
Abstract
Vibrio parahaemolyticus is one of the major pathogens responsible for vibriosis and zoonotic infections in teleosts, marine invertebrates, and also humans through consumption of contaminated or unprocessed seafood. Emergence of resistance against current accessible antibiotics and spread to the food chain and environment necessitate the development of safe and effective subunit vaccine against this bacterium. Many bacteria including V. parahaemolyticus produce extracellular curli fibrils, heteropolymeric filaments of major and minor subunit, which have been implicated in adhesion, biofilm formation, and virulence. Adhesins are the primary contact points with the host which help in establishing infection and colonization. CsgA, an adhesin, is the major structural component of the curli fiber that forms homopolymers of several hundred units. Due to their exposure on the cell surface, the curli fibers are recognized by the host’s immune system, would generate high immune response, and therefore can serve as effective vaccine candidate. In the present study, we describe characterization of the csgA gene, and preparation of recombinant soluble CsgA of V. parahaemolyticus (rVpCsgA), and evaluation of its vaccine potential. Immunization of BALB/c mice with the rVpCsgA mounted a strong immune response. Cellular immune assays such as antibody isotyping, in vitro splenocyte proliferation analysis, and cytokine profiling revealed mixed T-helper cell immune response. The anti-rVpCsgA antiserum was agglutination positive and specifically cross-reacted with the curli CsgA present on the outer membrane of V. parahaemolyticus cells, thus demonstrating its neutralization potential. One hundred percent survival of the immunized mice upon challenge with the lethal dosage of the bacterium established that the rVpCsgA could serve as an effective vaccine against the bacterium.
Key points
• Recombinant histidine-tagged CsgA of V. parahaemolyticus, rVpCsgA, was purified.
• The rVpCsgA immunization produced mixed immune response and agglutinating antibodies.
• Immunization with the rVpCsgA protected mice against V. parahaemolyticus challenge.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The emerging food and water-borne infectious diseases accentuate global public health and food safety concern. Amongst numerous pathogens, bacteria of Vibrionaceae family comprise some of the important pathogens of both humans and aquatic animals (Kim and Bang 2008). Several Vibrio species are accounted for causing vibriosis, the most prevalent infection in halophilic environment. The sharing of habitat by marine organisms and the microbial pathogen unexpectedly accounts for seafood-borne infection. The pathogenic Vibrio species are also responsible for infection in humans through various food-borne illnesses (Bhowmick et al. 2008). Recently, global warming and El Niño effect has stemmed the vibriosis infections (Ling et al. 2019). Abrupt rise in temperature has affected marine population due to physiological stress, changes in reproduction cycles, and infectivity pattern of pathogens, and strongly correlates with Vibrio population in the marine environment (Logar-Henderson et al. 2019). Hence, bacteria of Vibrio species pose significant threat to mankind and aqua-mariculture industry.
Vibrio parahaemolyticus, a Gram-negative zoonotic pathogen, is an obligate halophile and facultative anaerobe. According to Centers for Disease Control and Prevention report, more than 50% of food-borne gastrointestinal-vibriosis cases every year in the USA are due to V. parahaemolyticus infections (CDC 2019). Vibrio parahaemolyticus infects large marine population including several edible teleosts and marine invertebrates, resulting in acute hepatopancreatic necrosis syndrome and ultimately death (Letchumanan et al. 2015).
An alarming increase and global prevalence of V. parahaemolyticus infection has been observed worldwide ever since its discovery in 1950 (CDC 2019, Fujino et al. 1953). These contagions are conveyed to humans through consumption of contaminated seafoods, some of which are often consumed raw, thus leading to ingestion of infective dosage of these bacteria. The virulent strains cause acute dysentery, bloody diarrhea, vomiting, fever, and chills in humans (Shimohata and Takahashi 2010). The sporadic and severe cases of infection affect wound, ear, and also cause septicemia, which is life-threatening in immune-compromised individuals (Zhang and Orth 2013). Vibrio parahaemolyticus infection has been reported to adversely affect the gut microbiota community and the liver and kidney function in mice (Wang et al. 2020). Thus, V. parahaemolyticus causes severe losses to the marine fisheries sector and also is one of the leading causes of food-borne diseases in humans. Treatment of vibriosis with antibiotics leads to its spread to the food chain and environment via seafood. This has resulted in the failure of antibiotic treatment approaches against pandemic V. parahaemolyticus (WHO 2014). In addition, continued use of antibiotics, leading to evolution of multiple antibiotic-resistant Vibrio sp., poses serious health concern. Thus, vaccination remains the most effective measure to control food-borne diseases caused by V. parahaemolyticus.
Currently available commercial vaccines against vibriosis comprise attenuated live cultures of Vibrio anguillarum, Vibrio harveyi, V. parahaemolyticus, and Vibrio vulnificus (Powell et al. 2011). Heat-killed cultures of V. anguillarum and Vibrio ordalii have been shown to protect juvenile Atlantic halibut, salmon, and cobia (Lin et al. 2006; Bravo and Midtlyng 2007). However, these vaccine formulations suffer from severe drawbacks as the attenuated and killed vaccines need proper handling to avoid contamination of live bacteria. Also the preparation comprising a large number of bacterial proteins leads to non-focused immune response and antigenic load. Thus, there is a need to develop a subunit vaccine to overcome these drawbacks. Currently, no subunit vaccine is available against vibriosis, including that caused by V. parahaemolyticus.
An effective vaccine can be developed by targeting the molecules involved in adhesion and virulence. Adhesins present on the surface are essential at the primary stage of infection through adherence, colonization, and persistence in the host. Multiple fimbrial adhesins present on the extracellular surface such as long polar and lateral fimbriae, flagella, and components of pili (type I and type IV pilus) and curli have been characterized in Gram-negative bacteria (Langermann et al. 2000; Chapman et al. 2002; Maier and Wong 2015). Immunization with a number of fimbrial adhesins and outer membrane proteins (OMPs) of V. parahaemolyticus has been shown to confer different degrees of protection in mice (Zha et al. 2016), yellow croaker (Mao et al. 2007), crucian carp (Li et al. 2010, 2014), and zebrafish (Peng et al. 2016).
Besides flagella and fimbriae, many pathogenic bacteria such as Vibrio, Salmonella, Escherichia, Enterobacter, Shigella, Citrobacter, Altermonas, Pseudo altermonas, Shewanella, Halomonas, and Oceanicola species possess curli fibers (extracellular amyloid fibers) on the cell surface (Van Gerven et al. 2018). These entangled fibers have been implicated in adhesion, biofilm formation, and virulence in Escherichia coli and Salmonella (Dueholm et al. 2012; Van Gerven et al. 2018).
The curli fibers are amorphous and are made up of a heteropolymeric mixture of CsgA, a major component, and CsgB, a minor nucleator component (Chapman et al. 2002; Wang et al. 2008). Like other outer membrane proteins, and being abundantly present on the cell surface, the major component of the curli, CsgA is likely to be highly immunogenic as these would be easily recognized by the host (Wang et al. 2012). The CsgA can therefore be exploited as a potential vaccine candidate against this bacterium. The present study was undertaken to evaluate the immunogenic and protective potential of recombinant CsgA in murine model against V. parahaemolyticus, as use of mice for testing vaccine efficacy offers several advantages such as defined genetic background, well characterized immune system, low cost, and availability of immunological reagents thus allowing detailed analysis of immune response (Kiros et al. 2012).
Materials and methods
Bacterial strains
Expression host Escherichia coli BL21(λDE3)pLysS cells procured from GIBCO-BRL, USA were used for recombinant protein expression. A curli knock out strain E. coli LSR12 (E. coli C600 (ATCC No. 23274) with deletion of csgDEFG and csgBAC operons), used as negative control, was a kind gift from Prof. M. R. Chapman, University of Michigan, Ann Arbor, MI, USA.
Preparation of recombinant soluble CsgA of V. parahaemolyticus (rVpCsgA)
A synthetic construct (pET28.VpcsgA) harboring the mature CsgA of 402 bp designed on the basis of sequence information of V. parahaemolyticus csgA gene (protein id, KKF68034.1; Geninfo Identifier (GI), 814552847), cloned into pET28a(+) (Novagen, USA) at the NdeI and XhoI sites (obtained from GenScript, USA), was used for rVpCsgA production. The 159 amino acid residues long recombinant monomer (consisting of 26 amino acid residues contributed by the vector, including 6 histidine residues each at the N- and C-termini) produced from this construct would be of ~ 17 kDa.
Plasmid pET28.VpcsgA was transformed into competent E. coli BL21(λDE3)pLysS cells. Expression analysis of the rVpCsgA from the transformed cells was carried out as described earlier (Karan et al. 2020). The cells were induced with IPTG (1 mM) for additional 6–8 h and the expression of the rVpCsgA was analyzed by SDS-PAGE (15%) in the uninduced and induced cell lysates (prepared by boiling at 100 °C for 10 min in 6× gel loading buffer (0.375 M Tris-HCl, pH 6.8, 12% SDS, 60% glycerol, 12% β-mercaptoethanol, 0.06% bromophenol blue) to obtain 1× working concentration). The rVpCsgA was purified from the soluble fraction of the induced cell lysate by metal affinity chromatography using Co+2-NTA resin as described earlier (Karan et al. 2020). In purified rVpCsgA dialyzed against 50 mM Tris-HCl, pH 8.0 was concentrated using (3 kDa) Amicon ultra centrifugal filter, flash frozen in small aliquots, and stored at − 80 °C. Protein concentration was estimated by BCA protein estimation kit (Bio-Rad, USA). Purified recombinant protein was authenticated by MALDI-TOF-MS at the Sandor Proteomics, Hyderabad, India.
