
Abstract
1,3-Propanediol (1,3-PDO) is a monomer for the synthesis of various polyesters. It is widely used in industries including cosmetics, solvents, and lubricants. Here, the multi-modular engineering was used to improve the concentration and tolerance of 1,3-PDO in Klebsiella pneumoniae. Firstly, the concentration of 1,3-PDO was increased by 25 %, while the concentrations of by-products were reduced considerably through one-step evolution which focused on the glycerol pathway. In addition, the 1,3-PDO tolerance was improved to 150 g L−1. Secondly, co-substrate transport system was regulated, and the 1,3-PDO concentration, yield, and productivity of the mutant were improved to 76.4 g L−1, 0.53 mol mol−1, and 2.55 g L−1 h−1, respectively. Finally, NADH regeneration was introduced and the recombinant strain was successfully achieved with a high productivity of 2.69 g L−1 h−1. The concentration and yield of 1,3-PDO were also improved to 86 g L−1 and 0.59 mol mol−1. This strategy described here provides an approach of achieving a superior strain which is able to produce 1,3-PDO with high productivity and yield.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
1,3-Propanediol (1,3-PDO), a valuable bifunctional molecule, has several promising properties for many synthetic reactions, particularly for polymer and cosmetic industries (Anand et al. 2011; Saxena et al. 2009). The demand for 1,3-PDO-based polymers is constantly increasing, necessitating an increase in 1,3-PDO production (Hanna et al. 2015). The global demand for 1,3-PDO was 60.2 kt in year 2012 and is expected to reach approximately 150 kt by year 2019 (Lee et al. 2015). The production of 1,3-PDO by biotechnological process showed advantages over the conventional chemical one. Metabolic engineering could be used to build a new organism which could make use of cheaper and abundant substrates such as sugar and starch (Nakamura and Whited 2003; Yang et al. 2007; Zeng and Biebl 2002). The heterogeneous system using Escherichia coli to product 1,3-PDO was exploited by DuPont and Genencor International (Nakamura and Whited 2003). Although Dupont and Genencor had a super E.coli that reached a final 1,3-PDO concentration of 135 g L−1 using glucose as substrate, vitamin B12 was required in the fermentation process which impeded the large-scale production of 1,3-PDO.
As reported previously, process engineering and metabolic engineering approaches have been applied for producing 1,3-PDO. On one hand, the optimization of the culture medium components and fermentation process could enhance the 1,3-PDO titer of the natural producers such as Klebsiella pneumoniae (Jin et al. 2011), Lactobacillus diolivorans (Stefan et al. 2014), Clostridium pasteurianum (Kaeding et al. 2015), and Clostridium butyricum (Wilkens et al. 2012). The downstream separation process of 1,3-PDO production was difficult as these natural producers produced by-products of lactic acid and 2,3-butanediol (2,3-BD) (Kaur et al. 2012; Nemeth and Sevella 2008; Saxena et al. 2009). On the other hand, 1,3-PDO production using glycerol as a substrate was a coupled oxidation-reduction process. In the reductive branch, glycerol was reduced via 3-hydroxypropanal to 1,3-PDO. And, 3-hydroxypropanal was formed by dehydration of glycerol catalyzed by a B12-dependent enzyme. Then, 3-hydroxypropanal was reduced to 1,3-PDO by an NADH-oxidoreductase. ATP, NADH, and by-products were generated in the oxidative branch (Celinska 2010). There were various metabolic strategies, and dhaT & dhaB over-expression (Zhao et al. 2009a; Zhao et al. 2009b) or ldhA & budC deletion has been researched to enhance the productivity of 1,3-PDO (Kumar et al. 2012; Xu et al. 2009; Zhang et al. 2012). In addition, substrates uptake was an important infactor of biomass and products synthesis (Baek et al. 2013; Yang et al. 2007). The glucose PTS system was deleted to eliminate the Crabtree effect (Vieira et al. 2015). Furthermore, remodeling intracellular reducing power was also useful in optimizing synthesis of product and obtaining a strain with high productivity (Guo et al. 2011; Holm et al. 2010; Zhang et al. 2014). However, none of these work simultaneously targeted the global material, energy metabolism, and transport system to improve the productivity of 1,3-PDO product.
