
Abstract
To study the effect of proteases on pullulanase production, six protease-encoding genes (nprB, bpr, mpr, epr, vpr, and wprA) in the genome of Bacillus subtilis strain WS5, which already lacks the protease-encoding genes nprE and aprE, were sequentially disrupted using a CRISPR/Cas9 system. This created strains WS6–WS11, respectively. The strains WS3 (none) and WS4 (ΔnprE) were constructed earlier. After addition of expression plasmid pHYPULd4 into the strains WS3–WS11, the pullulanase production levels of the resulting strains (WS3PUL–WS11PUL, respectively) were investigated in shake-flask cultivations and recombinant strain WS5PUL produced the highest pullulanase activity (148.2 U/mL). Then, the scale-up pullulanase production levels of four recombinant strains WS5PUL, WS9PUL, WS10PUL, and WS11PUL were investigated in the 3-L fermenter cultivations. Strain WS9PUL produced the highest pullulanase activity (2449.6 U/mL) when fed an inorganic nitrogen source. However, the specific activity of the pullulanase obtained in a 3-L fermenter generally decreased as the number of protease deletions increased. Meanwhile, using pullulanase, α-cyclodextrin glucosyltransferase and β-cyclodextrin glucosyltransferase as reporter proteins, the protein production differences among strains WS3, WS9, and the widely used WB600 were investigated. Finally, the carbon to organic nitrogen source ratio of the feeding solution used in the 3-L fermenter was optimized. Recombinant strain WS9PUL fed with carbon and organic nitrogen sources in a ratio of 4:1 achieved a pullulanase activity of 5951.8 U/mL, the highest activity reported to date.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacillus subtilis, a non-pathogenic Gram-positive bacterium that has been widely used in the industrial production of heterologous proteins, is known to produce at least eight extracellular proteases (Nguyen et al. 2013; Tjalsma et al. 2004) including neutral proteases (NprE and NprB), serine proteases (Epr, Bpr and Vpr), an alkaline protease (AprE), a metalloprotease (Mpr), and a cell-wall protease (WprA). These proteases can degrade at least 43 kinds of native lipoproteins, membrane proteins, and secreted proteins to provide nutrients and perform quality control processes (Krishnappa et al. 2013; Pohl and Harwood 2010; Veening et al. 2008). Since native protein conformations are protease-resistant, the degradation of correctly folded proteins by extracellular proteases occurs much more slowly than that of misfolded proteins (Krishnappa et al. 2013). Besides degrading misfolded proteins, these proteases are also likely to degrade heterologous proteins that fold slowly upon membrane translocation (Krishnappa et al. 2014; Westers et al. 2006). This extracellular proteolytic processing can improve the overall quality of secreted protein folding, but it may somewhat decrease the production levels of target proteins. A series of protease-deficient strains derived from B. subtilis 168, including WB600, WB700, and WB800, has been constructed to increase heterologous protein secretion by reducing proteolysis (Wu et al. 2002; Wu et al. 1991).
In a previous study, we constructed a high-level protein production strain B. subtilis WS5, from which the two abundant protease-encoding genes nprE and aprE have been deleted (Wu et al. 2017). In this study, we investigated the effect of the deletion of six additional B. subtilis proteases on the production of heterologous proteins using Bacillus deramificans pullulanase as reporter protein. Pullulanase (EC 3.2.1.41) is a widely used debranching enzyme that catalyzes the hydrolysis of α-1,6 linkage in amylaceous polysaccharides such as pullulan, amylopectin, α- and β-dextrin, glycogen, and related oligosaccharides. Either alone or in conjunction with other amylolytic enzymes such as glucoamylase, α-amylase, β-amylase, or cyclodextrin glycosyltransferase (EC 2.4.1.19, CGTase), pullulanase can break down starch to produce small reducing sugars, including cyclodextrins and amylose during the saccharification process (Chen et al. 2013). It has been extensively used in the production of fuel ethanol, resistant starch, maltotriose syrup, and other products (Hii et al. 2012; Shi et al. 2013). Pullulanase has been produced in many host strains, including Escherichia coli (Duan et al. 2013), B. subtilis (Zhang et al. 2017), Bacillus flavothermus (Shankar et al. 2014), Brevibacillus choshinensis (Zou et al. 2016a), Pichia pastoris (Xu et al. 2006), and others. Perhaps due to its complicated structure and high molecular weight (Moller et al. 2016), pullulanase can easily become misfolded in the host strain during the production process (Duan et al. 2013). Previous reports have shown that pullulanase readily forms insoluble inclusion bodies in E. coli (Chen et al. 2014; Zou et al. 2014; Zou et al. 2016b), and that the pullulanase produced at a high rate in B. choshinensis is mainly thermolabile (Zou et al. 2016a). Among the traditional methods to improve pullulanase folding are controlling the fermentation temperature, optimizing the inducer concentration (Zou et al. 2014), and supplementing the growth medium with betaine (Duan et al. 2013). As shown in Fig. 1, since extracellular proteases more easily degrade misfolded pullulanase than the correctly folded protein, extracellular proteases can be used to improve the overall quality of enzyme folding. The proteolytic activity should be controlled at a relatively low level to minimize the degradation of correctly folded pullulanase. CGTases, which are members of glycohydrolase family 13, have been used primarily in the production of cyclodextrins (van der Veen et al. 2000). CGTases are classified as α-, β-, or γ-CGTases according to the number of glucose residues present in the product cyclodextrins (6, 7, and 8, respectively) (Kelly et al. 2009). In addition to pullulanase, Paenibacillus macerans α-CGTase, and Bacillus circulans 251 β-CGTase, two extracellular enzymes of high industrial value were used as reporter proteins in this study (Zhang et al. 2017).

Schematic representation of different extracellular protease-deficient strains and their extracellular pullulanase production
During fed-batch fermentation, the composition and component ratio of the feeding solution can have a large influence on cell growth and enzyme yield (Gupta et al. 2002; Kuo et al. 2009). Inorganic nitrogen can be assimilated rapidly and promote cell growth, which may be detrimental for protein production (Wu et al. 2007). There are also reports that optimizing the ratio of carbon to nitrogen sources in the feeding solution improves the production of nattokinase and poly(γ-glutamic acid) (Huang et al. 2011; Kwon et al. 2011). In this study, the level of extracellular pullulanase production in B. subtilis was optimized by screening multiple protease-deficient strains in shake-flask and 3-L fermenter cultivations, as well as optimizing the feeding solution used in fed-batch fermentation. This process led to a protease-deficient strain displaying the highest extracellular pullulanase production level reported to date. Indeed, this protease-deficient strain exhibits greater protein production capability than the frequently used strain WB600.