Immunogenicity of rVpCsgA
Experiments involving BALB/c mice were conducted after due approval of the Institutional Animal Ethics Committee (IAEC-JNU project code # 08/2017) of the Jawaharlal Nehru University, New Delhi, India. The animals were housed in the Animal House Facility of the University and all the experiments were performed as per the guidelines provided by the Institutional Animal Ethics Committee of the University (IAEC-JNU), in line with the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978).
Schematic representation of the immunization, sera collection, and challenge schedule is shown in Fig. S1. All operations (immunization, sera collection, and challenge) were performed at 11.30 am on the indicated day. After collecting pre-immune (PI) serum, BALB/c mice (female, 4–6 weeks, body weight 20.3 ± 1.2 g, n = 10 per group) were housed in the Central Laboratory Animal Resources, JNU, New Delhi. The mice were injected intraperitoneally (IP) with the purified rVpCsgA (15 μg, emulsified in alum, 1:1) at the age of 40–42 days, followed by two booster doses (15 μg) administered on days 15 and 28 of primary immunization. Control mice received PBS emulsified in alum. Immunized mice were bled on different days post-immunization (days 14, 21, and 35) through retro-orbital plexus under light anesthesia (anesthetic used avertin, 250 mg/g body weight. i.p.; 1.2% solution prepared as per the Cold Spring Harbor Protocol (http://cshprotocols.cshlp.org/content/2006/1/pdb.rec701.full)). The blood was allowed to clot for 1 h at 25 °C and the sera, collected by centrifugation at 7000 rpm in a microcentrifuge (Eppendorf, USA) at 4 °C for 7 min, were stored at − 20 °C until further use.
Enzyme-linked immunosorbent assay
Direct ELISA was used to determine end point titer of the anti-rVpCsgA antiserum using pre-immune (PI) serum as control as described earlier (Sharma and Dixit 2016). ELISA plates were coated with the rVpCsgA (500 ng/100 μl/well in 0.2 M carbonate-bicarbonate buffer, pH 9.2) and incubated at 4 °C for overnight. Non-specific sites were blocked by incubation in PBST (1× PBS, pH 7.4, 0.05% Tween-20) containing 2% BSA for 2 h at 37 °C. The plate was then incubated with different dilutions (1:500, 1:1000, 1:5000, 1:10,000, 1:25,000, 1:50,000, 1:75,000, 1:1,00,000) of anti-rVpCsgA antisera prepared in PBST containing 2% BSA at 37 °C for 1 h. After washing extensively with PBST (3 washes of 10 min each), horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG secondary antibody (1:5000 dilution in PBST containing 2% BSA) was added and color was developed by adding orthophenylenediamine (0.5 mg/ml in citrate phosphate buffer, pH 5.5 containing 30% H2O2 (1 μl/ml)).The reaction was terminated by the addition of 2 N H2SO4 and the absorbance was measured at 450 nm in an ELISA plate reader (Tecan, USA).
A similar ELISA was performed to understand class switching and determine the levels of different isotypes in the antisera using isotype (IgG1, IgG2a, IgG2b, and IgA)-specific rat anti-mouse mononoclonal antibodies as per the manufacturer’s directions (BD-Pharmingen mouse immunoglobulin isotyping ELISA kit).
Western blot analysis
Western blot analysis using anti-rVpCsgA antisera was performed as described earlier (Karan et al. 2016). The purified rVpCsgA and induced cell lysate of rVpCsgA resolved on the SDS-PAGE were transferred onto the nitrocellulose membrane. After overnight blocking with 3% bovine serum albumin (BSA) in 1× PBST at 4 °C, the membrane was immunoblotted with primary antibody (1:200), followed by incubation with HRP-conjugated anti-mouse secondary antibody (1:5000, 1 h) at room temperature (RT). After extensive washing with PBST between each incubation, the blot was developed using Pierce ECL Western blotting substrate as per the manufacturer’s direction (Thermo Scientific, USA). The image was acquired using a Biospectrum 500 Imaging system (UVP, UK).
Analysis of T cell response
Splenocytes from the rVpCsgA-immunized mice and PBS-immunized mice (control mice) were isolated a week after the second booster as described earlier (Yadav et al. 2016). Splenocytes were washed with 1× RPMI-1640 containing 10% fetal bovine serum medium (Gibco, Fisher Scientific, UK), followed by 0.9% NH4Cl wash to rupture the RBCs. Viable splenocytes were seeded in 96-well culture plates (1 × 105 cells/100 μl/well) and stimulated with rVpCsgA (20 μg/ml) for different time periods at 37 °C in 5% CO2 atmosphere. Splenocyte proliferation was measured using XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) cell proliferation kit (Biological Industries, USA). Absorbance was measured at 450 nm in a plate reader.
Determination of cytokines
The levels of IFN-γ and IL-4, marker cytokines of Th1 and Th2 response, in splenocyte culture supernatants collected at different time points post-stimulation, were measured using cytokine-ELISA kit (BD-Pharmingen, USA) as per the manufacturer’s instructions.
The global cytokine responses in the splenocyte culture supernatants (collected at 72 h post-stimulation) were analyzed using Proteome Profiler antibody array (ARY006, R&D Systems, USA) as per the manufacturer’s instructions. First and foremost, the array buffer 6 (provided by the manufacturer) was used to block the ready to use membrane pre-coated with analyte-specific capture antibodies at RT for 1 h. The blocking buffer was then drained followed by addition of the culture supernatants (1 ml of culture supernatant mixed well with 0.5 ml of array buffer 4 and 15 μl of reconstituted detection antibody cocktail) to the membrane. The membrane was allowed to incubate at 4 °C O/N on a rocking platform for 10 min followed by thorough washing with 1× wash buffer for 10 min. This was followed by the addition of streptavidin-HRP solution diluted in array buffer 6 (1:2000) to the membrane, incubation at RT for 1 h with shaking. After washing, the membrane with 1× wash buffer as before, 1 ml of Chemi Reagent mix (provided in the kit) was added for 1 min to detect the immunoreactive spots. Biospectrum 500 Imaging system (UVP, Cambridge, UK) was used to acquire the images. Densitometric analysis of the immunoreactive spots on the image acquired using Biospectrum 500 Imaging system (UVP, Cambridge, UK) was performed using ImageJ 1.52a, a freely available online tool (https://imagej.nih.gov/ij/).
In vitro agglutination assay
Agglutination ability of the anti-rVpCsgA antisera was determined by the method of Sharma and Dixit using V. parahaemolyticus cells (accession no. MTCC#451 from the Microbial Type Culture Collection, Institute of Microbial Technology, Chandigarh, India) (Sharma and Dixit 2016). Briefly, mid-log phase V. parahaemolyticus cells (1 × 108 colony-forming units (CFU)) were resuspended in different dilutions of either the anti-rVpCsgA or pre-immune antisera (1:100, 1:200, 1:300, 1:500 in 1 ml 1× PBS) and incubated at 37 °C for 2 h. The cells were collected by centrifugation at 4000×g for 10 min and washed with 1× PBS to remove any unbound antibodies, followed by resuspension in 100 μl of 1× PBS. The uniformly suspended cells were then smeared on glass slides, followed by air-drying, heat-fixing, and staining with 1% methylene blue (prepared in 95% ethanol containing 0.01% KOH). After washing with Milli-Q water, the cells were visualized under a bright-field microscope (Eclipse TE2000S, Nikon, USA).
Specificity of the anti-rVpCsgA antibody-mediated agglutination was determined by pre-incubating the antisera with rVpCsgA (1 μg per 1 μl of antisera at 1:300 dilution), prior to addition to the V. parahaemolyticus cells.
Cell surface CsgA recognition by anti-rVpCsgA antibodies
Whether or not the antibodies present in the anti-rVpCsgA are able to recognize the native CsgA present on the cell surface was analyzed by flow cytometry and immunoelectron microscopy as described below:
Flow cytometry
The ability of the anti-rVpCsgA antibodies to recognize the native CsgA present on the cell surface was analyzed by fluorescence-activated cell sorting (FACS) as described earlier (Sharma and Dixit 2016). Mid-log V. parahaemolyticus cells (1 × 106 CFU) were incubated for 1 h at 4 °C with anti-rVpCsgA antisera or the pre-immune sera (1 ml, 1:50) followed by washing and resuspension in 1× PBS. The cells were then pelleted by centrifugation and washed with 1× PBS to remove any unbound antibodies, followed by resuspension in small volume of 1× PBS. FITC-conjugated goat anti-mouse IgG secondary antibody (1:200) was added and incubated further (30 min, 4 °C). Fluorescence data collected using FACSVerse (BD Immunocytometry System, USA) was analyzed using the FACS Diva Version 8.0.1 software.
