of the samples collected had NO3 concentrations exceeding proposed natural baseline value; wells 22 and 35 had concentrations exceeding the Mexican Health Agency’s maximum recommended contaminant level of 44.2 mg/l as NO3, which also suggests an important impact of agricultural activities in groundwater.
Generally, there are no solubility controls for NO3, nor is it absorbed or exchanged significantly in aquifer materials under oxidizing conditions. The only method of nitrate removal from groundwater is by reducing it to N2 (Appelo and Postma 1996). Geochemical factors required for denitrification include, besides dissolved NO3 and an electron donor, suboxic or anoxic (reduced) groundwater conditions (DO concentrations less than 2.0 mg/l; Korom 1992) and the presence of denitrifying bacteria. Suitable electron donors that may reduce NO3 generally are organic matter, pyrite, manganese, hydrogen sulfide, or Fe(II) silicates. Reducing conditions in groundwater generally imply flow under confined conditions (Edmunds and others 2002). A number of factors such as (1) Fe (II) silicates being probably the only source for electron donors present in the aquifer material, (2) dissolved FeT is present in very low concentrations, and (3) groundwater flow in unconfined conditions prevails in SDID, mean that circumstances are not favorable for denitrification within the study area and the reduction of NO3 is not expected to occur considerably in groundwater along flow systems.
Discussion
Chromatography of cation exchange and its influence on the development of chemical groups
Direct cation-exchange reactions can be represented (Appelo and Postma 1996) as
where Exch indicates the exchanging solid surface: solids with a large specific surface, may adsorb ions. They can be related to the clay fraction (<2 μm), clay minerals, and oxy-hydroxides of iron, all of which can be found in the Salada Formation. This reaction takes place during re-freshening when freshwater flushes seawater in a coastal aquifer, displacing the freshwater/saltwater interface towards the ocean. Release of Na from the exchanger’s surface in return for Ca or Mg present in groundwater is related to the difference between aquifer material and groundwater Na/Ca ratios. During re-freshening, Na is released to the groundwater because, where seawater saturates the aquifer, the Na/Ca ratio in the exchanger’s surface is higher than in the freshwater aquifer. This direct cation-exchange reaction is considered to be important for samples 3, 5, 9,18, 22,23, 24, 30, 31, 34, and 35 of group 2.
The reverse cation-exchange reaction
generally results during seawater intrusion when freshwater flow towards the sea is reduced and the seawater wedge moves inland (Appelo and Postma 1996). In this condition, the Na/Ca ratio on the surface of the exchanger in the freshwater portion of the aquifer is lower than that of the seawater front moving inland. Reverse cation-exchange reactions are considered to control samples of groups 3–5.
Previous work on the study area has defined it as a coastal aquifer; however, typical geochemical conceptual models involving horizontal displacement of seawater by freshwater or seawater intrusion, fail to fully explain the observed water chemistry, such as the spatial distribution of salinity (Figs. 6 and 7). Hence, the development of cation-exchange reactions must be explained in a different context.
Cation exchange between water and exchanging solid surfaces in the aquifer is significant when relative cation concentrations become different along a groundwater flow line under non-steady situations. Cations strongly selected by exchanging solid surfaces will release other cations from the exchanger; therefore, they will be transported in a predictable manner throughout the flow line. Cations with the lowest selectivity to the exchanging solid surface will be transported first, followed by those with a higher affinity, and so on. This process is known as cation-exchange chromatography (Appelo and Postma 1996). This process is controlled by differences in the behavior of ions in relation to the adsorption capacity of the exchanging solid surface, dispersivity of the aquifer, and the contrast between the composition of the solution that displaces and the solution that is displaced (Manzano and Custodio 1998). The term displacement chromatography applies when the solution occupying the pores of a material has ions with a lower selectivity for the exchanging solid surface than the ions in the solution replacing it, a condition that applies when freshwater with a low Na/Ca ratio displaces seawater with higher Na/Ca and Mg/Ca ratios. In these conditions, Ca is preferred by the exchanging solid surface over Na and Mg. A typical pattern of re-freshening is present when freshwater flushes seawater, resulting in HCO3-Na type water with a Na/Cl ratio larger than 0.55 near the fresh water/salt water interface, which evolves in the direction of the freshwater source into HCO3-Mg type water, and eventually into a HCO3-Ca type water.