Materials and methods
Bacterial strains and plasmids
All bacterial strains and plasmids used in this study are listed in Table 1 and Table 2, respectively. E. coli JM109 was used to construct knockout plasmids. B. subtilis strain WS5 was constructed from an undomesticated B. subtilis strain in our library. It is kept in the China Center for Type Culture Collection with a deposit number of CCTCC M 2016536 (Wu et al. 2017; Zhang et al. 2017). Plasmid pMD18-T (Takara, Dalian, China) was used as a cloning vector. Knockout plasmid pHYcas9dsrf1, kept in our library, was used for the construction of six more knockout plasmids (Zhang et al. 2016). Plasmids pHYPULd4, pHYαCGTd4, and pHYCGTd4, kept in our library, were used for the heterologous production of B. deramificans pullulanase (Genbank accession number KT897705), P. macerans α-CGTase (Genbank accession number P04830.2), and B. circulans 251 β-CGTase (Genbank accession number P43379.1), respectively (Zhang et al. 2017).
Reagents and enzymes
The plasmid mini-prep kit, agarose gel DNA purification kit, and PCR purification kit were purchased from Tiangen Co. Ltd. (Beijing, China). PrimeSTAR polymerase, T4 DNA ligase, restriction enzymes, and DL2000 DNA Marker were purchased from Takara (Dalian, China). Medium molecular weight protein marker for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis was purchased from Shanghai Generay Biotech Co. Ltd. (Shanghai, China). Pullulan was purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan). α-cyclodextrin and β-cyclodextrin were purchased from Sigma-Aldrich (Milwaukee, WI, USA). Other reagents were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China).
Culture media and transformation
The LB medium used in this study contained 5 g/L (w/v) yeast extract, 10 g/L (w/v) tryptone, and 10 g/L (w/v) NaCl. The shake-flask fermentation medium contained 18 g/L (w/v) corn steep powder, 10 g/L (w/v) yeast extract, 12.54 g/L (w/v) K2HPO4, 2.31 g/L (w/v) KH2PO4, and 10 g/L (w/v) glucose. The medium used for 3-L fermenter cultivation consisted of 18 g/L (w/v) corn steep powder, 10 g/L (w/v) yeast extract, 2 g/L (w/v) Na2SO3, 15 g/L (w/v) maltose, 1 g/L (w/v) (NH4)2-H-citrate, 1 g/L (w/v) MgSO4·7H2O, 14.6 g/L (w/v) K2HPO4, 4 g/L (w/v) NaH2PO4·H2O, 2.68 g/L (w/v) (NH4)2SO4, and 3 mL/L (v/v) trace element solution (Wenzel et al. 2011). The original feeding solution contained 500 g/L (w/v) glucose, 7.89 g/L (w/v) MgSO4·7H2O, 63.36 g/L (w/v) (NH4)2HPO4, and 40 mL/L (v/v) trace element solution. The media described above were supplemented with 20 mg/L (w/v) tetracycline or 100 mg/L (w/v) ampicillin as needed. The protein production and gene knockout plasmids were inserted into B. subtilis using the method of Anagnostopoulos and Spizizen (Anagnostopoulos and Spizizen 1961).
Plasmids construction
All primers used in this study are listed in Table S1. The knockout plasmids pHYcas9dnprB, pHYcas9dbpr, pHYcas9dmpr, pHYcas9depr, pHYcas9dvpr, and pHYcas9dwpr were constructed by exchanging the 20-nt guide sequence and homologous repair templates of knockout plasmid pHYcas9dsrf1 for those of the corresponding protease genes. Through inverse PCR, the 20-nt guide sequence of plasmid pHYcas9dsrf1 was altered using the following six primer pairs: P1/P2 (for pHYcas9dnprB), P3/P4 (for pHYcas9dbpr), P5/P6 (for pHYcas9dmpr), P7/P8 (for pHYcas9depr), P9/P10 (for pHYcas9dvpr), and P11/P12 (pHYcas9dwpr). The modified pHYcas9dsrf was digested with XbaI for subsequent ligation. Using overlap extension PCR, the homologous repair templates of the six knockout plasmids were amplified from the genome of B. subtilis strain WS5 using the following 12 primer pairs: P13/P14 and P15/P16 (for pHYcas9dnprB), P17/P18 and P19/P20 (for pHYcas9dbpr), P21/P22 and P23/P24 (for pHYcas9dmpr), P25/P26 and P27/P28 (for pHYcas9depr), P29/P30 and P31/P32 (for pHYcas9dvpr), and P33/P34 and P35/P36 (pHYcas9dwpr). An XbaI site was present at both ends of each of these constructs. In the repair template junction of upstream and downstream, 6 bp of native sequence was replaced by an XhoI site and 5 bp of random sequence, which inserts an XhoI site and causes gene frameshift mutation after homology-directed repair (Zhang et al. 2016). After ligation with cloning vector pMD18-T and XbaI digestion, the six template sequences were ligated into the six correspondingly modified linear pHYcas9dsrf1 constructs, yielding the six knockout plasmids pHYcas9dnprB, pHYcas9dbpr, pHYcas9dmpr, pHYcas9depr, pHYcas9dvpr, and pHYcas9dwpr.
Protease genes disruption
Knockout plasmid transformants were screened using tetracycline resistance and confirmed through colony PCR of the cas9 gene using primers P37 and P38. The sequences outside the homologous repair template were amplified from the transformant genomes using the following corresponding verification primer pairs: P39/P40 (for nprB), P41/42 (for bpr), P43/P44 (for mpr), P45/P46 (for epr), P47/P48 (for vpr), and P49/P50 (for wprA). PCR products were digested with XhoI to screen mutants. The mutants were further verified with DNA sequencing. Knockout plasmids were cured through overnight cultivation of the strains at 51 °C (Dempsey and Dubnau 1989).
Culture conditions
Shake-flask cultivations
Cultures used for routine plasmid construction and seed cultures were grown in 50-mL shake flasks containing 10 mL of LB medium at 37 °C with shaking at 200 rpm on a rotary shaker. After 10 h of cultivation, the seed cultures were diluted into 250-mL shake flasks containing 50 mL of shake-flask fermentation medium at a ratio of 7.5% (v/v) and cultured at 37 °C with shaking at 200 rpm for 2 h. The resulting cultures were fermented for 48 h at 33 °C with shaking at 200 rpm.