Immunoelectron microscopy
Interaction of the anti-rVpCsgA antibodies with the CsgA on V. parahaemolyticus cells was confirmed by immunogold labeling as described by Chatrath et al. (2014). Mid-log phase V. parahaemolyticus cultures were incubated in TEM fixative (4% paraformaldehyde and 0.5% glutaraldehyde in 1× PBS) initially for 60 min at RT, followed by re-incubation in fresh TEM fixative for another 10 min at 4 °C. The cells were then treated with aldehyde quenching solution (100 mM ammonium chloride in 1× PBS) for 10 min at 4 °C, followed by two washes with 1× PBS at 4 °C (15 min each). Thin (100 nm) sections of the agar-encased cells, embedded in London resin (LR) white resin (Electron Microscopy Sciences, USA), were obtained on nickel grids (200 mesh, Electron Microscopy Sciences, USA) using Leica Ultracut (Leica, Germany). After blocking with 2% BSA in 1× PBS, the grids were incubated O/N with primary antibody (1:20) and washed thrice with PBST for 10 min each. The grids were then incubated (60 min, RT) with colloidal (10 nm) gold-labeled anti-mouse antibody (Sigma-Aldrich, USA). After five washes with PBST, the grid was rinsed with Milli-Q water and stained with saturated uranyl acetate solution (1–2%). The immunolabeled cells were visualized by transmission electron microscopy (Tecnai F20 TEM, USA) at the National Institute of Immunology, New Delhi.
Detection of curli fibers on V. parahaemolyticus cells
Congo red staining
Congo red staining (Sigma-Aldrich, USA) and Congo red fluorescence measurement were used to detect the curli system in V. parahaemolyticus by the method of Kan et al. (2019). Congo red (2 mg/ml) was added to the mid-log phase V. parahaemolyticus culture for 10 min at 37 °C and centrifuged at 4300×g. The color of the pellet was observed and compared with negative control. For Congo red fluorescence analysis, the cells from overnight culture were grown in the presence of Congo red (25 μg/ml) for 4–5 h. Congo red fluorescence (excitation at 300 nm, slit width 10 nm; emission at 510 nm, slit width 5 nm) was measured using a fluorimeter (Carry Eclipse, Varian Optical Spectroscopy Instruments, USA) at 25 °C.
Negative staining
Mid-log phase grown V. parahaemolyticus cells, washed in PBS, were adsorbed on the copper grids (200 mesh, Electron Microscopy Sciences, USA) and stained with 2% uranyl acetate and visualized by transmission electron microscopy (Chatrath et al. 2014).
In vitro adhesion and invasion assay of V. parahaemolyticus cells on colorectal carcinoma cells
Ability of anti-rVpCsgA antisera to inhibit adherence and subsequently internalization of the host cell was assessed using in vitro adhesion assay using human colorectal carcinoma cells HCT116 (ATCC#CCL247) essentially as described earlier (Luna-Pineda et al. 2019). The HCT116 cells (procured from National Centre for Cell Science, Pune, India) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) medium in a T25 cell culture flask at 37 °C in 5% CO2 atmosphere. Upon attaining confluency, the HCT116 cells were harvested and seeded in a 24-well culture plate (1 × 105 cells/1000 μl/well in fresh DMEM supplemented with 10% FBS) and incubated at 37 °C for 16 h in a 5% CO2 incubator. Simultaneously, a primary culture of V. parahaemolyticus was inoculated in LB broth and allowed to grow overnight at 37 °C with shaking (200 rpm). The next day, the secondary culture of bacterial cells (inoculated with 1% primary culture) grows until late log phase. Vibrio parahaemolyticus cells from the late log phase culture (1 × 107 colony-forming units (CFU)) were incubated with pre-immune antisera (1:50 in 1 ml 1× PBS) or anti-rVpCsgA antiserum (1:50 and 1:100 in 1 ml PBS) at 37 °C for 2 h (in triplicate). The HCT116 cells treated with PBS were included as negative control. The bacterial cells were harvested by centrifugation (4300×g for 10 min). After washing with 1× PBS, the V. parahaemolyticus cells (pre-incubated with PBS or antisera) resuspended in DMEM containing 10% fetal bovine serum were added to the HCT116 monolayers (multiplicity of infection, 1:100; in quadruplicates) and incubated at 37 °C for 2 h in 5% CO2 atmosphere. The medium was removed and the cells were washed three times with DMEM to remove any non-adhered bacterial cells. The monolayers were then treated with lysis buffer (100 μl, 0.1% Triton X-100 in 1× PBS) for 10 min at RT. LB broth (900 μl) was then added to each well and the adhered and intracellular bacteria were collected by repeated up- and down-pipetting. Different dilutions of the suspension thus obtained were made in LB broth, plated on LB agar, and incubated overnight at 37 °C. The CFU/ml (corresponding to the adhered and intracellular bacteria) in the suspension was determined by plating different dilutions on LB-agar plates.
Effect of rVpCsgA immunization on liver and kidney function
To determine if the rVpCsgA immunization is safe and does not cause any adverse effect on mice physiology, commercial services from the PATHLAB, East of Kailash, New Delhi, India were availed to carry out liver and kidney function analyses in the serum of immunized mice. The serum samples harvested from the rVpCsgA-immunized mice (n = 4/group) after 16 h after the administration of the protein were subjected to biochemical analysis for hepatic function markers (aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and albumin) and renal function markers (creatinine and blood urea nitrogen (BUN)), using assay kits from Medicon, India in automated biochemical analyzer (Accute, TBA-40 FR, Toshiba, Japan). To assess the tissue damage, the liver, lung, and kidney from the immunized mice were excised immediately after sacrifice, washed with chilled PBS, and fixed in PBS-buffered formaldehyde for further processing (embedding in paraffin, hematoxylin-eosin staining, sectioning, and microscopy) by the commercial service provider PATHLAB, East of Kailash, New Delhi, India. Samples (sera and tissue) from the PBS-immunized mice were included as control.
Bacterial challenge studies
Prior to challenge studies, LD50 dose of the V. parahaemolyticus was determined by the method of Reed and Muench (1938). The LD50 dose was determined to be 2.5 × 108 CFUs. Protective efficacy of the anti-rVpCsgA antisera was determined by passive challenge of unimmunized BALB/C mice (n = 10) with 2× LD50 dose of V. parahaemolyticus cells in 50 μl 1× PBS (IP), pre-incubated with equal volume of anti-rVpCsgA antisera at 37 °C for 1 h. Control group was challenged with the same amount of cells pre-incubated with neat pre-immune serum. The mice were monitored for survival for 10 days.
Another set of PBS- and the rVpCsgA-immunized mice (n = 10 per group) was used for challenge studies. These mice were given a 3rd booster on day 60. In vivo challenge of these mice was performed by challenging them with the 2× LD50 dose of V. parahaemolyticus (IP) 7 days after the 3rd booster at 11.30 am. Challenged mice were observed for survival for 10 days.
Statistical analysis
The significance (p value) was calculated using paired Student’s t test. The data represent mean ± standard deviation of three independent experiments, performed in triplicates.
Gene sequence
The nucleotide sequence of the synthetic csgA gene of V. parahaemolyticus has been submitted to the National Center for Biotechnology Information (NCBI) GenBank (Accession no. MN530930).
Results
Preparation of rVpCsgA
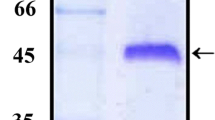
For recombinant CsgA expression, a synthetic VpcsgA gene was designed and cloned into expression vector pET28a(+) which was transformed into E. coli BL21(DE3)pLysS cells. Induction of the E. coli BL21(λDE3)pLysS cells harboring the pET28.rVpCsgA showed the expression of the histidine-tagged rVpCsgA in the soluble fraction (data not shown). The rVpCsgA from the soluble fraction was purified to near homogeneity (~ 98%) and appeared to be in oligomeric forms (Fig. 1; putative dimer and tetramer). The authenticity of the purified rVpCsgA was confirmed by MALDI-TOF-MS analysis (Fig. S2).

Preparation of the recombinant CsgA of V. parahaemolyticus (rVpCsgA). SDS-PAGE (15%) analysis of rVpCsgA purified from the soluble fraction (P). M indicates migration of protein molecular weight (kDa). Arrows point to the oligomeric forms of the histidine-tagged rVpCsgA
Analysis of immune response
Immunogenicity of the rVpCsgA
Immunization with the rVpCsgA generated a highly effective immune response, evident from high end point titers of the anti-rVpCsgA antisera (1:100,000, dilution of the anti-rVpCsgA antiserum, at which the absorbance became almost equal to pre-immune serum) (Fig. 2a). A significant increase in the IgG levels was observed in the antisera collected on day 21, after first booster dose. No further increase was observed after the second booster.

Determination of antigenicity of the rVpCsgA. a Antibody titer determination. Sera from the immunized mice, collected on different days post-immunization, were analyzed for the rVpCsgA-specific antibody titer by ELISA. Primary antibody was used at different dilutions in 2% BSA in 1× PBS containing 0.05% Tween-20 and secondary antibody was used at 1:5000 dilution. The rVpCsgA-specific antibody titers were determined to be greater than 1:100,000. PI indicates pre-immune serum. Significant changes with respect to the absorbance obtained with PI are shown ($, p ≤ 0.001; *, p ≤ 0.005; #, p ≤ 0.05). b Immunoblot analysis using anti-rVpCsgA antiserum. Both the induced cell lysates of the E. coli BL21(DE3)pLysS cells harboring the pET28.VpcsgA and the purified rVpCsgA, resolved on SDS-PAGE (15%), were immunoblotted with anti-rVpCsgA antiserum (1:200 in 1× PBS containing 0.05% Tween-20). Immunoreactive bands at the positions seen in the purified protein (P) and also at the expected size of the monomer (~ 17 kDa) are seen in the induced cell lysate (I). Migration of the molecular weight markers is shown (kDa)
Western blot analysis using anti-rVpCsgA antisera showed specific immunoreactive bands at the expected size (i.e., monomeric and oligomeric positions) in the induced cell lysate as well as the two bands in the purified protein confirm the antigenicity of the purified rVpCsgA, as well as specificity of the anti-rVpCsgA antibodies (Fig. 2b).