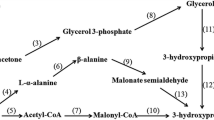
In this work, the multi-modular engineering was applied to regulate the 1,3-PDO biosynthesis system in K. pneumoniae based on mixtures of glucose and crude glycerol deriving from biodiesel production (Fig. 1). A one-step evolution strategy was used to improve the tolerance and enhance the concentration of 1,3-PDO while reducing the by-products. The substrate transport system and NADH regeneration system were optimized. Finally, the recombinant strain eliminated the glucose inhibition and achieved a higher glycerol consumption rate. The concentration, productivity, and yield of 1,3-PDO were further increased.

Scheme of multi-modular engineering of 1,3-PDO biosynthesis system
Materials and methods
Media, strains, plasmids, and materials
E. coli Top 10 and its derivatives were cultured in LB medium, which contained 5-g yeast extract, 5 g NaCl, and 10 g tryptone per liter. Ampicillin or kanamycin was added to screen the recombinant strains with a final concentration of 100 or 50 mg L−1, respectively. The kits for genomic DNA isolation, plasmid extraction, and DNA recovery were purchased from Omega Bio-Tek (Norcross, GA), and all the enzymes were purchased from New England Biolabs (Ipswich, MA). KP-WT(screened from KCTC2242) was used as parental strain. All the strains and plasmids used are listed in Table 1.
Plasmid construction
All primers used for gene cloning are listed in Supplementary Table S1. The genes budC (KPN2242_13265), ldhL (KPN2242_13685), lldD (KPN2242_22800), dld (KPN2242_15905), ldhA (KPN2242_10020), dhaD (KPN2242_20560), dhaK (KPN2242_20565), and dhaT (KPN2242_20540) were amplified by PCR using the genomic DNA of K. pneumoniae as a template. The PCR products were subcloned into plasmid pMD 18-T to generate the plasmids T-budC, T-ldhL, T-lldD, T-dld, T-ldhA, T-dhaD, T-dhaK, and T-dhaT. Then, the random mutant genes were obtained by error-prone PCR using the plasmids T-budC, T-ldhL, T-lldD, T-dld, T-ldhA, T-dhaD, T-dhaK, and T-dhaT as a template, respectively.
The glycerol uptake facilitator protein coding gene glpF from E. coli K12 (EF, X15054.1), FPS1 from S. cerevisiae S288c (X54157.1), glpF from Lactobacillus rhamnosus LA-04-1 (Wang et al. 2015b) (LF, AP011548.1), glpF from K. pneumoniae (KF, KPN2242_23120), aquaporin Z coding gene from K. pneumoniae (AZ, KPN2242_07615), glycerol diffusion protein coding gene from K. pneumoniae (gd, KPN2242_20510), and propanediol utilization protein: propanediol diffusion facilitator coding gene from K. pneumoniae (pdu, KPN2242_18960) were amplified by PCR using each genomic DNA as template. These PCR products were subcloned into appropriate vector to generate PK-EF-EGFP, PK-FPS1-EGFP, PK-LF-EGFP, PK-KF-EGFP, PK -AZ-EGFP, PK-gd-EGFP, PK-pdu-EGFP, PK-EF, PK-FPS1, PK-LF, PK-KF, PK-AZ, PK-gd, PK-pdu; tac-EF-EGFP, tac-FPS1-EGFP, tac-LF-EGFP, tac-KF-EGFP, tac-AZ-EGFP, tac-gd-EGFP, tac-pdu-EGFP, tac-EF, tac-FPS1, tac-LF, tac-KF, tac-AZ, tac-gd, and tac-pdu.
The formate dehydrogenase encoding gene from Pichia pastoris GS115 (fdh, PAS_chr3_0932), soluble pyridine nucleotide transhydrogenase encoding gene from K. pneumoniae (udh, KPN2242_24300), and glucose dehydrogenase encoding gene from Bacillus subtilis subsp. Subtilis str. 168 (gdh, BSU03930) were amplified using each genomic DNA as template. The PCR products were subcloned into plasmid PK-KF to generate the plasmid KFUG.
Deletion of the crr
The coding sequences of crr neighboring gene PdxK and EI were amplified and inserted into the vector pRSFDuet to generate plasmid pRSF-PdxK-EI. Then, the chloromycetin resistance coding gene Cm was inserted into pRSF-PdxK-EI. The resulting plasmid, pRSF-PdxK-Cm-EI, was used as a template for the further PCR reaction.