Bioreactor cultivations
A frozen glycerol stock (100 μL) was used to inoculate 50 mL of LB medium supplemented with 20 mg/L tetracycline in a 250-mL shake flask. The resulting culture was incubated at 37 °C, with shaking at 200 rpm, for 12 h. For fed-batch fermentation, a 10% (v/v) seed culture was used to inoculate a 3-L fermenter (BioFlo 110, New Brunswick Scientific Co., Edison, NJ) containing 0.9 L of scale-up fermentation medium. During the entire fermentation process, the pH was maintained at 6.5 using 20% (v/v) H3PO4 and NH4OH; the temperature was maintained at 37 °C; the dissolved oxygen content (DO) was maintained at 30% with agitation speed (300 to 700 rpm) and pure oxygen as needed. Antifoam was added manually to control the foam height, usually one drop at a time. Different recombinant strains produced different amounts of foam during fermentation, and the feeding solutions also affected the amount of foam produced. The total amount of antifoam added was about 100 to 500 μL. Tetracycline (20 mg/L) was added every 24 h. After inoculation, the DO decreased slowly and the agitation speed increased correspondingly (from 300 to 700 rpm) when the DO fell below 30%. The DO suddenly increased about 5 h after inoculation, indicating that the maltose in the medium had been completely consumed and fed-batch cultivation should be started. Detected using an SBA-40C biosensor (Biology Institute of Shandong Academy of Sciences, Jinan, China), the glucose concentration was maintained at < 0.5 g/L with feeding at a speed of 2 to 9 mL/h. The culture medium was sampled at the times indicated for each experiment.
Determination of biomass
The dry cell weight (DCW) was determined by centrifuging a 5-mL sample of culture broth at 12000×g for 10 min at 4 °C, resuspending the precipitate in 0.9% (w/v) NaCl and re-pelleting it through centrifuging at 12000×g for 10 min at 4 °C, and finally drying the precipitate to constant weight at 105 °C. Centrifugation was conducted with an Eppendorf 5427 R centrifuge (Eppendorf Co., Hamburg, Germany).
Enzyme activity assays
Pullulanase activity
Pullulanase activity was measured by determining the reducing sugar released from pullulan (Zou et al. 2014). The reaction was initiated by adding 0.1 mL of appropriately diluted enzyme solution to a mixture containing 0.9 mL of sodium acetate buffer (100 mM, pH 4.5) and 1 mL of 1% (w/v) pullulan, and then incubating the mixture at 60 °C for 10 min. The reaction was stopped by adding 3 mL of 3,5-dinitrosalicylic acid solution and boiling for 7 min. Finally, the mixture was diluted to 15 mL with deionized water and the absorbance at 540 nm was measured with a spectrophotometer (BioPhotometer plus, Eppendorf Co., Hamburg, Germany). One unit of pullulanase activity was defined as the amount of enzyme that released 1 μmol of reducing sugars per minute from pullulan under the conditions described above.
α-CGTase activity
Reactions were initiated by adding 0.1 mL of appropriated diluted enzyme solution to 2 mL of preheated starch solution (2% [w/v] dissolved in 25 mM phosphate buffer, pH 5.5) and incubated at 50 °C for 10 min. The reactions were stopped by adding 0.2 mL of 3 M HCl and keeping the mixtures at room temperature for 5 to 10 min. Product formation was assessed by adding 0.2 mL of 0.44 mM methyl orange to the mixture, incubating it at 16 °C for 15 min, and then measuring the absorbance of the mixture at 505 nm. One unit of α-CGTase cyclization activity was defined as the amount of enzyme that produces 1 μmol of α-cyclodextrin per minute from starch under the conditions described above (Lejeune et al. 1989).
β-CGTase activity
Reactions were initiated by adding 0.1 mL of appropriately diluted enzyme solution to 2 mL of preheated starch solution (1% [w/v] dissolved in 25 mM phosphate buffer, pH 5.5) and incubated at 50 °C for 10 min. The reactions were stopped by adding 0.2 mL of 0.6 M HCl and incubating for 5 to 10 min. Product formation was assessed by adding 0.5 mL of 0.6 M Na2CO3 and 0.2 mL of 1.2 mM phenolphthalein to the mixture, sequentially, keeping at room temperature for 15 min, and then measuring absorbance of mixture at 550 nm. One unit of β-CGTase cyclization activity was defined as the amount of enzyme that produced 1 μmol of β-cyclodextrin per minute from starch under the conditions described above (Penninga et al. 1996).
SDS-PAGE gel electrophoresis
The 20 μL culture supernatants containing reporter protein were mixed with 5 μL SDS-PAGE buffer (5×) and denatured at 100 °C for 5 min, and then 8 μL of the mix was added to the SDS-PAGE gel. Electrophoresis was performed with a 5% stacking and a 12% separating gel (Laemmli 1970). Protein bands were stained with 0.25% Coomassie Brilliant Blue R-250.
Statistical analysis
All experiments were performed independently three times. The results are shown as the mean ± standard deviation. Statistical analyses were performed using Student’s t test. Differences yielding P values < 0.05 were considered statistically significant.
Results
Construction of multiple protease-deficient B. subtilis strains
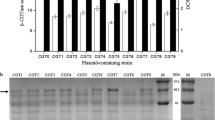
The effort to construct new B. subtilis production strains with reduced proteolytic activity began with strain WS5, from which the genes encoding the most abundant proteases, NprE and AprE, have been deleted (Li et al. 2004). Six additional protease genes were deleted from the genome of this strain, sequentially, using the CRISPR/Cas9 system. To do this, the B. subtilis CRISPR/Cas9 knockout plasmid pHYcas9dsrf1 was modified to produce the six knockout plasmids pHYcas9dnprB, pHYcas9dbpr, pHYcas9dmpr, pHYcas9depr, pHYcas9dvpr, and pHYcas9dwpr. As described in Materials and methods, the sgRNA sequence and homologous repair template of knockout plasmid pHYcas9dsrf1 were replaced with those of the protease genes to be knocked out. Using an established B. subtilis CRISPR/Cas9 system manipulation protocol (Zhang et al. 2016) and the six knockout plasmids described above, the six protease genes nprB, bpr, mpr, epr, vpr, and wprA were sequentially knocked out of the WS5 genome. The resulting B. subtilis strains (WS6, WS7, WS8, WS9, WS10, and WS11, respectively) were, therefore, deficient in three (WS6) to eight (WS11) proteases (Table 1). As shown in Fig. 2, PCR products obtained from the genomes of these protease-deficient strains using verification primers can be digested with XhoI, confirming the frameshift mutation within the gene. The results were also confirmed using DNA sequencing (Fig. S1).