Immune response analysis by antibody isotyping
The switch of immunoglobulin expression and differentiation of isotypes in the anti-rVpCsgA antisera collected on different days post-immunization were analyzed by measuring the levels of different IgG isotypes. Isotyping of the anti-rVpCsgA antisera showed a significant increase in the levels of all the isotypes on all the antisera collected on different days, when compared to pre-immune serum with maximum increase in IgG1 followed by IgG2b (Fig. 3). The ratio of IgG1:IgG2a and IgG1:IgG2b was found to be > 1, indicating a Th2-biased mixed immune response (Bielinska et al. 2016).

Antibody isotyping of the anti-rVpCsgA antisera. Sera collected at different days (day 14, day 21, and day 35) post-immunization from the rVpCsgA-immunized mice were analyzed for the levels of different isotypes by ELISA using isotype-specific secondary antibodies. PI indicates pre-immune serum. Significant changes with respect to the levels of respective isotype in pre-immune serum are shown ($, p ≤ 0.001; *, p ≤ 0.005; #, p ≤ 0.05)
In vitro T cell response by the rVpCsgA
Figure 4a shows that stimulation with the rVpCsgA resulted in significantly enhanced proliferation of splenocytes at all the intervals (p ≤ 0.001 to p ≤ 0.005) post-stimulation when compared to the rVpCsgA-stimulated splenocytes of the PBS-immunized mice and unstimulated splenocytes, evidenced by higher absorbance in XTT cell proliferation assay. The proliferation index (A450 stimulated/unstimulated) was determined to be 1.94, 2.81, and 2.9 at 24, 48, and 72 h respectively for the splenocytes from the rVpCsgA-immunized mice at the respective time point.

In vitro T cell response assessment. a Stimulation of splenocyte proliferation by the rVpCsgA. Splenocytes isolated from the PBS- and rVpCsgA-immunized mice were stimulated with the rVpCsgA (stimulated) or with the corresponding volume of the PBS (unstimulated) and monitored for proliferation by incubation in a humidified CO2 (5%) incubator for 24 h, 48 h, and 72 h using XTT cell proliferation assay. Significant changes are determined by comparing the rVpCsgA-stimulated splenocytes with PBS-stimulated splenocytes. $, p ≤ 0.001; *, p ≤ 0.005. b and c Culture supernatants from the PBS-stimulated or rVpCsgA-stimulated splenocytes from rVpCsgA-immunized mice collected at different time intervals were analyzed using cytokine-specific antibodies (1:250) and detection antibodies for IFN-γ (1:500, panel b) and IL-4 (1:250, panel c) levels using cytokine-specific ELISA employing the use cytokine-specific capture antibodies. Significant changes with respect to the levels of respective cytokine in PBS-stimulated splenocytes are shown ($, p ≤ 0.001; *, p ≤ 0.005)
The culture supernatants of the splenocytes from proliferation assay, collected at different time points post-stimulation, were analyzed for the IFN-γ and IL-4 levels, which are indicative of type I and type II immune response. As shown in Fig. 4b and Fig. 4c, a significant increase in the levels of both IFN-γ (p ≤ 0.005–0.001) and IL-4 (p ≤ 0.001) was observed, respectively in the culture supernatant collected at all the three time intervals post-stimulation indicating a mixed T-helper cell immune response. Though a slight decrease in the IFN-γ level was noticed at 72 h when compared to that at 48 h post-stimulus, the IFN-γ levels still remained significantly higher than that of the control supernatant. Culture supernatants from control splenocytes (PBS-immunized mice and stimulated with rVpCsgA) did not show any change in either cytokine’s level.
As maximum proliferation of splenocytes was noted at 72 h post-stimulation with the rVpCsgA, the culture supernatant collected at this time point was analyzed to have a deeper insight into the T-helper cell immune response generated against rVpCsgA. The changes in the level of the numerous cytokines and chemokines, significant players in CD4+ T cell differentiation, were analyzed using antibody array comprising the specific capture antibodies against a panel of cytokines/chemokines of different T helper cells’ pathways (Fig. S3). Amongst the test cytokines, significantly increased levels of 26 cytokines/chemokines (p ≤ 0.05 to 0.001) were observed in the culture supernatants of rVpCsgA-stimulated splenocytes from rVpCsgA-immunized mice when compared to that from PBS-stimulated splenocytes (Fig. 5). Maximum change at the level of p ≤ 0.001 was observed in the levels of IP-10, TIMP-1, TNF-α, KC, G-CSF, TREM-1, GM-CSF, JE, IL-13, IL-10, sICAM-1, CCL-4, IL-16, IL-1α, CXCL2, IL-17, IL-1β, and CCL3. The TARC, IL-3, IL-6, IFN-γ, IL-1F3, and IL-2 showed an increase at p ≤ 0.005. Minimum yet significant increase (p ≤ 0.05) was observed in IL-4 and CCL-5. Thus, an increase in different molecules belonging to Th1, Th2, and Th17 differentiation pathways was observed.

Cytokine array analysis to assess global cytokine response. Cytokine array kit from the R&D systems was used to determine the relative levels (pixel density) of different cytokines in the culture supernatants in the PBS-stimulated and rVpCsgA-stimulated splenocytes isolated from the rVpCsgA-immunized mice (Fig. S2). Only cytokines/chemokines showing significant change with respect to PBS-stimulated splenocytes are graphically shown. $, p ≤ 0.001; *, p ≤ 0.005; #, p ≤ 0.05
Neutralization potential of the anti-rVpCsgA antisera and specificity of anti-rVpCsgA antibody interaction with V. parahaemolyticus CsgA
Since presence of curli amyloid fibers on the bacterial cells is essential for interaction of the anti-rVpCsgA antibodies to bring about agglutination and neutralization, presence of curli fibers on the V. parahaemolyticus (#451) cells used in the study was first confirmed using Congo red staining and negative staining (Fig. S4). As evident from Fig. S4a, the V. parahaemolyticus (#451) cells showed deep positive Congo red staining indicating the presence of curli fibers, whereas the E. coli LSR12 cells lacking the curli fibers showed negligible staining. This was further confirmed by approximately 5-fold higher Congo red fluorescence of the stained V. parahaemolyticus (#451) in comparison to that of E. coli LSR12 cells (Fig. S4b). TEM analysis of the negatively stained V. parahaemolyticus (#451) cells clearly showed the presence of non-uniform, amorphous, entangle curli fibers on its cell surface (Fig. S4c). Thus, these cells were used to assess the agglutination ability of the anti-rVpCsgA antisera.
The neutralization potential of the anti-rVpCsgA antisera was assessed by its ability to agglutinate live V. parahaemolyticus cells. Agglutination analysis showed that incubation of the V. parahaemolyticus cells with anti-rVpCsgA antisera resulted in positive agglutination, whereas no clumping was observed with pre-immune sera (Fig. 6). As expected, an increase in antisera dilution showed decreased agglutination. A decrease in agglutination of the cells upon incubation with the anti-rVpCsgA antisera, pre-incubated with the rVpCsgA (lower most panels), confirmed the specificity of the clumping due to specific interaction between the anti-rVpCsgA antibodies with the CsgA present on the cells.

In vitro neutralization ability of the anti-rVpCsgA antisera by agglutination assay. a Mid-log phase cells (1 × 108 CFU) of V. parahaemolyticus (MTCC#451) were resuspended in different dilutions of anti-rVpCsgA antisera (in 1× PBS) and incubated at 37 °C for 2 h. b The V. parahaemolyticus (MTCC#451) cells were resuspended in anti-rVpCsgA antisera pre-incubated with the rVpCsgA (1 μg per 1 μl of antisera at 1:300 dilution) and incubated further for 2 h at 37 °C. Extent of agglutination was visualized under light microscopy (magnification × 40)
Qualitative specific interaction of the anti-rVpCsgA antibodies with the CsgA present on the cell surface is evident from the appearance of black dots on the V. parahaemolyticus cells when anti-rVpCsgA antisera was used in colloidal gold immunolabeling followed by TEM analysis. No such dots are visible on the cells incubated with pre-immune sera (Fig. 7a). Quantitative analysis of interaction of anti-rVpCsgA antibodies with the cell surface CsgA was analyzed by FACS (Fig. S5). Analysis of fluorescence data showed a significantly increased fluorescent cell population (~ 27 folds, p ≤ 0.001) with anti-rVpCsgA antisera in comparison to pre-immune sera (Fig. 7b).