The deposition of sediments of the Salada Formation could be related to the last marine transgression and be correlated with the maximum interglacial (IS 5e) that took place in this region approximately 85,000 years ago (Ortlieb 1991). After this maximum, the ocean coastline moved away to its present position (approximately 5,000 years ago). After this time, low salinity water associated with natural recharge has been flushing seawater from the Salada Formation to produce an interaction between water and sediments, resulting in diagenetic modifications of the sediments. The Na/Ca ratio in rainfall that occurs near the coast will be very similar to that of the ocean (Appelo and Postma 1996). However, during the natural infiltration of rainfall, addition of CO2 in the soil zone and dissolution of carbonates take place and lower the fresh-water Na/Ca ratio. The displacement chromatography associated with the infiltration of freshwater through a sediment of marine origin with a high Na/Ca ratio causes the direct cation-exchange reactions. The water types identified in groups 1 and 2 show that the expected evolution trend is incomplete (Fig. 8) because the HCO3-Ca water type, indicating the last evolutionary stage of the process, is not detected. The coast line is more than 20 km from most wells; however, it is likely that in the last 5,000 years the total flushing of seawater is incomplete and the overall groundwater quality, therefore, is controlled by mixing and cation-exchange reactions. This could also imply limited natural recharge conditions, low groundwater head potentials, and/or partial flushing of still present entrapped seawater in local low hydraulic conductivity zones within the Salada Formation.
The displacement of freshwater by higher salinity water produces the opposite effects to those described earlier. When the concentration of the displacing solution increases, the selectivity of the exchange solid surface is modified to such an extent that bivalent cations (Ca, Mg, Sr, Ba) reduce their preference for the exchange solid surface in relation to monovalent cations (Li, Na, K), producing reverse cation-exchange reactions that release bivalent cations to solution. In field conditions, groundwater extraction increases the natural hydraulic gradients within the drawdown cone, producing mixing of low salinity groundwater with (1) some entrapped seawater that has not been totally flushed from the sediment and/or (2) irrigation-return water that reached the saturated zone. The relation of Cl vs. Na/Cl, shown in Fig. 9, suggests that mixing (in these conditions) produces water with a Cl concentration >230 mg/l. This effect is similar to the displacement of freshwater by seawater obtained within the drawdown cone of the well: the affinity of bivalent cations to the exchanger solid surface declines in relation to the monovalent cations, thereby resulting in the development of reverse cation-exchange reactions. The higher the percentage of entrapped seawater and/or irrigation-return present in the mixture, the more frequently water quality types of groups 3–5 will appear in extracted groundwater.
According to laboratory experiments (Beekman and Appelo 1990), at the start of these processes, Ca and Mg are released to the solution in accordance to the ratio of the content of both ions in the low salinity water. When, as a result of the exchange processes, the Ca content in the exchange solid surface is depleted, the behavior of Ca and Mg ions changes: the former reduces its concentration in the solution while the concentration of the latter increases in high salinity water. Figure 9 shows that both the concentrations of Ca and Mg increase in relation to Cl, which corresponds to the first stage of the behavior identified by these authors.
The development of this reaction can also be used to identify various aspects related to the groundwater flow systems in the area. Recognized reactions, for example direct cation exchange, should be related to preferential infiltration of natural recharge. Arid zones are often characterized by localized recharge along preferential flow paths that take place in topographical depressions on the surface of the land (Scanlon and others 2002), as in some parts of the riverbeds that cross the study area transversally. In this arid region, natural recharge is extremely limited, probably associated with exceptional wet periods with intense precipitation (most of them linked to tropical cyclonic events). These preferential zones correspond to zones of the riverbeds with relatively high hydraulic conductivity where water infiltration takes place. In the northern zone, runoff infiltration in some parts of the Santo Domingo stream produces the groundwater of group 2 (samples 3, 22, and 31).
Samples from groups 1 and 2 predominate in the area between the Santo Domingo and Bramonas streams, suggesting that this zone constitutes the region where the largest amount of natural recharge appears to be taking place. Groups 3, 4, and 5 predominate on the left side of the Bramonas stream as well as in the westernmost part of the SDID. In this zone, the mixing between detected end members includes a small proportion of low salinity end members (group 1).