Confirmation of the disruptions of nprB, bpr, mpr, epr, vpr, and wprA through digestion with XhoI. PCR products amplified from the genomes of wild-type (lane WT) and mutant (lane MT) strains using verification primer pairs were digested with XhoI. Lane M—DNA marker (DL 2000, Takara Bio Inc.)
Pullulanase production levels of multiple protease-deficient strains
To investigate pullulanase production in protease-deficient strains, we transformed strains WS3 (none), WS4 (ΔnprE), WS5 (ΔnprE, ΔaprE), and WS6 through WS11 (Table 1) with the pullulanase gene expression plasmid pHYPULd4, yielding the strains WS3PUL through WS11PUL, respectively. The pullulanase production levels of the nine strains were determined using shake-flask cultivations. Strain WS3PUL showed the highest DCW (3.7 g/L) and strain WS11PUL showed the lowest (3.2 g/L) (Fig. 3a). The extracellular pullulanase activities of recombinant strains WS3PUL through WS11PUL were 137.4, 147.6, 148.2, 138.9, 118.9, 125.2, 138.7, 135.4, and 101.6 U/mL, respectively (Fig. 3a). Strain WS5PUL, deficient in two proteases, showed the highest pullulanase activity (148.2 U/mL), while strain WS11PUL, deficient in eight proteases, showed the lowest pullulanase activity. Perhaps because the gene encoding WprA protease was deleted, the pullulanase activity of strain WS11PUL decreased significantly compared with that of strain WS10PUL. The pullulanase activities of strain WS3PUL (not protease-deficient) and WS4PUL (deficient in NprE) were slightly below or similar to that of strain WS5PUL (deficient in both NprE and AprE), while the pullulanase bands in the SDS-PAGE analysis of strains WS3PUL and WS4PUL were much thinner than that seen in the analysis of strain WS5PUL (Fig. 3b), suggesting that the specific activities of the pullulanase proteins produced by strains WS3PUL and WS4PUL were higher than that of the pullulanase protein produced by strain WS5PUL. This difference may result from the degradation of misfolded pullulanase by the abundant proteases NprE and AprE. Multiple background bands in the analysis of strains WS3PUL and WS4PUL were also much thinner than those of the other strains, suggesting a greater extent of degradation of the corresponding native exported proteins by higher levels of extracellular proteases. The SDS-PAGE band of strains WS5PUL through WS11PUL was in good agreement with the pullulanase activity values (Fig. 3b).

Comparison of cell growth and pullulanase activity in the culture supernatant (a) for multiple recombinant strains in shake-flask cultivations and SDS-PAGE analysis (b) of the culture supernatants. Error bars represent standard deviations from the means of three replicates. An asterisk indicates a statistically significant difference between two activity values (P < 0.05). DCW (black), pullulanase activity (white). Lane M—protein molecular weight markers (medium molecular weight, Shanghai Generay Biotech Co. Ltd.). The arrow to the right indicates the band corresponding to pullulanase
Scale-up of pullulanase production in a 3-L fermenter
The ideal pullulanase production strain would exhibit low proteolysis while guaranteeing the quality of pullulanase folding. Given that strain WS5PUL produced the highest pullulanase activity in shake-flask cultivations and strains WS9PUL, WS10PUL, and WS11PUL should have relatively low proteolytic activity, fermentation of these four recombinant strains was performed in a 3-L fermenter. In this experiment, the carbon and nitrogen sources present in the feeding solution were 500 g/L glucose and 63.36 g/L (NH4)2HPO4, respectively. The glucose concentration was kept below 0.5 g/L to avoid glucose repression of the expression promoter used in this study (Zhang et al. 2017). Before 78 h, the DCWs of strains WS5PUL, WS9PUL, WS10PUL, and WS11PUL increased continually, reaching 62.6, 67.0, 67.2, and 56.1 g/L at 78 h, respectively. After 78 h, the DCWs of recombinant strains WS9PUL and WS11PUL decreased, while the DCWs of recombinant strains WS5PUL and W10PUL increased continually, reaching 64.6 and 70.5 g/L at 85 h, respectively (Fig. 4a).

Comparison of cell growth (a) and pullulanase activity in the culture supernatant (b) among recombinant strains WS5PUL (■), WS9PUL (●), WS10PUL (○), and WS11PUL (Δ) in 3-L fermenter cultivation. Error bars represent standard deviations from the mean of three independent replicates
Before 51 h, the pullulanase activities of strains WS5PUL, WS9PUL, WS10PUL, and WS11PUL in a 3-L fermenter increased at different rates, reaching 1490.0, 1804.0, 1653.3, and 1047.6 U/mL at 51 h, respectively. After 51 h, the pullulanase activity of recombinant strain WS11PUL decreased slowly, while the pullulanase activities of strains WS5PUL, WS9PUL, and WS10PUL increased continually until about 75 h. The strains WS5PUL, WS9PUL, and WS10PUL reached their highest activities of 2095 U/mL at 75 h, 2449.6 U/mL at 78 h, and 2177.1 U/mL at 72 h, respectively (Fig. 4b).
Purification, kinetic parameters, and aggregation state of pullulanase
To further investigate the effect of proteases on pullulanase production during high-density fermentation, the pullulanase proteins obtained from the cultivation of strains WS5PUL, WS9PUL, WS10PUL, and WS11PUL in a 3-L fermenter were purified to homogeneity using Q-Sepharose anion exchange and size-exclusion chromatography (Fig. S2).
All four of these pullulanase proteins showed the same elution volume (13.85 mL) during size-exclusion chromatography. Through the calibration curve of molecular weight and elution volume measured earlier in our laboratory (Zou et al. 2016b), the molecular weight of the native protein was determined to be 150 kDa, demonstrating that the pullulanase proteins obtained from strains WS5PUL, WS9PUL, WS10PUL, and WS11PUL all exist as dimers in solution.
As shown in Table 3, the pullulanase protein with the highest specific activity was obtained from strain WS5PUL, while the pullulanase protein with the second-highest specific activity was obtained from strain WS9PUL. The specific activities of the pullulanase proteins obtained from strains WS10PUL and WS11PUL were much lower than those of the pullulanase proteins obtained from strains WS5PUL and WS9PUL. The Km values of the enzymes obtained from strains WS5PUL, WS9PUL, and WS10PUL were similar, but lower than that of the enzyme obtained from strain WS11PUL. The Vmax, kcat, and kcat/Km values of the enzymes obtained from strains WS5PUL, WS9PUL, WS10PUL, and WS11PUL showed a decreasing trend.