Analysis of cross-reactivity of the antibodies present in the anti-rVpCsgA antisera with the surface exposed CsgA on V. parahaemolyticus cells. a Transmission electron microscopy of the immunogold-labeled V. parahaemolyticus cells treated with pre-immune (PI) and anti-rVpCsgA antisera. Mid-log phase grown V. parahaemolyticus (MTCC#451) cells were fixed and thin sections of the cells stained with the pre-immune or the anti-rVpCsgA antisera (1:10 dilution) were processed for immunogold labeling using colloidal gold-labeled anti-mouse antibody (1:5). The stained grids were visualized under transmission electron microscope. Specific presence of defined black dots in the cells incubated only with the anti-rVpCsgA antisera indicates the specificity of the reactivity of the antibodies present in the anti-rVpCsgA antisera with the native CsgA on the cell surface of V. parahaemolyticus (MTCC#451) cells. b Flow cytometry analysis to assess the interaction of the antibodies present in the anti-rVpCsgA antisera with the native CsgA present on V. parahaemolyticus (MTCC#451) cells. The cells were incubated with either pre-immune (PI) or anti-rVpCsgA antisera (1:50) followed by FITC-conjugated secondary antibody (1:200 in 1× PBS). Percentage of FITC-stained fluorescent cell population is plotted. Data represent mean ± SD of three independent experiments, shown in Fig. S3. *, p ≤ 0.001 with respect to cells treated with PI
Besides assessing the agglutination ability of the anti-rVpCsgA antisera, the anti-rVpCsgA antisera was also assessed for its ability to inhibit adherence of V. parahaemolyticus in vitro using HCT116 cells as the curli fibers composed of CsgA also play an important role in its adhesion to the host cells (Fig. 8). The HCT116 cells treated with the V. parahaemolyticus cells pre-incubated with the anti-rVpCsgA antiserum showed significantly (p ≤ 0.001) reduced mean of adhered and intracellular bacteria (6.38 × 105 CFU/ml and 1.69 × 106 CFU/ml with1:50 and 1:100 dilutions of the anti-rVpCsgA antisera, respectively), when compared to that treated with the bacterial cells pre-incubated with the control pre-immune serum (8.365 × 106 CFU/ml at 1:50 dilution of the pre-immune serum).

In vitro neutralization of adhesion and invasion of V. parahaemolyticus on HCT116 cells by the anti-rVpCsgA antiserum. Vibrio parahaemolyticus cells (1 × 107 CFU) pre-incubated with either pre-immune serum (PI, 1:50; 1 ml 1× PBS) or anti-rVpCsgA antiserum (1:50 and 1:100 in 1 ml 1× PBS) at 37 °C for 2 h were allowed to adhere and invade the HCT116 cells (1 × 105 cells/1000 μl/well; multiplicity of infection, 1:100) for 2 h at 37 °C in 5% CO2 atmosphere. The adhered and intracellular V. parahaemolyticus cells were determined using microdilution method. The graph shows Bee-Swarm plot of CFU/ml of four replicates of each experimental sample. Statistical significance (***, p ≤ 0.001) is calculated with respect to bacterial cells pre-incubated with PI
Protective efficacy of rVpCsgA immunization against V. parahaemolyticus
Challenge of normal BALB/c mice with the 2 × LD50 dose of live V. parahaemolyticus cells (5 × 108 CFU) pre-incubated with the anti-rVpCsgA antisera did not result in any mortality during the observation period (10 days). On the other hand, all the mice administered with the cells pre-incubated with pre-immune sera died within 12 h (Fig. 9a).

Bacterial challenge assay. a Protective efficacy of the anti-rVpCsgA antiserum. Naïve mice (n = 10 per group) were challenged with 2 × LD50 (5 × 108 CFUs in 50 μl 1× PBS, IP) of live V. parahaemolyticus (MTCC#451) cells pre-incubated with equal volume of either the pre-immune (PI) or anti-rVpCsgA antisera. b In vivo challenge of immunized mice. Mice immunized with the rVpCsgA were challenged with 2 × LD50 dose of V. parahaemolyticus (MTCC#451) cells. PBS-immunized mice challenged with the same dose of bacterial cells were included as control. The challenged mice were monitored for survival for 10 days
Figure 9b shows the results of in vivo challenge assay of the rVpCsgA-immunized BALB/c mice with 2 × LD50 dose of V. parahaemolyticus cells. Mice immunized with the rVpCsgA showed 100% survival, whereas all the PBS-immunized mice died within 12 h only. The rVpCsgA-immunized mice did not show any sign of sickness and remained healthy with normal intake of food and water. No mortality was observed even beyond the observation period.
Effect of the rVpCsgA immunization on liver and kidney function
Since mice infected with V. parahaemolyticus have been shown to have disturbed liver and kidney function (Wang et al. 2020), we evaluated if the rVpCsgA immunization affected the liver and kidney function and caused tissue damage. As shown in Table 1, no significant difference in the serum levels of SGPT, SGOT, ALP, and albumin (indicative of hepatic function) was noted between the PBS-immunized and rVpCsgA-immunized mice. Likewise, no significant change was observed in the serum creatinine and blood urea nitrogen (BUN) levels of the rVpCsgA-immunized mice when compared to PBS-immunized mice. Histological examination of the liver, kidney, and lung tissue sections (hematoxylin and eosin stained) of the rVpCsgA-immunized mice was carried out to assess if the rVpCsgA immunization caused any tissue damage (Fig. 10). Like the kidneys of the control PBS-immunized mice, the kidneys from the rVpCsgA-immunized mice revealed intact glomeruli and tubules in the renal cortex. No tubular cell sloughing or necrosis or foci of inflammation were observed. Similarly, the liver tissue from the rVpCsgA-immunized mice revealed intact central vein with normal endothelial cells with no evidence of coagulative necrosis of hepatocytes, and no sinusoidal oedema or inflammation. Sections from the lung tissue of the rVpCsgA-immunized mice also showed normal morphology. No alveolar oedema or interstitial neutrophilic infiltration, congestion, or alveolar/pulmonary hemorrhage was evident. Thus, the rVpCsgA immunization did not cause any tissue damage.

Histological evaluation of the liver, kidney, and lung tissues of the rVpCsgA-immunized mice. Representative microscopic images of the hematoxylin-eosin-stained sections of the liver, lung, and kidney tissues isolated from the immunized mice. a PBS-immunized control mice tissue. b rVpCsgA-immunized mice tissue. Magnification ×40
Discussion
Due to the development of multidrug-resistant V. parahaemolyticus, the most commonly reported Vibrio species responsible for vibriosis in humans, the bacterium poses a major problem to both the aquaculture and food export industry. Vaccination remains the most effective method to combat this infection in aquaculture. Generation of an effective immune response and antigen-specific memory is key to develop an effective vaccine. Like other enteropathogenic bacteria, a number of OMPs and adhesins of V. parahaemolyticus have been assessed for their immunogenicity in a variety of test organisms, however with a varied degree of protection. Bacterial cell surface is the primary point of interaction with the host; hence, a protein which is abundantly present on the surface is likely to confer a most effective protective immune response (Wang et al. 2012).
CsgA, the major subunit of curli fibers, in E. coli and Salmonella enterica as csg (curli-specific gene) and tafi (thin aggregative fimbriae), respectively has been reported to be responsible for colonization and biofilm formation (Dueholm et al. 2012). It is one of the most abundant proteins present on the cell surface and can be an effective vaccine candidate. However, the reports of CsgA as a potential vaccine are scarce and remain limited to enteropathogenic E. coli (Luna-Pineda et al. 2019). In the present study, we have explored the vaccine potential of CsgA of V. parahaemolyticus, an important pathogen.
As the well-studied E. coli CsgA amyloid fiber resembled other bacterial, fungal, and mammalian amyloids, based on tinctorial Congo red staining, and Thioflavin T binding properties (Van Gerven et al. 2015), we therefore tested if the V. parahaemolyticus strain used in the present study possessed CsgA curli amyloids or not. Positive Congo red staining of the V. parahaemolyticus (MTCC #451) established the presence of CsgA curli amyloid fibers on its surface, which was further confirmed by the presence of entangled fibers when observed under electron microscopy (Fig. S4). Therefore, this bacterium was used for agglutination assay and challenge studies of mice immunized with the recombinant CsgA of V. parahaemolyticus.
To evaluate the vaccine potential, the rVpCsgA (without the signal sequence) was expressed from a synthetic gene construct under the control of T7 promoter and purified from soluble cytoplasmic fraction. Very high antigen-specific end point titers showed the rVpCsgA to be highly immunogenic. Emulsion with alum, acceptable for human use and has been used in a number of commercial vaccines, aided in optimizing the stability and assisted in adsorption of antigen. The antibody titers against the rVpCsgA administered with alum were comparable to that obtained with a number of adhesins, fimbrial proteins, and OMPs emulsified with CFA/IFA (Yadav et al. 2016; Khalouie et al. 2017) that is known to generate a very high immune response. An increase in antibody titer upon the first booster administration is in agreement with earlier reports put forth for the OmpF and OmpC of Aeromonas hydrophila and OmpW of Vibrio alginolyticus due to humoral immune response (Qian et al. 2007; Yadav et al. 2014, 2016). Subsequent exposure of mature B lymphocytes with antigenic rVpCsgA consequently results in the differentiation, and proliferation of rVpCsgA-activated B cell for immunological memory (Gioia et al. 2005, Hauge et al. 2007). The generation of memory B cells induces heightened level of rVpCsgA-specific IgG antibody with higher sustenance and slower decline kinetics (several folds higher than primary IgG level generated after initial immunization) with no further increase in the IgG levels upon subsequent booster administration (Yadav et al. 2014).
Immunoblotting with the polyclonal antisera confirmed the antigenicity of the rVpCsgA and specificity of the anti-rVpCsgA antibodies as immunoreactive bands at the expected monomeric and oligomeric positions were detected in the induced cell lysates of E. coli cells harboring the pET28.VpcsgA. Positive agglutination of V. parahaemolyticus cells by the anti-rVpCsgA antiserum indicates that the anti-rVpCsgA antibodies present in the antisera are able to recognize the native CsgA on V. parahaemolyticus surface. Loss of agglutination upon pre-incubation of the antisera with the rVpCsgA confirmed that the anti-rVpCsgA antibodies specifically interacted with the cell surface CsgA. Immunogold labeling and FACS analysis of the V. parahaemolyticus cells using anti-rVpCsgA antisera further validated specific interaction of the anti-rVpCsgA antibodies with the bacterial CsgA. Significant reduction in the number of adhered and intracellular V. parahaemolyticus cells upon pre-incubation with the anti-rVpCsgA antibodies in the adhesion and invasion assay using HCT116 cells in vitro further confirmed the neutralizing ability of the anti-rVpCsgA antiserum. The anti-CsgA antibodies present in the anti-rVpCsgA antiserum are able to coat the curli fibers present on the bacterial cells, thus inhibiting its interaction, adherence, and subsequently invasion with the host HCT116 cells.