Geochemistry of carbonates
Because chromatography of cation exchange has a significant influence on Ca and Mg and other alkali-earth elements, such as Ba and Sr, it also has an effect on the geochemistry of carbonates. Carbonate minerals are present in the Salada Formation, and groundwater chemistry is presumably also controlled by carbonate reaction. Mineral equilibrium calculations for groundwater samples were made using the PHREEQC geochemical code (Parkhurst 1995). Most groundwaters are in equilibrium with carbonate minerals, such as calcite, dolomite, and magnesite, but are under-saturated with respect to celestite (not shown) and witherite (Fig. 10). The samples representing groups 1 and 2 are in equilibrium with calcite and dolomite, meaning that the reduction of Ca and Mg as a result of direct cation-exchange reactions modifies the equilibrium established with carbonates accordingly with the following reaction:

Saturation Index for various mineral in groundwater samples of Santo Domingo Irrigation District
This shows that there is a tendency to dissolve more Ca and Mg (on the basis of calcite, dolomite, or another mineral) ions by ion exchange, shifting the direction of the reaction to the right, increasing the pH of the solution, and the concentration of HCO3, a condition present in group 2 waters (Table 3). This geochemical behavior is congruent with the evolution of pH, as the concentration of hydrogen ions has the lowest values in group 2 as well as the highest concentration of HCO3. The relatively high pH values of two samples from group 1 could explain the presence of the proposed reactions (cation exchange and dissolution) alone (without the mixture of salt-water).
The process of mixing and reverse cation-exchange reactions produces an increase in Ca and Mg concentrations in groups 3, 4, and 5 (Fig. 9), and the following reaction is produced:
The addition of Ca and Mg during reverse cation-exchange reactions requires adjustments in pH and HCO3 to maintain the conditions of equilibrium with carbonates. As equilibrium with carbonates is maintained, the reaction must shift towards the right, reducing the alkalinity and generating hydrogen ions.
Geochemistry of trace elements
The geochemical behavior of trace elements (Sr and Li) shows similar linear tendencies to those detected for mayor cations (Fig. 9). The origin of these elements may be related to the weathering of feldspars and mafic minerals included in the rock fragments that constitute the Salada Formation. In groups 3, 4, and 5, Sr as well as Ca and Mg can be mobilized by reverse cation-exchange reactions. The origin of Ca and Mg may also be linked to the carbonates cementing the sandstone. Carbonate dissolution during direct cation-exchange process releases Ca and Mg, as well as Sr and Ba found as impurities in carbonates. Samples are subsaturated with respect to strontianite (Fig. 10) and celestite (not shown), indicating, as with Li, whose behavior is conservative, a mixture between defined end members.
Because the average B concentration is ~0.15 mg/l in the water samples and the groundwater temperature is relatively homogeneous and close to the annual environmental mean, this reduces the possibility that the origin of B could be linked to hydrothermal sources. The most important B species in natural waters are anions or uncharged complexes, meaning that they are probably not significantly absorbed onto the surface of minerals (Hem 1985). Concentrations of Sr, Li, and B in relation to the salinity of groundwater (Fig. 9) suggest that the extreme saline member involved in the blend has a different composition to that of modern seawater.
The distribution of F and Ba, as related to Cl, is different from the tendencies observed for other elements (Fig. 9), as high concentrations generally occur for low values of Cl, and a small content is observed as salinity increases. This behavior suggests the presence of solubility controls with regard to minerals containing F and Ba. In the case of F, its origin must be associated with the clasts of the volcanic rocks derived from the Comondu Formation. The principal mineral that usually controls F presence in groundwater is fluorite (CaF2); however, the saturation indices (not shown) indicate sub-saturation with regard to this mineral. Another mineral that may control the dissolved F is fluorapatite, although, in this case, evaluations of PO4 species are not available to calculate the respective saturation index and to discuss a theory on the presence of a solubility control regarding this mineral. The tendency of F suggests that the extreme saline member has a minimum amount of this element (probably similar to average seawater) and that there are variable concentrations in recently infiltrated groundwater (between 0.15 and 0.6 mg/l). The mixture between end members with a conservative behavior of F is ruled out due to the relative availability of Ca for F control.
As far as Ba is concerned, the mineral controlling its presence in groundwater appears to be barite (BaSO4), as suggested by a higher SO4 content as related to Ba. The relation of Ba to salinity (as Cl) suggests a tendency for groups with high Ba content to evolve to low Ba concentration as salinity increases (Fig. 9). This could incorporate possible Ba precipitation when a mixture of water with high salinity occurs. Samples range from unsaturated to equilibrium with respect to barite and are unsaturated with respect to gypsum, suggesting that gypsum can be dissolved and SO4 concentration could increase as compared with Ba. Such process could be explained by the higher activity of Ca compared with Ba.