In order to delete chromosomal crr gene, the linear DNA fragment (PdxK-Cm-EI) was amplified from pRSF-PdxK-Cm-EI using glucosinolates phosphorylated modification primers PdxK-up*/EI-down*. This amplified DNA fragment was transformed into competent cells using electrical transformation, and the recombinants were screened on LB plates containing chloromycetin (final concentration of 34 mg L−1).
Methods of strain screen and 1,3-PDO tolerance test
The mixture of mutant fragments generated by error-prone PCR was transformed into the competent cells to obtain mutant KP*. Mutant with improved phenotypes was used as the starting strain for another round of mutant fragments homologous recombination if necessary. The recombinants were screened on LB plates with Bromocresol green and KMnO4-KBr which also contained 130 g L-1 of 1,3-PDO. The well-grown colonies with low acid productivity were transferred into the LB medium for the further selection.
In order to test 1,3-PDO tolerance, the cell growth rate of 95 mutants was determined under different 1,3-PDO concentrations. KP-WT and seven mutants were cultured overnight and inoculated into 96-well plates (initial OD600 = 0.1). Each well contained 200-μL LB medium supplemented with 0 to 160 g L-1 1,3-PDO, and the cell density of each strains was measured every 30 min.
Intracellular NADH/NAD+, enzyme activity, and gene expression analysis
Recombinant strains were grown overnight in 4-mL LB medium and then inoculated in the fresh medium (initial OD600 = 0.1) containing kanamycin for 16 h. The EGFP fluorescence intensity was measured by spectrophotofluoro-meter and flow cytometric analysis (FACS). The spectrophotometry kit for total intracellular NAD+ or NADH concentration determination was purchased from COMIN (Suzhou, China).
Acetoin reductase (AR), lactate dehydrogenase (LDH), and 1,3-PDO oxidoreductase (PDOR) activity were assayed by measuring the absorbance of NADH at 340 nm according to the research of (Jeyakanthan et al. 2010; Marcal et al. 2009; Xu et al. 2009).
Measurement of biomass, substrate consumption rate, and metabolic products
Cell density was determined by measuring the turbidity of culture medium at 600 nm using a spectrophotometer (Thermo Scientific, Waltham, MA), and the growth curve was drew based on the cell density data.
The concentration of glucose, glycerol, 1,3-PDO, 2,3-BD, and lactic acid were determined by UltiMate 3000 HPLC (Thermo Scientific, Waltham, MA) using the following apparatus and operating conditions: Bio-Rad Aminex HPX-87H column (Bio-Rad Laboratories, Hercules, CA) with RID and UV detectors, column temperature of 65 °C, and 0.6 mL min−1 5 mM sulfuric acid as mobile phase.
Fed-batch cultivation condition
K. pneumoniae was cultured in KP medium containing (per liter) 1.3 g KH2PO4, 3.4 g K2HPO4·3H2O, 0.2 g MgSO4·7H2O, 3.0 g (NH4)2SO4, 20 g glycerol, 5 g glucose, 1 g yeast extract, and 5 mL trace elements. Trace elements contained (per liter) 2.5 g CoCl2·6H2O, 15 g MnCl2·4H2O, 2.1 g Na2MoO4·2H2O, and 100.8 g Fe (II) citrate. The final pH of KP medium was adjusted to 7.0 by 4 mM KOH. Fed-batch fermentations were carried out in a 5-L fermentor (BIOTECH-2002, Shanghai Baoxing Company, China) containing 2-L KP medium supplemented with trace elements. The glycerol concentration was maintained at ∼15 g L−1 by regulating the feeding rate of the glycerol solution (700 g L−1) manually, and glucose (15 g L−1) was added to enhance cell growth. For cultivating K. pneumoniae strains, the pH of the medium was automatically adjusted by adding 4 M KOH and Ca(OH)2 together. In order to eliminate the influence of impurities in crude glycerol on the experimental accuracy, we used the pure glycerol in this study.
Results
Glycerol metabolic pathway engineering
The mutant genes generated by error-prone PCR were introduced to strain KP-WT and lead to the homologous recombination. And then, it was inoculated into LB liquid medium. After overnight culture, KP-WT and 95 mutants with small color zone and clear zone were transferred into the fresh LB medium containing different concentrations of 1,3-PDO and were used to measure the cell growth for 10 h. Half of the mutants showed lower cell growth rate than KP-WT strain. For 1,3-PDO that was a growth-associated product, strains with higher cell growth rate labeled KP-M-2#, 10#, 15#, 22#, 56#, 72#, and 94# were collected for further research.