Ability of the rVpCsgA to induce Th2-biased mixed immune response, established by antibody isotyping of anti-rVpCsgA antisera with an IgG1 to IgG2a/IgG2b ratio much greater than 1, indicated that it is able to stimulate both humoral and cell-mediated response. Significantly higher isotype titers in comparison to the pre-immune sera, at all time points, suggest induction of broad and steady immune response, comprising both the arms of immune response. This is of significance as it is a desirable characteristic of an antigen for providing long-term protection against an infection.
Highly immunogenic antigens stimulate humoral and cellular immune response through B or T cell-mediated pathway by promoting the secretion of cytokines from antigen-presenting and other cells (Spellberg and Edwards 2001). Validation of generation of mixed immune response by the rVpCsgA was further confirmed by measurement of IFN-γ (a Th1 cytokine) and IL-4 (a Th2 cytokine) responsible for isotype switching to IgG2a and IgG1, respectively, in the culture supernatant of the rVpCsgA-stimulated splenocytes isolated from the mice immunized with the rVpCsgA. An increase in proliferation of the rVpCsgA-primed splenocytes isolated from the rVpCsgA-immunized mice, upon re-stimulation with the rVpCsgA, demonstrates that the rVpCsgA is able to generate T cell memory. Increased proliferation of splenocytes has also been reported with other pilli, fimbrial, and flagellin-based vaccine candidates against Vibrio cholerae (Molaee et al. 2017). During splenocyte culture, the diverse cell populations of lymphocytes and antigen-presenting cells undergo maturation upon exposure to the antigenic rVpCsgA and the mature antigen-presenting cells signal the naïve T cells to stimulate T cell proliferation. The T cells are able to recall the previous encounter with rVpCsgA leading to their rapid proliferation and secretion of various cytokines to further assist in averting the apparent infection. Generation of T cell memory by the rVpCsgA, a hallmark of adaptive immune response and absolute requirement for a vaccine to potentiate immune response upon exposure to the pathogen, further confirms the potential of the rVpCsgA as an effective subunit vaccine.
In line with the observed increase in IFN-γ and IL-4 in primary cytokine ELISA, significantly increased levels of signature cytokines of both Th1 response (IL-2, IFN-γ) and Th2 response (IL-4, IL-10, and IL-13), together with the chemokines/cytokines secreted by Th17 cells, in the culture supernatants of the splenocytes stimulated with the rVpCsgA, further validated generation of mixed immune response.
The crucial role of protection against bacterial infection is conciliated through inflammatory mediators (IL-1β, IL-6, G-CSF, GM-CSF, TNF-α) as well as assimilated by chemotactic factors (JE/CCL2, CCL7, KC/CXCL1, CXCL2, CXCL5, CXCL8, and adhesion molecules sICAM-1/CD54) (Kolls et al. 2008). The noted increase in CCL5 is in line with mixed immune response as it is known to co-stimulate the Th1 response cytokine IL-2, and also helps in differentiation of Th2 cells (Liao et al. 2011). An observed increase in the levels of IL-1β/IL-1F2 and other cytokines of type 1 superfamily (IL-2, IL-3, IL-4, IL-6, GM-CSF), reported to assist neutrophils to the site of inflammation, also indicate proinflammatory response (Carson and Kunkel 2017). Together with IL-6, an increase in the levels of another member of IL-6 superfamily, i.e., CSF, was also observed, which has been reported to further stimulate inflammatory response, differentiation of B cell, and regulation of T cells including maturation and differentiation of Th17 cells (Tanaka et al. 2014). An increase in the IFN-γ levels is in agreement with the observed increase in IL-17 levels, released from recently gleamed Th17 cells that regulate the Th1 cells (Shi et al. 2008). Likewise, TIMP-1 (tissue inhibitor of mettaloprotease-1) known to resist multiple infection and secreted by Th1 and Th17 cells were also upregulated (Adamson et al. 2013). Th17 cells have been reported to direct the antigen-presenting cells to the site of infection and protect against a number of extracellular and intracellular pathogens (Wu et al. 2007; Schulz et al. 2008). Stimulation of Th1 response is also evident from enhanced levels of other proinflammatory chemokines, namely JE/CCL2, CCL3, CCL4, CCL5, CXCL2, and IP10 (Lebre et al. 2005). The CCL3 and CCL4 released from the natural killer cells mediate the recruitment of CD8+ T cells and are involved in the acute inflammatory state (Amati et al. 2017), whereas elevated IFN-γ levels stimulate antigen presentation to T cells, differentiation of B cells, thus help in combating the infection (Carson and Kunkel 2017). A concurrent increase in the TNF-α is in agreement with the increased splenocyte proliferation and increased levels of IFN-γ, known to increase secretion of TNF-α, together with MHCI and MHCII (Carson and Kunkel 2017). Elevated levels of multi-functional IL-16, involved in both innate and adaptive immune response with a Th1-mediated inflammatory response, were also noted. Elevated IL-4 levels together with increased IL-13 levels indicate upregulated differentiation of B cell which produce IgG1 and IgE and stimulate macrophages, and thus are critical in clearing the infection (Wynn 2003). Increased levels of IL-13 have also been reported to stimulate cell-mediated immunity and provide resistance to intracellular pathogens like Leishmania major and Listeria monocytogenes (Wynn2003). The culture supernatants of stimulated splenocytes also showed increased levels of TARC (thymus and activation-regulated chemokine), expressed on Th2 cells, which is in line with the observed increase of IL-4, known to induce differentiation of naïve T cells to Th2 cells, which in turn produce more IL-4 (Scheu et al. 2017).
Together with these proinflammatory cytokines, an increase in some of the anti-inflammatory cytokines/chemokines such as IL-10 and IL-1F3 was also observed in the array analysis. IL-F3 blocks the IL-1 receptor binding and perhaps acts antagonistically to the proinflammatory cytokines to keep the excess inflammatory damage in control. Moderately increased levels of IL-16 in the culture supernatants of stimulated splenocytes show modulation of immune response as it is known to control the T cell cytotoxic damage (Wilson et al. 2004). Likewise, moderate increase in the levels of TERM-1 (triggering receptor expressed on myeloid cells), involved in the production of iNOS, NF-κB, Cox-2, and sICAM (soluble intracellular adhesion molecule, constitutively expressed on epithelial cells), would regulate inflammation (Schaller et al. 2017). Thus, overall analysis of the global cytokine profile of the culture supernatant of the stimulated splenocytes points towards mixed type of immune response characterized on the basis of the secreted cytokines and their abilities to assist other immune cells.
It is well known that better protection is obtained by vaccine preparations which are able to stimulate all the arms of immune response and that a balanced mixed immune response comprising both Th1 and Th2 type immune response is necessary for any successful vaccine and clearing of extracellular pathogens (Spellberg and Edwards 2001). Our results show that IFN-γ levels were highest at 48 h post-stimulation, whereas IL-4 levels were highest at 72 h post-stimulation. Thus, switching of immune response towards Th2-biased mixed immune response would prove beneficial in clearing V. parahaemolyticus infection as it is primarily an extracellular pathogen.
In vitro protective efficacy of the anti-rVpCsgA antisera established by agglutination assay was validated by passive challenge of normal mice, wherein pre-incubation of the 2 × LD50 dose of V. parahaemolyticus cells with the antisera resulted in complete protection. One hundred percent survival of the rVpCsgA-immunized mice with the lethal dose of V. parahaemolyticus further validated these data. Though a number of surface-associated afimbrial/fimbrial adhesins/OMPs (VP0802, VP1243, and VP0966) have been found to generate an effective immune response in mice, immunization with only VP0802 conferred ~ 67% protection against the bacterial challenge (Li et al. 2014). In different fish species, immunization with other OMPs of V. parahaemolyticus conferred relatively lower protection (~ 30 to ~ 75%) against the bacterial challenge (Mao et al. 2007; Li et al. 2010, 2014; Peng et al. 2016). Thus, the protection conferred by the rVpCsgA immunization in the present study in mouse model is much greater than that reported by other OMPs of V. parahaemolyticus. Better protection with the rVpCsgA immunization could be attributed to greater and exposed abundance of the CsgA on V. parahaemolyticus cells and therefore the anti-rVpCsgA antibodies could interact with the cells more effectively than the other OMPs.
It is important to note that the present study is aimed at developing a CsgA-based subunit vaccine. Unlike the whole V. parahaemolyticus which adversely affects the kidney and liver function of the infected mice (Wang et al. 2020), the rVpCsgA immunization did not cause any tissue damage as evident from the serum biochemical parameters indicative of liver and kidney function, and tissue histopathology. The idea of using the rVpCsgA, a single non-toxic protein as a subunit vaccine in the present study, is that it does not cause the disease or put the organism receiving at risk of complications associated with the live bacteria or attenuated bacteria with a risk of reversion or heat-killed bacteria which generates non-focussed immune response. In the present study, we have used the rVpCsgA for immunization and the generated immune response clearly shows that it has been processed by the immune system like any other foreign non-self molecule either through phagocytosis mediated by macrophages, dendritic cells, or B cell receptor mediated endocytosis and processed further (Kindt et al. 2007). Subsequently, the exhibition of the peptides generated by processing of the rVpCsgA on antigen-presenting cells helped activate humoral and cellular immune responses. Therefore, though V. parahaemolyticus damages different organs, the recombinant soluble CsgA of V. parahaemolyticus (rVpCsgA), a single antigenic protein administered with an immune adjuvant in BALB/c mice, that is processed by the immune system, was unlikely to cause such systemic damage, as there was no live V. parahaemolyticus present in the same.