The difference in production among B. subtilis strains WS3, WS9, and WB600
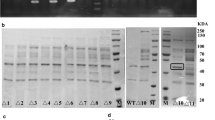
The proteolytic activity of B. subtilis strain WS3, which is not a protease-deficient strain, was higher than those of the protease-deficient strains. B. subtilis strain WS9 is deficient in six extracellular proteases (NprE, AprE, NprB, Bpr, Mpr, and Epr), as is the B. subtilis protease-deficient strain WB600. To assess the differences in heterologous protein production among B. subtilis strains WS3, WS9, and WB600, two additional reporter enzymes besides pullulanase, α-CGTase and β-CGTase, were employed. Three gene expression plasmids, one for pullulanase (pHYPULd4), one for α-CGTase (pHYαCGTd4), and one for β-CGTase (pHYCGTd4) were used to transform B. subtilis strain WB600, yielding recombinant strains WBPUL, WBαCGT, and WBβCGT, respectively (Table 1). Then, expression plasmids pHYαCGTd4 and pHYCGTd4 were used to transform strain WS3, yielding recombinant strains WS3αCGT and WS3βCGT, respectively (Table 1). Finally, expression plasmids pHYαCGTd4 and pHYCGTd4 were used to transform strain WS9, yielding recombinant strains WS9αCGT and WS9βCGT, respectively (Table 1). The protein production levels of these recombinant strains were determined using shake-flask cultivations. As shown in Fig. 5 for pullulanase, β-CGTase, and α-CGTase recombinant strains, the DCWs obtained using host strain WS3 were a little higher than those obtained using host strains WS9 and WB600. The DCW of WS9PUL was a little higher than that of WBPUL, while the DCW of WS9αCGT and WBαCGT, and WS9βCGT and WBβCGT were approximately the same. As shown in Fig. 5, the extracellular pullulanase activity obtained using host strain WS3 was similar to that obtained using host WS9, while the extracellular α-CGTase and β-CGTase activities obtained using host strain WS3 were 1.15-fold and 1.26-fold greater than those obtained using host strain WS9, respectively. The extracellular pullulanase, α-CGTase, and β-CGTase activities obtained using host strain WS9 were 2.04-fold, 1.61-fold, and 1.17-fold greater than those obtained using host WB600, respectively. The differences with pullulanase and α-CGTase, but not β-CGTase, were statistically significant. An SDS-PAGE analysis of the culture supernatants was in good agreement with the activity values (Fig. 5d).

Comparison of cell growth and enzyme activity in the culture supernatant among host strains WS3, WS9, and WB600 with reporter proteins pullulanase (a), α-CGTase (b), and β-CGTase (c) in shake-flask cultivations, as well as SDS-PAGE analysis (d) of the culture supernatants. Error bars represent standard deviations from the means of three independent replicates. An asterisk indicates a statistically significant difference between the two activity values (P < 0.05). DCW (black), pullulanase activity (white), Lane M—protein molecular weight markers (medium molecular weight, Shanghai Generay Biotech Co. Ltd.)
Enhancing pullulanase production through feeding solution optimization
To enhance the production of recombinant pullulanase during the 3-L fermentation of strain WS9PUL, experiments were performed to optimize the feeding solution. The inorganic nitrogen source (NH4)2HPO4 present in the original feeding solution can be assimilated rapidly by the host strain, promoting rapid bacterial growth. However, rapid bacterial growth creates nutritional and metabolic pressures that are not conducive to pullulanase production (Kwon et al. 2011). Therefore, we replaced the (NH4)2HPO4 in the original feeding solution with the organic nitrogen sources corn steep powder and yeast extract. Initial shake-flask experiments suggested that a 1.8:1 (w/w) ratio of corn steep powder to yeast extract would be the optimal nitrogen source for pullulanase production (data not shown), so this ratio was present in the media used for both the shake-flask and 3-L fermenter cultivations described above and the feeding solution in this experiment. To further optimize the carbon (glucose) to nitrogen (corn steep powder and yeast extract) source ratio of the feeding solution, we prepared feeding solutions containing a total of 500 g/L of carbon and organic nitrogen sources at ratios (w/w) of 2:1, 4:1, and 9:1. As shown in Fig. 6a, the carbon to organic nitrogen source ratio of 9:1 achieved the highest DCW value of 63.45 g/L at 103 h, which was 1.1-fold greater than that obtained with a ratio of 2:1 (57.44 g/L at 99 h) and 1.03-fold greater than that obtained with a ratio of 4:1 (61.88 g/L at 99 h). As shown in Fig. 6b, the carbon to organic nitrogen source ratio of 4:1 achieved the highest pullulanase activity of (5951.8 U/mL) at 99 h, which was 1.07-fold greater than that obtained with a 2:1 ratio (5562.9 U/mL at 99 h) and 1.25-fold greater than that obtained with a 9:1 ratio (4742.53 U/mL at 103 h).

Comparison of cell growth (a) and pullulanase activity in the culture supernatant (b) for recombinant strain WS9PUL in 3-L fermenter cultivations fed with carbon to organic nitrogen source ratios of 2:1(■), 4:1(●), and 9:1(Δ). Comparisons of pullulanase activity and pullulanase activity per unit biomass for recombinant strain WS9PUL in 3-L fermenter cultivations fed with an inorganic nitrogen source or fed with carbon and organic nitrogen sources in a ratio of 4:1 (c). Error bars represent standard deviations from the means of three independent replicates. An asterisk indicates a statistically significant difference between the two activity values (P < 0.05). Pullulanase activity (white), pullulanase activity per unit biomass (black)
As shown in Fig. 6c, the pullulanase activity obtained by feeding with a carbon to organic nitrogen source ratio of 4:1 was 2.43-fold greater than that obtained using fed-batch cultivation with an inorganic nitrogen source (5951.8 versus 2449.6 U/mL; P < 0.05). The pullulanase activity per unit biomass obtained with carbon to organic nitrogen source ratio of 4:1 was 2.63-fold greater than that obtained during fed-batch with inorganic nitrogen source (96.18 × 103 versus 36.56 × 103 U/g DCW; P < 0.05).