The parent strain KP-WT could not grow when the 1,3-PDO concentration was 110 g L−1. However, the mutants, except for KP-M-10#, survived in the medium containing 1,3-PDO ranging from 110 to 150 g L−1. The tolerance of mutants to 1,3-PDO was increased by 36 % (Supplementary Fig. S1a–h). When these seven mutants were cultured in KP medium at 37 °C for 24 h, five mutants produced more 1,3-PDO than KP-WT. As shown in Fig. 2, KP-M-15# had the highest final concentration of 1,3-PDO of among these mutants. 1,3-PDO and acetic acid concentration were increased ∼5 and ∼1 g L−1, while 2,3-BD and lactic acid concentration were reduced ∼2 and ∼0.9 g L−1, respectively. The results indicated that reducing the concentration of by-products could effectively enhance the concentration of 1,3-PDO.

The shake flask fermentation results of mutants and KP-WT
To further characterize genetic mechanism to improve 1,3-PDO production in strain KP-M-15#, sequencing and enzyme assay analysis were conducted. Genome sequencing was performed by BGI (Beijing, China). Among the mutant genes, dhaT was directly related to 1,3-PDO production, and other mutant genes were directly related to by-products production (Table 2).
The activity of three key enzymes in KP* was determined. The results showed that the PDOR (encoded by dhaT gene) activity was increased by 18.9 %, but the AR (encoded by budC gene) activity and LDH (encoded by ldhA, lldD, Dld and ldhL gene) activities were decreased by 87.5 and 25.4 %, respectively (Supplementary Table S2). The results of fermentation and enzyme activity of KP* indicated that the one-step evolution method was helpful to redirect glycerol metabolic pathway.
Optimization of transport system
Glucose was shown to be indispensable to enhance biomass in the production process of 1,3-PDO from glycerol with K. pneumoniae. However, glycerol utilization was inhibited in the presence of glucose. In order to eliminate the glucose effect, PTS transport system was disrupted. To determine the effect of crr deletion on glycerol metabolism, the crr mutant strain KP** was grown in five glucose-glycerol media containing 20 g L−1 glycerol and different concentrations of glucose (5 ∼ 25 g L−1). Normally, glycerol could not be used by pathway engineered strain KP* with the glucose concentration around 10 g L−1. However, in the fermentation process of KP**, glycerol and glucose could be consumed simultaneously. The results of these cultivation proved that KP** was capable to use glycerol and glucose as co-substrate for biomass formation and 1,3-PDO production.
In the co-substrate fed-batch fermentation process (Fig. 4a, b), 1,3-PDO concentration of the pathway engineered and crr deletion strain KP** achieved 68.1 g L−1 with an increase of 25 %, while the concentration of 2,3-BD and lactic acid were decreased by 56.7 and 88.3 %, respectively. Moreover, 1,3-PDO productivity and conversion rate were improved from 1.82 to 2.26 g L−1 h−1 and 0.47 to 0.52 mol mol−1, respectively.
Efficient uptake of glycerol was also important to increase conversion rate. Accordingly, we expressed many kinds of channel protein in KP** including glycerol uptake facilitator protein, aquaporin Z protein, glycerol diffusion protein, and propanediol diffusion facilitator. Besides, inducible and constitutive expression system with different gene expression levels was used to express the channel proteins. The gene expression level was measured through fusion protein with GFP. As shown in Fig. 3a, the fluorescence intensity of all the strains was higher than that of the control. The results indicated that all the channel proteins were successfully expressed with different expression levels in KP**.

a Fluorescence intensity of transport system optimized strains fusing GFP expression with tac and PK promoter. b The glycerol consumption of transport system optimized strains with tac and PK promoter
Recombinant strains with constitutive expression system showed overall lower expression level than strains with inducible expression system. When the glycerol consumption rate of all recombinant strains was determined (Fig. 3b), compared with the control strain KP**, most of the high expression level strains had an even lower glycerol consumption rate similarly. It indicated that the expression level of channel protein was not proportional to glycerol consumption rate. Taken together, recombinant strain PK-KF was screened to be a better producer for the production of 1,3-PDO with a twofold glycerol consumption rate than the control strain KP**. In the fed-batch fermentation, the maximum 1,3-PDO concentration of PK-KF was improved to 76.4 g L−1 with a productivity of 2.55 g L−1 h−1. PK-KF also produced 13 g L−1 2,3-BD and 5 g L−1 lactic acid. The concentration of 1,3-PDO was increased by 12.2 % (Fig. 4c).