Thus, the present study clearly demonstrated that the recombinant CsgA generated a potent immune response, and neutralizing antibodies in BALB/c mice against V. parahaemolyticus. We also demonstrated that immunization with recombinant CsgA of V. parahaemolyticus activated all the three arms of T cell response and conferred protection against the bacterial challenge. Thus, the rVpCsgA of V. parahaemolyticus can be considered a potential subunit vaccine candidate against vibriosis based on mouse model used in the study. While use of mouse model offers several advantages, there exist certain limitations. For example, different routes of vaccine delivery cannot be attempted. Also, several mucosal surfaces and immune compartments are not easily accessible (Kiros et al. 2012). Unlike large animals who are often infected with the same or closely related pathogens (Elahi et al. 2007), mice are physiologically and immunologically less related to humans. Further, the inbred laboratory mice do not allow distribution analysis of vaccine responders and non-responders, which is possible with outbred large animals. Nonetheless, the present study has laid the groundwork establishing the vaccine potential of rVpCsgA of V. parahaemolyticus for further studies to be undertaken to test its efficacy in other animal models.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its supplementary information files.
References
Adamson A, Ghoreschi K, Rittler M, Chen Q, Sun H-W, Vahedi G, Kanno Y, Stetier-Stevenson WG, O’Shea JJ, Laurence A (2013) Tissue inhibitor of metalloproteinase 1 is preferentially expressed in Th1 and Th17 T-helper cell subsets and is a direct stat target gene. PLoS ONE 8:e59367. https://doi.org/10.1371/journal.pone.0059367
Amati AL, Zakrzewicz A, Siebers K, Wilker S, Heldmann S, Zakrzewicz D, Hecker A, McIntosh JM, Padberg W, Grau V (2017) Chemokines (CCL3, CCL4, and CCL5) inhibit ATP-induced release of IL-1β by monocytic cells. Mediat Inflamm 2017:1434872. https://doi.org/10.1155/2017/1434872
Bhowmick R, Ghosal A, Das B, Koley H, Saha DR, Ganguly S, Nandy RK, Bhadra RK, Chatterjee NS (2008) Intestinal adherence of Vibrio cholerae involves a coordinated interaction between colonization factor GbpA and mucin. Infect Immun 76:4968–4977. https://doi.org/10.1128/IAI.01615-07
Bielinska AU, O’Konek JJ, Janczak KW, Baker JR Jr (2016) Immunomodulation of TH2 biased immunity with mucosal administration of nanoemulsion adjuvant. Vaccine 34:4017–4024. https://doi.org/10.1016/j.vaccine.2016.06.043
Bravo S, Midtlyng PJ (2007) The use of fish vaccines in the Chilean salmon industry 1999-2003. Aquaculture 270:36–42
Carson WF, Kunkel SF (2017) Type I and II cytokine superfamilies in inflammatory responses. In: Cavaillon JM, Singer M (eds) Inflammation: from molecular and cellular mechanisms to the clinic. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, pp 587–618. https://doi.org/10.1002/9783527692156.ch24
CDC (2019) Centers for disease control and prevention, national center for emerging and zoonotic infectious diseases (NCEZID), division of foodborne, waterborne, and environmental diseases. Vibrio species causing vibriosis. https://www.cdc.gov/vibrio/faq.html/
Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammer M, Nomark S, Hultgren S (2002) Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295:851–855. https://doi.org/10.1126/science.1067484
Chatrath S, Gupta VK, Garg LC (2014) The PGRS domain is responsible for translocation of PE_PGRS30 to cell poles while the PE and the C-terminal domains localize it to the cell wall. FEBS Letters 588:990–994. https://doi.org/10.1016/j.febslet.2014.01.059
Dueholm MS, Albertsen M, Otzen D, Nielsen PH (2012) Curli functional amyloid systems are phylogenetically widespread and display large diversity in operon and protein structure. PLoS One 7:e51274. https://doi.org/10.1371/journal.pone.0051274
Elahi S, Holmstrom J, Gerdts V (2007) The benefits of using diverse animal models for studying pertussis. Trends Microbiol 15:462–468. https://doi.org/10.1016/j.tim.2007.09.003
Fujino T, Okuno Y, Nakada D, Aoyama A, Mukai T, Ueho T (1953) On the bacteriological examination of shirasu food poisoning. Med J Osaka Univ 4:299–304
Gioia CAC, de Sousa AB, Cruz SC, Junior FCS, Andrade AFB, Sassi RM, Frasch CE, Milagres LG (2005) Effect of a booster dose of serogroup B meningococcal vaccine on antibody response to Neisseria meningitidis in mice vaccinated with different immunization schedules. FEMS Immunol Med Microbiol 44:35–42. https://doi.org/10.1016/j.femsim.2004.11.013
Hauge S, Madhun A, Cox RJ, Haaheim LR (2007) Quality and kinetics of the antibody response in mice after three different low-dose influenza virus vaccination strategies. Clin Vaccine Immunol 14:978–983. https://doi.org/10.1128/CVI.00033-07
Kan A, Birnbaum DP, Praveschotinunt P, Joshi NS (2019) Congo red fluorescence for rapid in situ characterization of synthetic curli systems. Appl Environ Microbiol 85:e00434–e00419. https://doi.org/10.1128/AEM.00434-19
Karan S, Dash P, Kaushik H, Sahoo PK, Garg LC, Dixit A (2016) Structural and functional characterization of recombinant interleukin-10 from Indian major carp Labeo rohita. J Immunol Res 2016:3962596–3962511. https://doi.org/10.1155/2016/3962596
Karan S, Mohapatra A, Sahoo PK, Garg LC, Dixit A (2020) Structural-functional characterization of recombinant apolipoprotein A-I from Labeo rohita demonstrates heat-resistant antimicrobial activity. Appl Microbiol Biotechnol 104:145–159. https://doi.org/10.1007/s00253-019-10204-7
Khalouie F, Mousavi SL, Nazarian S, Amani J, Pourfarzam P (2017) Immunogenic evaluation of chimeric recombinant protein against ETEC, EHEC and Shigella. Mol Biol Res Commun 6:101-112. doi: https://doi.org/10.22099/mbrc.2017.4081
Kim MN, Bang HJ (2008) Detection of marine pathogenic bacterial Vibrio species by multiplex polymerase chain reaction (PCR). J Environ Biol 29:543–546
Kindt TJ, Goldsby RA, Osborne BA, Kuby J (2007) The major histocompatibility complex and antigen presentation. In: Ahr K (ed) Kuby immunology, 6th edn. WH Freeman and Company, New York, pp 189–244
Kiros TG, Levast B, Auray G, Strom S, van Kessel J, Gerdts V (2012) The importance of animal models in the development of vaccines. In: Baschieri S (ed) Innovation in vaccinology. Springer, Dordrecht, pp 251–264. https://doi.org/10.1007/978-94-007-4543-8_11
Kolls JK, McCray PB Jr, Chan YR (2008) Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol 8:829–835. https://doi.org/10.1030/nri2433
Langermann S, Mollby R, Burlein JE, Palaszynski SR, Auguste CG, DeFuscoA Strouse R, Schenerman MA, Hultgren SJ, Pinkner JS, Winberg J, Guldevall L, Soderhall M, Ishikawa K, Normark S, Koenig S (2000) Vaccination with FimH adhesin protects cynomolgus monkeys from colonization and infection by uropathogenic Escherichia coli. J Infect Dis 181:774–778. https://doi.org/10.1086/315258
Lebre MC, Burwell T, Vieira PL, Lora J, Coyle AJ, Kapsenberg ML, Clausen BE, De Jong EC (2005) Differential expression of inflammatory chemokines by Th1- and Th2-cell promoting dendritic cells: a role for different mature dendritic cell populations in attracting appropriate effector cells to peripheral sites of inflammation. Immunol Cell Biol 83:525–535. https://doi.org/10.1111/j.1440-1711.2005.01365.x
Letchumanan V, Yin WF, Lee LH, Chan KG (2015) Prevalence and antimicrobial susceptibility of Vibrio parahaemolyticus isolated from retail shrimps in Malaysia. Front Microbiol 6:33. https://doi.org/10.3389/fmicb.2015.00033
Li C, Ye Z, Wen L, Chen R, Tian L, Zhao F, Pan J (2014) Identification of a novel vaccine candidate by immunogenic screening of Vibrio parahaemolyticus outer membrane proteins. Vaccine 32:6115–6121. https://doi.org/10.1016/j.vaccine.2014.08.077
Li H, Ye MZ, Peng B, Wu HK, Xu CX, Xiong XP, Wang C, Wang SY, Peng XX (2010) Immunoproteomic identification of polyvalent vaccine candidates from Vibrio parahaemolyticus outer membrane proteins. J Proteome Res 9:2573–2583. https://doi.org/10.1021/pr1000219
Liao W, Lin JX, Leonard WJ (2011) IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol 23:598–604. https://doi.org/10.1016/j.coi.2011.08.003
Lin JHY, Chen TY, Chen MS, Chen HE, Chou RL, Chen TI, Su MS, Yang HL (2006) Vaccination with three inactivated pathogens of cobia (Rachycentron canadum) stimulates protective immunity. Aquaculture 255:125–132