Discussion
The pullulanase from B. deramificans has a high molecular weight (79 kDa) and misfolds easily (Zou et al. 2016b). During shake-flask cultivations, the DCWs of strains WS3PUL through WS11PUL (except WS8PUL) somewhat showed a decreased trend, indicating that protease deficiency may have a somewhat negative effect on cell growth. The pullulanase production levels of multiple protease-deficient strains showed that the presence of small amounts of extracellular proteases (NprB, Bpr, Mpr, Epr, Vpr, and WprA) was favorable for pullulanase production. Proteolysis by the proteases NprE and AprE, which were much stronger than that of the other proteases (Krishnappa et al. 2013), may influence the level of pullulanase production, yielding similar pullulanase activity despite higher pullulanase specific activity. The pullulanase activities of strain WS3PUL (137.4 U/mL) and WS4PUL (147.6 U/mL) were slightly below or similar to that of strain WS5PUL (148.2 U/mL), while the SDS-PAGE analysis showed that the pullulanase contents of strains WS3PUL and WS4PUL were much lower than that of strain WS5PUL (Fig. 3b). Compared with the other eight strains, there is no specific thicker band in the background bands of WS3PUL (Fig. 3b). In WS3PUL, the pullulanase may be degraded to peptides or amino acids, which can be reabsorbed as nutrients. The proteases NprB, Bpr, Mpr, Epr, and Vpr can mildly degrade misfolded pullulanase in the extracellular space (Pohl et al. 2013), which may reduce secretion stress and promote nutrient recycling. WprA, a cell-wall-associated protease produced constitutively during the entire growth period (Stephenson and Harwood 1998a), can degrade pullulanase prior to release into the medium, which might promote correct pullulanase folding inside the cell and increase extracellular pullulanase activity. The intracellular folding of pullulanase might be crucial in B. subtilis, as it is in B. choshinensis (Zou et al. 2016b).
When cultured in a 3-L fermenter fed with 500 g/L glucose and 63.36 g/L (NH4)2HPO4, the DCW results were consistent with those seen in shake-flask cultivations, where strain WS11PUL displayed the lowest DCW. It has been indicated that protease deficiency can promote cell lysis, which starts at the beginning of the post-exponential growth phase (Westers et al. 2004). Cell-wall-associated protease WprA is composed of cell-wall-binding proteins (CWBPs) 23 and 52 (Margot and Karamata 1996). CWBP52 possesses serine protease activity, while the role of CWBP23 may be related to folding and the regulation of CWBP52 activity (Harwood and Cranenburgh 2008). Protease WprA deficiency can increase the production of native cell-wall proteins (Stephenson and Harwood 1998b) that have not had the quality of their folding maintained by proteolysis of misfolded proteins. The resulting accumulation of misfolded proteins may result in cell lysis. After cell lysis, the intracellular proteases that have leaked into the medium can also degrade extracellular proteins (Stephenson et al. 1999). The activity results showed that the effects of proteases on pullulanase activity in a 3-L fermenter differ from those observed in shake flasks. In the 3-L fermenter, the presence of proteases NprB, Bpr, Mpr, and Epr had a negative effect on pullulanase activity, while the presence of protease Vpr, and especially protease WprA, contributed to pullulanase activity. These results may be due to a higher level of protease accumulation during scale-up fermentations than during shake-flask cultivations. The cultivation time at which strain WS11PUL exhibited its highest pullulanase activity was much shorter than those of strains WS5PUL, WS9PUL, and WS10PUL. This may result from an earlier transition from exponential to post-exponential growth and cell lysis for strain WS11PUL.
Extracellular proteases have both a positive impact on pullulanase production (they improve pullulanase folding) and a negative impact on pullulanase production (they degrade correctly folded pullulanase). The protease activity present during cultivation of strain WS5PUL in a 3-Lfermenter may be excessive, and that may decrease the pullulanase production level despite increasing its specific activity. As for strains WS10PUL and WS11PUL, the lack of proteases Vpr and WprA greatly weakened the control of pullulanase folding, which resulted in the secretion of pullulanase with low specific activity and poor Km and kcat values. Compared with strains WS5PUL, WS10PUL, and WS11PUL, the recombinant strain WS9PUL, which is deficient in proteases NprE, AprE, NprB, Bpr, Mpr, and Epr, showed a suitable balance of pullulanase degradation during 3-Lfermenter cultivation. This resulted in high-level extracellular pullulanase activity with acceptable characteristics.
During shake-flask cultivations, the extracellular pullulanase activities obtained using host strains WS3 and WS9 were similar, while the extracellular α-CGTase and β-CGTase activities obtained using host strain WS3 were a little higher than those obtained using host strain WS9. The pullulanase band in the SDS-PAGE analysis of strain WS3PUL was much thinner than that seen in the analysis of strain WS9PUL (Fig. 5d), suggesting a great extent of degradation by extracellular proteases. The α-CGTase and β-CGTase bands in the SDS-PAGE analysis of strains WS3αCGT and WS3βCGT were similar with those seen in the analysis of strains WS9αCGT and WS9βCGT, respectively, suggesting a small extent of degradation by extracellular proteases. The results showed that pullulanase was more sensitive to proteolysis than α-CGTase and β-CGTase. The ideal pullulanase production strain WS9, optimized through shake-flask and 3-L fermenter cultivations, may be not the ideal strain for α-CGTase and β-CGTase production. As shown in the pullulanase production that a higher level of protease may be accumulated during scale-up fermentations than during shake-flask cultivations, the ideal α-CGTase and β-CGTase production strain remained to be optimized from strains WS3 through WS11 by shake-flask and 3-L fermenter cultivations.
During the shake-flask cultivations, the growth characteristics of strains WS9 and WB600 were similar, while the heterologous protein production level of strain WS9 was greater than that of WB600. The results suggest that strain WS9 may have great industrial value in heterologous protein production. WB600 was derived from model strain B. subtilis 168, which is auxotrophic for tryptophan (Wu et al. 1991). Besides being deficient in protease genes, strain WS9 was deficient in srfC, spoIIAC, and amyE genes, for which the strain produces less foam, shows no spore formation, and does not release α-amylase activity, respectively. The deficiency of srfC gene in strain WS9 can reduce the amphiphilic molecule surfactin production, which may not be related to heterologous production (Coutte et al. 2010). The deficiency of spoIIAC gene in strain WS9 would slightly increase the dry cell weight of fermentation, while it does not affect the heterologous production (Kabisch et al. 2013). During the post-exponential growth phase, α-amylase is one of the major extracellular proteins released by B. subtilis (Gupta and Rao 2014). The deficiency of amyE gene in strain WS9 would reduce the secretion stress and nutrition consumption, which can improve the heterologous production (Waldeck et al. 2007). There may exist other sequence polymorphisms in the genome of strains WS9 and WB600, which are not associated with deficient genes described above. The glutamate dehydrogenase in strain B. subtilis 168 was reported to be inactive that it cannot efficiently use amino acids of the glutamate family (Kabisch et al. 2013). Overall, the production difference between strains WS9 and WB600 may be related to the sequence polymorphisms in the genome of their original strains and the genotype modified by gene deficient.