Conclusions
Groundwater management in the area of study has been carried out with the assumption that one of the main sources of salinization is seawater intrusion from the coastline. The results of this study, however, suggest that this situation is not responsible for the salinization observed: groundwater management should be carried out differently. Thus, installing wells increasingly far from the coast, a policy that has been implemented to date, will not solve the problem unless the importance of irrigation-return flow and seawater that remains in the geological units are taken into consideration. When these additional sources of salinization are recognized, controls should be imposed when installing wells to keep contact with the clayey units that form the base of the aquifer. Modifications to the flow rate of extraction and/or the length of operation of the well should also be taken into account. To achieve optimal results, specific field tests, such as a continuous salinity determination of groundwater in relation to pumping time, should be carried out during step-drawdown and constant drawdown pumping tests.
A definition of potential zones where direct ion-exchange processes take place could be useful for recommending facility sites for artificial recharge works because they provide zones where the natural recharge flow is quickly and easily infiltrated. Recognition of reverse ion-exchange reactions indicates zones where groundwater withdrawals must be controlled to prevent the salinity problem from increasing over time. Groundwater management could be more effective if the percentage of water that returns to the saturated zone in the form of irrigation-return flow is reduced. This would produce higher crop yields and use less water for irrigation than with present practices, and, therefore, a low irrigation-return infiltration that could improve groundwater quality in the long term.
The increase in salinity of groundwater may be controlled if these processes are understood and integrated into a suitable management plan for this resource. This hydrogeochemical interpretation has permitted the identification of the main sources of salinization that modify the quality of groundwater in this area of interest. The identified potential sources of salinization include (1) seawater remaining in the sediments that comprise the developed aquifer and (2) infiltration of irrigation-return flow. The composition and location of seawater were not directly identified, but the mixture lines detected suggest that inflowing saline water differs in composition to that of modern seas, particularly when the major ions (Ca, Mg, Na, SO4) and trace elements (Li, Sr, B) are considered. The infiltration of irrigation-return flow associated with farming practices was identified by the concentration of NO3 and SO4 and their relation to salinity of extracted groundwater. The concentrations of trace elements can be considered to represent the release of impurities during the incongruent dissolution produced when groundwater interacts with feldspars, carbonates, and probably clayey minerals. These reactions may be reflected in diagenetic processes that modify the sediments of the Salada Formation that comprise the aquifer.
An explanation of the removal of seawater from the aquifer as a result of natural recharge in this arid area could be accomplished from a hydrogeochemical point of view using a chromatographic pattern in which cations with a greater selectivity for clays displace other ions, encouraging the development of direct ion-exchange reactions and the generation of HCO3-Na type water with a high Na/Cl ratio (group 2). Although a complete chromatographic sequence was not observed, an expected increase in Mg was detected in samples from group 1. According to local hydrogeological conditions, direct ion-exchange reactions indicate that there are zones where the replacement of seawater by low salinity water is efficient, meaning that they are linked to preferential recharge areas. The development of a chromatographic pattern depends on the amount of exchangeable cations and their concentration in solution, where the relation between these concentrations controls the speed of the displacement front. This relationship is small in the case of seawater intrusion that displaces groundwater with low salinity; the resulting chromatographic process is swift, with limited lateral extension and, therefore, difficult to observe in field conditions (Appelo and Postma 1996).
In the study area, when, as a result of extraction through wells, irrigation-return infiltrates and/or salt-water remaining in the aquifer displaces low salinity groundwater, the chromatographic pattern shifts to one of salinization, producing reactions of reverse ion exchange that create water with the chemistry of groups 3, 4, and 5, generating well-defined chromatographic patterns of salinization. The fact that this salinization pattern can be clearly identified in this case suggests that a typical seawater intrusion process is not occurring in the area of interest. The development of a chromatographic pattern for a situation of seawater being replaced by groundwater with less salinity has been described by Lawrence and others (1976), Chapelle and Knobel (1983), Stuyfzand (1999), Beekman (1991), Manzano and Custodio (1998), among others. The salinization pattern, however, has only been described in laboratory experiments (Beekman and Appelo 1990). In this study, the salinization pattern is a broad and well-defined situation that is probably related to the fact that the difference between the salinity levels of the water responsible for the displacement and the water that is actually displaced is relatively small.