Ling R, Logar-Henderson C, Fisman D (2019) El Nino southern oscillation predicts vibriosis risk in the United States. Int J Infect Dis 79:18. doi:https://doi.org/10.1016/j.ijid.2018.11.060
Logar-Henderson C, Ling R, Tuite AR, Fisman DN (2019) Effects of large-scale oceanic phenomena on non-cholera vibriosis incidence in the United States: implications for climate change. Epidemiol Infect 147:e243. https://doi.org/10.1017/S0950268819001316
Luna-Pineda VM, Moreno-Fierros L, Cázares-Domínguez V, Ilhuicatzi-Alvarado D, Ochoa SA, Cruz-Cordova A, Valencia-Mayoral P, Rodríguez-Leviz A, Xicohtencatl-Cortes J (2019) Curli of uropathogenic Escherichia coli enhance urinary tract colonization as a fitness factor. Front Microbiol 10:2063. https://doi.org/10.3389/fmicb.2019.02063
Maier B, Wong GCL (2015) How bacteria use type IV pili machinery on surfaces. Trends Microbiol 23:775–788. https://doi.org/10.1016/j.tim.2015.09.002
Mao Z, Yu L, You Z, Wei Y, Liu Y (2007) Cloning, expression and immunogenicity analysis of five outer membrane proteins of Vibrio parahaemolyticus zj2003. Fish Shellfish Immunol 23:567–575. https://doi.org/10.1016/j.fsi.2007.01.004
Molaee N, Mosayebi G, Amozande-Nobaveh A, Soleyman MR, Abtahi H (2017) Evaluation of the immune response against recombinant proteins (TcpA, TcpB, and FlaA) as a candidate subunit cholera vaccine. J Immunol Res 2017:2412747–2412748. https://doi.org/10.1155/2017/2412747
Peng B, Ye JZ, Han Y, Zeng L, Zhang JY, Li H (2016) Identification of polyvalent protective immunogens from outer membrane proteins in Vibrio parahaemolyticus to protect fish against bacterial infection. Fish Shellfish Immunol 54:204–210. https://doi.org/10.1016/j.fsi.2016.04.012
Powell A, Pope EC, Eddy FE, Roberts EC, Shields RJ, Francis MJ, Smith P, Topps S, Reid J, Rowley AF (2011) Enhanced immune defences in Pacific white shrimp (Litopenaeus vannamei) post-exposure to a Vibrio vaccine. J Invertebr Pathol 107:95–99. https://doi.org/10.1016/j.jip.2011.02.006
Qian R, Chu W, Mao Z, Zhang C, Wei Y, Yu L (2007) Expression, characterization and immunogenicity of a major outer membrane protein from Vibrio alginolyticus. Acta Biochim Biophys Sin 39:194–200. https://doi.org/10.1111/j.1745-7270.2007.00268.x
http://cshprotocols.cshlp.org/content/2006/1/pdb.rec701.full
Reed LJ, Muench H (1938) A simple method of estimating fifty per cent end points. Am J Hyg 27:493–497
Schaller TH, Batich KA, Suryadevara CM, Desai R, Sampson JH (2017) Chemokines as adjuvants for immunotherapy: implications for immune activation with CCL3. Expert Rev Clin Immunol 13:1049–1060. https://doi.org/10.1080/1744666X.2017.1384313
Scheu S, Ali S, Ruland C, Arolt V, Alferink J (2017) The C-C chemokines CCL17 and CCL22 and their receptor CCR4 in CNS autoimmunity. Int J Mol Sci 18:2306. https://doi.org/10.3390/ijms18112306
Schulz SM, Kohler G, Holscher C, Iwakura Y, Alber G (2008) IL-17A is produced by Th17, γδ T cells and other CD4− lymphocytes during infection with Salmonella enterica serovar enteritidis and has a mild effect in bacterial clearance. Int Immunol 20:1129–1138. https://doi.org/10.1093/intimm/dxn069
Sharma M, Dixit A (2016) Immune response characterization and vaccine potential of a recombinant chimera comprising B cell epitope of Aeromonas hydrophila of outer membrane protein C and LTB. Vaccine 34:6259–6266. https://doi.org/10.1016/j.vaccine.2016.10.064
Shi G, Cox AC, Vistica BP, Tan C, Wawrousek EF, Gery I (2008) Phenotype switching by inflammation-inducing polarized Th17 cells, but not by Th1 cells. J Immunol 181:7205–7213. https://doi.org/10.4049/jimmunol.181.10.7205
Shimohata T, Takahashi A (2010) Diarrhea induced by infection of Vibrio parahaemolyticus. J Med Investig 57:179–182. https://doi.org/10.2152/jmi.57.179
Spellberg B, Edwards JE (2001) Type1/Type2 immunity in infectious diseases. Clin Infect Dis 32:76–102. https://doi.org/10.1086/317537
Tanaka T, Narazaki M, Kishimoto T (2014) IL-6 in inflammation, immunity, and disease. Cold spring Harb Perspect biol. 6:1-16. Doi:https://doi.org/10.1101/cshperspect.a016295
Van Gerven N, Klein RD, Hultgren SJ, Remaut H (2015) Bacterial amyloid formation: structural insights into curli biogenesis. Trends Microbiol 11:693–706. https://doi.org/10.1016/j.tim.2015.07.010
Van Gerven N, Van der Verren SE, Reiter DM, Remaut H (2018) The role of functional amyloids in bacterial virulence. J Mol Biol 430:3657–3684. https://doi.org/10.1016/j.jmb.2018.07.010
Wang R, Deng Y, Deng Q, Sun D, Fang Z, Sun L, Wang Y, Gooneratne R (2020) Vibrio parahaemolyticus infection in mice reduces protective gut microbiota, augmenting disease pathways. Front Microbiol 11:73. https://doi.org/10.3389/fmicb.2020.00073
Wang X, Hammer ND, Chapman MR (2008) The molecular basis of functional bacterial amyloid polymerization and nucleation. J Biol Chem 283:21530–21539
Wang C, Liu Y, Li H, Xu WJ, Zhang H, Peng XX (2012) Identification of plasma-responsive outer membrane proteins and their vaccine potential in Edwardsiella tarda using proteomic approach. J Proteome 75:1263–1375. https://doi.org/10.1016/j.jprot.2011.11.001
Wilson KC, Center DM, Cruikshank WW (2004) The effect of interleukin-16 and its precursor on T lymphocyte activation and growth. Growth Factors 22:97–104. https://doi.org/10.1080/08977190410001704679
WHO (2014) Antimicrobial resistance global report on surveillance. World Health Organization. http://www.who.int/drugresistance/documents/surveillancereport/en/
Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, Chu HW (2007) IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumonia infection. Microbes Infect 9:78–86. https://doi.org/10.1016/j.micinf.2006.10.012
Wynn TA (2003) IL-13 effector functions. Annu Rev Immunol 21:425–456. https://doi.org/10.1146/annurev.immunol.21.120601.141142
Yadav SK, Sahoo PK, Dixit A (2014) Characterization of immune response elicited by the recombinant outer membrane protein OmpF of Aeromonas hydrophila, a potential vaccine candidate in murine model. Mol Biol Rep 41:1837–1848
Yadav SK, Meena JK, Sharma M, Dixit A (2016) Recombinant outer membrane protein C of Aeromonas hydrophila elicits mixed immune response and generates agglutinating antibodies. Immunol Res 64:1087–1099. https://doi.org/10.1007/s12026-016-8807-9
Zha Z, Li C, Li W, Ye Z, Pan J (2016) LptD is a promising vaccine antigen and potential immunotherapeutic target for protection against Vibrio species infection. Sci Rep 6:38577. https://doi.org/10.1038/srep38577
Zhang L, Orth K (2013) Virulence determinants for Vibrio parahaemolyticus infection. Curr Opin Microbiol 16:70–77. https://doi.org/10.1016/j.mib.2013.02.002
Acknowledgments
The Indian Council of Medical Research, New Delhi is acknowledged for research fellowship to SK. Prof. M. R. Chapman, Molecular, Cellular and Developmental Biology, University of Michigan, Ann Arbor, MI, USA is gratefully acknowledged for kindly providing with the E. coli C600 csg knockout strain, E. coli LSR12.
Funding
The present study has been carried out with the financial support from the Department of Science and Technology PURSE grant (SR/PURSE/Phase2/11(C) 2015) to the Jawaharlal Nehru University, New Delhi and the intramural funding from the JNU, New Delhi, India.
Author information
Authors and Affiliations
Contributions
AD, DC, and SK conceived and designed the study. SK performed the experiments and acquired the data. SK, DC, and AD analyzed and interpreted the data. SK, DC, and AD drafted and revised the manuscript. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The work presented in this manuscript does not contain any study comprising human participants. The use of animals (BALB/c mice) for immunization was approved by the Institutional Animal Ethics Committee (IAEC) of the University (IAEC project code 08/2017), New Delhi. The guidelines prescribed by the IAEC were followed for the use of animals.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(PDF 2.06 mb)
Rights and permissions
About this article

Cite this article
Karan, S., Choudhury, D. & Dixit, A. Immunogenic characterization and protective efficacy of recombinant CsgA, major subunit of curli fibers, against Vibrio parahaemolyticus. Appl Microbiol Biotechnol 105, 599–616 (2021). https://doi.org/10.1007/s00253-020-11038-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-020-11038-4