Concentrations of main metabolites in 5-L fermentor of KP-WT, KP**, PK-KF, and KP**-KFUG
NADH regeneration
Since 1,3-PDO synthesis pathway was a NADH-dependent process, the intracellular ratio of NADH to NAD+ of the mutant strains was measured in logarithmic growth phase (4.5 h) and stationary phase (16 h) during the fermentation process and the results were listed in Table 3.
The ratio of NADH to NAD+ of KP** was lower than that of KP-WT at 4.5 h caused by the decline of glucose consumption rate reduction. During the product synthesis process, the ratio of NADH to NAD+ of KP** was a little higher than that of KP-WT. Glycerol channel protein was introduced to enhance glycerol consumption, and the ratio of NADH to NAD+ in recombinant strain PK-KF was increased from 0.998 to 1.015 at 4.5 h and from 0.869 to 0.988 at 16 h. It indicated that one-step evolution strategy and transporter engineering strategy could not only redistribute the carbon metabolism but also influence intracellular energy level.
Based on the promising results obtained from strategies of one-step evolution pathway engineering and transporter engineering for the production of 1,3-PDO, we found that intracellular NADH level was an important factor to increase the yield of 1,3-PDO. In order to enhance the concentration of 1,3-PDO, another strategy (NADH regeneration) was performed to increase intracellular ratio of NADH to NAD+. The ratio of NADH to NAD+ was obviously increased by NADH regeneration system including three enzymes. Finally, the ratio of NADH to NAD+ of KP**-KFUG was 1.259, the highest among the four strains.
As expected, the fed-batch fermentation results of KP**-KFUG showed that the concentration of 1,3-PDO, lactic acid, and 2,3-BD reached 86 (with a productivity of 2.69 g L−1 h−1), 18, and 14.8 g L−1, respectively. Compared with the production of PK-KF, 1,3-PDO production was increased by 9.6 g L−1. 1,3-PDO conversion reached 0.59 mol mol−1. Simultaneously, the concentrations of 2,3-BD and lactic acid were increased by 5 and 9.8 g L−1, respectively (Fig. 4d).
Discussion
In order to produce 1,3-PDO effectively, multi-modular engineering, including one-step evolution, transporter engineering, and NADH regeneration, was applied in 1,3-PDO biosynthesis system of K. pneumoniae. Blocking the by-product pathways was a simple but effective method to improve the productivity and final concentration of 1,3-PDO (Zhang et al. 2012; Yang et al. 2007). 2,3-BD and lactic acid were the main competitors to the production of 1,3-PDO by KP-WT. Moreover, due to the similarity between 2,3-BD and 1,3-PDO, it would be a challenge to purify the 1,3-PDO from the mixture, making it a bottleneck in the bioprocess. 2,3-BD from pyruvate was considered as a three-step process in K. pneumoniae under anaerobic conditions. In the last step, acetoin was converted to 2,3-BD by NADH-dependent AR. Lactic acid was formed by LDH. According to the K. pneumoniae Genome Sequencing Project, K. pneumoniae had many isozymes, such as LDH. There were four LDHs: two D-LDHs and two L-LDHs. D-LDH encoded by ldhA mainly contributed to the synthesis of lactic acid along with the consumption of NADH (Tarmy and Kaplan 1968; Xu et al. 2009). Another D-LDH was FAD protein and NADH independent, contributing a little to the D-lactic acid formation. The two L-LDHs, known as membrane-bound flavoprotein, were coupled to the respiratory chain (Jiang et al. 2001).
It was reported that error-prone whole genome amplification could increase the tolerance and final concentration of the product (Ye et al. 2013; Luhe et al. 2011). However, the mutant was not easily to find out the mutation sites by error-prone whole genome. The multiplex genome engineering (MAGE) was also an effective method to optimize genome and obtain target strains (Wang et al. 2009). However, this method could not be used in our original K. pneumoniae with high efficiency. Here, a one-step evolution strategy through multi-genes homologous recombination was applied to improve the concentration of 1,3-PDO and reduce the concentrations of by-products. This method, with a high mutation efficiency, did not use toxic mutagens and was easy to be analyzed. Moreover, the mutants were stable.