References
ACSA (Ariel Construcciones SA) (1969) Estudio geohidrológico completo de los acuíferos del Valle de Santo Domingo, Baja California Sur. Informe técnico T-1. Secretaría de Agricultura y Recursos Hidráulicos, México
Appelo CAJ, Postma D (1996) Geochemistry, groundwater and pollution. Balkema, Rotterdam
Bear J, Dagan G (1962) The steady interface between two immiscible fluids in a two-dimensional field of flow. Hydraulic Lab, Technion, Haifa Israel, IASH, P,N. Prog. Report 2
Beekman HE (1991) Ion chromatography of fresh and salt water intrusions. PhD Thesis, Free University, Amsterdam
Beekman HE, Appelo D (1990) Ion chromatography of fresh-and salt-water displacement: laboratory experiments and multicomponent transport modeling. J Cont Hydrol 7:21–37
Calvache ML, Pulido-Bosch A (1994) Modeling the effects of salt-water intrusion dynamics for a coastal karstified block connected to a detrital aquifer. Groundwater 32(5):767–771
Calvache ML, Pulido-Bosch A (1997) Effects of geology and human activity on the dynamics of salt-water intrusion in three aquifers in southern Spain. Environ Geol 30:215–223
Chapelle FH, Knobel LL (1983) Aqueous geochemistry and the exchangeable cation composition of glauconite in the Aquia aquifer, Maryland. Groundwater 21:343–352
Chiocchini U, Gisotti G, Macioce A, Manna F, Bolasco A, Lucarini C (1997) Environmental geology problems in the Tyrrhenian coastal area of Santa Marinella, province of Rome, central Italy. Environ Geol 32(1):1–8
Cserna de Z (1989) An outline of the geology of Mexico. In: The geology of North America: an overview, ch 9. Geol Soc Am Bull A, pp 233–264
Custodio E, Llamas RM (1983) Hidrología subterránea, 2nd edn. Omega, Barcelona
DESISA (Desarrollo y Sistemas S A) (1997) Actualización del estudio geohidrológico del Valle de Santo Domingo, Baja California Sur. Informe técnico para Comisión Nacional del Agua
Díaz-Jiménez G (1981) La sobreexplotación al acuífero del valle de Santo Domingo, BCS, sus consecuencias y posibles soluciones. MSc Thesis, Colegio Posgraduados Chapingo, México
Edmunds WM, Carrillo-Rivera JJ, Cardona A (2002) Geochemical evolution of groundwater beneath Mexico city. J Hydrol 258(1–24)
Freeze RA, Cherry JA (1979) Groundwater. Prentice–Hall, Englewood Cliffs
Giménez E, Morell I (1997) Hydrogeochemical analysis of salinization processes in the coastal aquifer of Oropesa (Castellón, Spain). Environ Geol 29:118–131
Glover RE (1959) The pattern of fresh water flow in a coastal aquifer. J Geophys Res 64:457–459
Graniel-Castro E, Cardona A, Carrillo-Rivera JJ (1999) Hidrogeoquímica en el acuífero calcáreo de Mérida Yucatán; elementos traza. Ingeniería Hidráulica México 14(3):19–28
Hem JD (1985) Study and interpretation of the chemical characteristics of natural water, 3rd edn. US Geol Surv Water-Supply Paper 2254
Korom SF (1992) Natural denitrification in the unsaturated zone: a review. Water Resour Res 28(6):1657–1668
Lambrakis N, Kallergis G (2001) Reaction of subsurface coastal aquifers to climate and land use changes in Greece; modeling of groundwater refreshening patterns under natural recharge conditions. J Hydrol 245:19–31
Lawrence AR, Lloyd JW, Marsh JM (1976) Hydrochemistry and groundwater mixing in part of the Lincolnshire limestone aquifer, England. Groundwater 14:12–20
Lloyd JW, Heathcotte JA (1985) Natural inorganic hydrochemistry in relation to groundwater. Clarendon Press, Oxford
Manzano M, Custodio E (1998) Origen de las aguas salobres en sistemas acuíferos deltaicos: Aplicación de la teoría de la cromatografía iónica al acuífero del Delta del Llobregat. Procc IV Congreso Latinoamericano de Hidrología Subterránea, Montevideo, Uruguay, pp 973–996
Martínez DE, Bocanegra EM (2002) Hydrogeochemistry and cation-exchange processes in the coastal aquifer of Mar Del Plata, Argentina. Hydrogeol J 10(3):393–408