For recombinant strain WS9PUL in a 3-L fermenter, the cultivation times yielding the highest pullulanase activity when feeding with organic nitrogen source were longer than that seen when feeding with inorganic nitrogen source, which may result from the abundant nutrients present in corn steep powder and yeast extract. The highest pullulanase activity produced using strain WS9PUL was much higher than that obtained from E. coli (1567.9 U/mL) (Zou et al. 2014), B. choshinensis (1005.8 U/mL) (Zou et al. 2016a), or P. pastoris (350.8 U/mL) (Xu et al. 2006). And the average pullulanase productivity produced using WS9PUL (60.1 U/mL·h−1) was much higher than that obtained from E. coli (33.4 U/mL·h−1), B. choshinensis (14.0 U/mL·h−1), or P. pastoris (7.3 U/mL·h−1).
In summary, the positive and negative impacts of extracellular proteases on the production and biological characteristics of recombinant pullulanase were investigated in this work. Through shake-flask and 3-L fermenter cultivations, host strain WS9 was found to produce the highest pullulanase activity among the protease-deficient mutants tested. Meanwhile, using pullulanase, α-CGTase, and β-CGTase as reporter proteins, the protein production differences among host strains WS3, WS9, and WB600 were investigated. Finally, the inorganic nitrogen source (NH4)2HPO4 in the original feeding solution was replaced with the organic nitrogen sources corn steep powder and yeast extract, and the carbon to organic nitrogen source ratio was optimized. With a carbon to organic nitrogen source ratio of 4:1, recombinant strain WS9PUL achieved a pullulanase activity value of 5951.8 U/mL, which represents the highest activity reported to date.
References
Anagnostopoulos C, Spizizen J (1961) Requirements for transformation in Bacillus subtilis. J Bacteriol 81:741–746
Chen WB, Nie Y, Xu Y (2013) Signal peptide-independent secretory expression and characterization of pullulanase from a newly isolated Klebsiella variicola SHN-1 in Escherichia coli. Appl Biochem Biotechnol 169:41–54
Chen A, Li Y, Liu X, Long Q, Yang Y, Bai Z (2014) Soluble expression of pullulanase from Bacillus acidopullulyticus in Escherichia coli by tightly controlling basal expression. J Ind Microbiol Biotechnol 41:1803–1810
Coutte F, Leclere V, Bechet M, Guez JS, Lecouturier D, Chollet-Imbert M, Dhulster P, Jacques P (2010) Effect of pps disruption and constitutive expression of srfA on surfactin productivity, spreading and antagonistic properties of Bacillus subtilis 168 derivatives. J Appl Microbiol 109:480–491
Dempsey LA, Dubnau DA (1989) Localization of the replication origin of plasmid pE194. J Bacteriol 171:2866–2869
Duan X, Chen J, Wu J (2013) Optimization of pullulanase production in Escherichia coli by regulation of process conditions and supplement with natural osmolytes. Bioresour Technol 146:379–385
Gupta M, Rao KK (2014) Phosphorylation of DegU is essential for activation of amyE expression in Bacillus subtilis. J Biosci 39:747–752
Gupta R, Beg QK, Khan S, Chauhan B (2002) An overview on fermentation, downstream processing and properties of microbial alkaline proteases. Appl Microbiol Biotechnol 60:381–395
Harwood CR, Cranenburgh R (2008) Bacillus protein secretion: an unfolding story. Trends Microbiol 16:73–79
Harwood CR, Wipat A (1996) Sequencing and functional analysis of the genome of Bacillus subtilis strain 168. FEBS Lett 389:84–87
Hii SL, Tan JS, Ling TC, Ariff AB (2012) Pullulanase: role in starch hydrolysis and potential industrial applications. Enzyme Res 2012:921362
Huang J, Du Y, Xu G, Zhang H, Zhu F, Huang L, Xu Z (2011) High yield and cost-effective production of poly(gamma-glutamic acid) with Bacillus subtilis. Eng Life Sci 11:291–297
Kabisch J, Thuermer A, Huebel T, Popper L, Daniel R, Schweder T (2013) Characterization and optimization of Bacillus subtilis ATCC 6051 as an expression host. J Biotechnol 163:97–104
Kelly RM, Dijkhuizen L, Leemhuis H (2009) The evolution of cyclodextrin glucanotransferase product specificity. Appl Microbiol Biotechnol 84:119–133
Krishnappa L, Dreisbach A, Otto A, Goosens VJ, Cranenburgh RM, Harwood CR, Becher D, van Dijl JM (2013) Extracytoplasmic proteases determining the cleavage and release of secreted proteins, lipoproteins, and membrane proteins in Bacillus subtilis. J Proteome Res 12:4101–4110
Krishnappa L, Monteferrante CG, Neef J, Dreisbach A, van Dijl JM (2014) Degradation of extracytoplasmic catalysts for protein folding in Bacillus subtilis. Appl Environ Microbiol 80:1463–1468
Kuo CC, Lin CA, Chen JY, Lin MT, Duan KJ (2009) Production of cyclodextrin glucanotransferase from an alkalophilic Bacillus sp. by pH-stat fed-batch fermentation. Biotechnol Lett 31:1723–1727
Kwon EY, Kim KM, Kim MK, Lee IY, Kim BS (2011) Production of nattokinase by high cell density fed-batch culture of Bacillus subtilis. Bioprocess Biosyst Eng 34:789–793
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lejeune A, Sakaguchi K, Imanaka T (1989) A spectrophotometric assay for the cyclization activity of cyclomaltohexaose (alpha-cyclodextrin) glucanotransferase. Anal Biochem 181:6–11
Li WF, Zhou XX, Lu P (2004) Bottlenecks in the expression and secretion of heterologous proteins in Bacillus subtilis. Res Microbiol 155:605–610
Margot P, Karamata D (1996) The wprA gene of Bacillus subtilis 168, expressed during exponential growth, encodes a cell-wall-associated protease. Microbiology 142:3437–3444
Moller MS, Henriksen A, Svensson B (2016) Structure and function of alpha-glucan debranching enzymes. Cell Mol Life Sci 73:2619–2641
Nguyen TT, Quyen TD, Le HT (2013) Cloning and enhancing production of a detergent- and organic-solvent-resistant nattokinase from Bacillus subtilis VTCC-DVN-12-01 by using an eight-protease-gene-deficient Bacillus subtilis WB800. Microb Cell Factories 12:79
Penninga D, vanderVeen BA, Knegtel RMA, vanHijum S, Rozeboom HJ, Kalk KH, Dijkstra BW, Dijkhuizen L (1996) The raw starch binding domain of cyclodextrin glycosyltransferase from Bacillus circulans strain 251. J Biol Chem 271:32777–32784
Pohl S, Harwood CR (2010) Heterologous protein secretion by Bacillus species from the cradle to the grave. Adv Appl Microbiol 73:1–25
Pohl S, Bhavsar G, Hulme J, Bloor AE, Misirli G, Leckenby MW, Radford DS, Smith W, Wipat A, Williamson ED, Harwood CR, Cranenburgh RM (2013) Proteomic analysis of Bacillus subtilis strains engineered for improved production of heterologous proteins. Proteomics 13:3298–3308
Shankar R, Madihah MS, Shaza EM, Aswati NKO, Suraini AA, Kamarulzaman K (2014) Application of different feeding strategies in fed batch culture for pullulanase production using sago starch. Carbohydr Polym 102:962–969
Shi M, Chen Y, Yu S, Gao Q (2013) Preparation and properties of RS III from waxy maize starch with pullulanase. Food Hydrocolloid 33:19–25
Stephenson K, Harwood CR (1998a) Influence of a cell-wall-associated protease on production of alpha-amylase by Bacillus subtilis. Appl Environ Microbiol 64:2875–2881
Stephenson K, Harwood CR (1998b) Influence of a cell-wall-associated protease on production of alpha-amylase by Bacillus subtilis. Appl Environ Microbiol 64:2875–2881
Stephenson K, Bron S, Harwood CR (1999) Cellular lysis in Bacillus subtilis; the affect of multiple extracellular protease deficiencies. Lett Appl Microbiol 29:141–145
Tjalsma H, Antelmann H, Jongbloed JD, Braun PG, Darmon E, Dorenbos R, Dubois JY, Westers H, Zanen G, Quax WJ, Kuipers OP, Bron S, Hecker M, van Dijl JM (2004) Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol Mol Biol Rev 68:207–233
van der Veen BA, Uitdehaag JC, Dijkstra BW, Dijkhuizen L (2000) Engineering of cyclodextrin glycosyltransferase reaction and product specificity. Biochim Biophys Acta 1543:336–360
Veening JW, Igoshin OA, Eijlander RT, Nijland R, Hamoen LW, Kuipers OP (2008) Transient heterogeneity in extracellular protease production by Bacillus subtilis. Mol Syst Biol 4:184
Waldeck J, Meyer-Rammes H, Wieland S, Feesche J, Maurer KH, Meinhardt F (2007) Targeted deletion of genes encoding extracellular enzymes in Bacillus licheniformis and the impact on the secretion capability. J Biotechnol 130:124–132
Wenzel M, Mueller A, Siemann-Herzberg M, Altenbuchner J (2011) Self-inducible Bacillus subtilis expression system for reliable and inexpensive protein production by high-cell-density fermentation. Appl Environ Microbiol 77:6419–6425
Westers L, Westers H, Quax WJ (2004) Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochim Biophys Acta 1694:299–310
Westers H, Westers L, Darmon E, van Dijl JM, Quax WJ, Zanen G (2006) The CssRS two-component regulatory system controls a general secretion stress response in Bacillus subtilis. FEBS J 273:3816–3827
Wu XC, Lee W, Tran L, Wong SL (1991) Engineering a Bacillus subtilis expression-secretion system with a strain deficient in six extracellular proteases. J Bacteriol 173:4952–4958
Wu SC, Yeung JC, Duan Y, Ye R, Szarka SJ, Habibi HR, Wong SL (2002) Functional production and characterization of a fibrin-specific single-chain antibody fragment from Bacillus subtilis: effects of molecular chaperones and a wall-bound protease on antibody fragment production. Appl Environ Microbiol 68:3261–3269
Wu Q-L, Chen T, Gan Y, Chen X, Zhao X-M (2007) Optimization of riboflavin production by recombinant Bacillus subtilis RH44 using statistical designs. Appl Microbiol Biotechnol 76:783–794
Wu J, Zhang K, Su L, Huang Y (2017) A Bacillus subtilis strain for efficient heterologous protein expression and high-density cultivation. China Patent CN106754466A
Xu B, Yang Y-J, Huang Z-X (2006) Cloning and overexpression of gene encoding the pullulanase from Bacillus naganoensis in Pichia pastoris. J Ind Microbiol Biotechnol 16:1185–1191
Zhang K, Duan X, Wu J (2016) Multigene disruption in undomesticated Bacillus subtilis ATCC 6051a using the CRISPR/Cas9 system. Sci Rep 6:strep27943
Zhang K, Su L, Duan X, Liu L, Wu J (2017) High-level extracellular protein production in Bacillus subtilis using an optimized dual-promoter expression system. Microb Cell Factories 16:32
Zou C, Duan X, Wu J (2014) Enhanced extracellular production of recombinant Bacillus deramificans pullulanase in Escherichia coli through induction mode optimization and a glycine feeding strategy. Bioresour Technol 172:174–179
Zou C, Duan X, Wu J (2016a) Efficient extracellular expression of Bacillus deramificans pullulanase in Brevibacillus choshinensis. J Ind Microbiol Biotechnol 43:495–504
Zou C, Duan X, Wu J (2016b) Magnesium ions increase the activity of Bacillus deramificans pullulanase expressed by Brevibacillus choshinensis. Appl Microbiol Biotechnol 100:7115–7123
Funding
This work was funded by grants from the National Science Fund for Distinguished Young Scholars (31425020), the National Natural Science Foundation of China (31501419), the 111 Project (No. 111-2-06), and the Postgraduate Research & Practice Innovation Program of Jiangsu Provence (KYCX17_1416).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 495 kb).
Rights and permissions
About this article

Cite this article
Zhang, K., Su, L. & Wu, J. Enhanced extracellular pullulanase production in Bacillus subtilis using protease-deficient strains and optimal feeding. Appl Microbiol Biotechnol 102, 5089–5103 (2018). https://doi.org/10.1007/s00253-018-8965-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-8965-x