Mina UF (1957) Bosquejo geológico del territorio sur de la Baja California. Asoc Mex Geol Petrol IX:139–269
Moran-Zenteno D (1994) The geology of the Mexican Republic. Am Assoc Petrol Geol Studies in Geology, no 39, USA
Nordstrom DK, Ball JW, Donahoe RJ, Whittemore D (1989) Groundwater chemistry and water–-rock interactions at Stripa. Geochim Cosmochim Acta 53:1727–1740
Ortlieb L (1991) Quaternary shorelines along the northeastern Gulf of California, Geochronological data and neotectonic implications. In: Pérez-Segura E, Jacques-Ayala C (eds) Studies of Sonora geology. Geol Soc Am Spec Paper 254:95–120
Parkhurst DL (1995) User’s guide to PHREEQC: a computer program for speciation, reaction-path, advective-transport and inverse geochemical calculations. US Geol Surv Water-Res Invest Rep 95-4227
Petalas CP, Diamantis IB (1999) Origin and distribution of saline groundwaters in the upper Miocene aquifer system, coastal Rhodope area, northeastern Greece. Hydrogeol J 7(3):305–316
Richter BC, Kreitler CW (1993) Geochemical techniques for identifying sources of ground-water salinization. CRC Press, Boca Raton
Rivera A, Ledoux E, Sauvagnac S (1990) A compatible single-phase/two-phase numerical model. 2. Application to a coastal aquifer in Mexico. Ground Water 28(2):215–223
Rodvang SJ, Simpkins WW (2001) Agricultural contaminants in Quaternary aquitards: a review of occurrence and fate in North America. Hydrogeol J 9(1):44–59
Sadeg AS, Karahanoðlu N (2001) Numerical assessment of seawater intrusion in the Tripoli region, Libya. Environ Geol 40:1151–1168
Sakr S (1999) Validity of a sharp-interface model in a confined coastal aquifer. Hydrogeol J 7(2):155–160
Scanlon BR, Healy RW, Cook PG (2002) Choosing appropriate techniques for quantifying groundwater recharge. Hydrogeol J 10(1):18–39
Schmorak S (1967) Saltwater encroachment in the Coastal Plain of Israel. Int Assoc Sci Hydrol Symp 72:305–318
Steinich B, Escolero O, Marín LE (1998) Salt-water intrusion and nitrate contamination in the Valley of Hermosillo and El Sahuaral coastal aquifers, Sonora, Mexico. Hydrogeol J 6(4):518–526
Stiff HA (1951) The interpretation of chemical water by means of patterns. J Petrol Technol 3:15–17
Stuyfzand PJ (1999) Patterns in groundwater chemistry resulting from groundwater flow. Hydrogeol J 7(1):15–27
TMI (Técnicas Modernas de Ingeniería, SA) (1979) Estudio Integral para la rehabilitación del Valle de Santo Domingo, Estado de Baja California Sur. Informe técnico. Secretaría de Agricultura y Recursos Hidráulicos
Zhou X, Chen M, Ju X, Ning X, Wang J (2000) Numerical simulation of sea water intrusion near Beihai, China. Environ Geol 40(1–2):223–233
Acknowledgements
We would like to thank the Santo Domingo Irrigation District personnel for allowing access to information files and well sites in the study area; to Gerencia de Aguas Subterráneas, CNA, for the financial support provided through a contract to Desarrollo y Sistemas SA; help during some stages of the investigation from Ing. Octavio del Conde and Ing. Juan Pablo del Conde is also appreciated. We also thank the Earth Sciences Water and Soil Chemistry Laboratory staff of Facultad de Ingeniería-UASLP for the water chemical analyses. Related investigations were partially supported by CONACyT and the Academic Exchange Program 2003 UNAM-UASLP (projects G-0705, G-0706, G-0707).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cardona, A., Carrillo-Rivera, J.J., Huizar-Álvarez, R. et al. Salinization in coastal aquifers of arid zones: an example from Santo Domingo, Baja California Sur, Mexico. Env Geol 45, 350–366 (2004). https://doi.org/10.1007/s00254-003-0874-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00254-003-0874-2