The mutant genes generated by error-prone PCR were introduced to strain KP-WT, and the mutant genes amplified by modified primers were not easy to be degraded. Through many cycles of homologous recombination and different screen methods, KP* was screened to be a fine producer to produce 1,3-PDO. The mutant, with a better 1,3-PDO tolerance, had a potential to obtain a higher concentration of 1,3-PDO. The changes of enzyme activity were caused by gene sequences mutations. Since only half of the target genes were changed, this method could be used for further evolution and was expected to provide a better strain for 1,3-PDO production.
Glucose added to the fermentation process was beneficial to cell growth and 1,3-PDO production. However, the glucose inhibited glycerol utilization by K. pneumoniae and prevented the synthesis of 1,3-PDO from glycerol (Yang et al. 2007). Deletion of glucose transporter encoding gene could eliminate the glucose repression (Baek et al. 2013). When gene crr was deleted in KP*, glycerol was consumed normally at a high glucose level, but the cell growth rate of KP** was lower than that of KP*. The effect of glucose catabolism in K. pneumoniae was supplement of reducing power and biomass formation. Thus, the cell density (OD600nm) and intracellular ratio of NADH to NAD+ of mutant strain KP** were reduced when the glucose metabolized slowly. Another approach to increase cell density and 1,3-PDO synthetic efficiency was to enhance the glycerol metabolism, by strengthening the glycerol transport system. And, this approach could also provide reducing power and the precursor of 1,3-PDO synthesis. KP**-KF could produce higher concentration of 1,3-PDO and faster glycerol consumption rate in the glucose-glycerol fermentation process.
Propanediol diffusion facilitator, which was a propanediol utilization protein, had little effect on enhancing glycerol catabolism. To the best of our knowledge, there is little research about its definite effect in K. pneumoniae. Although the mechanism was unclear now, the reason could be related to the expression level of glycerol channel protein. And, glycerol channel proteins had a reversible effect and could also transport glycerol from intracellular to extracellular.
In order to verify the effect of NADH regeneration, the main products and intracellular ratio of NADH to NAD+ of KP**-KFUG were determined. 1,3-PDO concentration and the ratio of NADH to NAD+ were higher than those of PK-KF. Since glycerol metabolism node was rigid node, glycerol distribution was not easily to be affected by related enzymes expression level. The catalyst of PDOR needs NADH as its cofactor. If intracellular ratio of NADH to NAD+ was at a low level, 1,3-PDO could be catalyzed to 3-hydroxypropanal. Since the 3-hydroxypropanal was a toxic compound, it would inhibit the activities of GDHt and PDOR. Therefore, we could improve the 1,3-PDO yield by increasing the intracellular ratio of NADH to NAD+. Nonetheless, the highest intracellular ratio of NADH to NAD+ was not the best situation. As the intracellular ratio of NADH to NAD+ was further increased, the concentration of NADH-dependent by-products was also increased. 1,3-PDO was a growth coupled product, but the results in Fig. 4c, d were not consistent. In the stationary phase, the 1,3-PDO concentration increased significantly but the cell grew slowly. The possibility was that citrate synthase was inhibited by high ratio of NADH to NAD+. When the intracellular ratio of NADH to NAD+ increased, the TCA circle was inhibited and the cell stopped growing. In order to achieve higher concentration of 1,3-PDO with lower concentrations of by-products, we needed to insure that the intracellular ratio of NADH to NAD+ was at an appropriate level. We could also further increase the 1,3-PDO end concentration by increasing the cell density.
As shown in Table 4, the comparison results indicated that the multi-modular engineering strategy described here provide a new approach to achieve a superior strain which had high concentration and productivity of 1,3-PDO production.
References
Anand P, Saxena RK, Marwah RG (2011) A novel downstream process for 1,3-propanediol from glycerol-based fermentation. Appl Microbiol Biot 90:1267–1276
Baek RO, Won KH, Sun YH, Lian HL, Akihiko K, Jeong WS, Chul HK (2013) The production of 1,3-propanediol from mixtures of glycerol and glucose by a Klebsiella pneumoniae mutant deficient in carbon catabolite. Bioresour Technol 130:719–724
Celinska E (2010) Debottlenecking the 1,3-propanediol pathway by metabolic engineering. Biotechnol Adv 28:519–530
Guo ZP, Zhang L, Ding ZY, Shi GY (2011) Minimization of glycerol synthesis in industrial ethanol yeast without influencing its fermentation performance. Metab Eng 13:49–59
Hanna P, Joanna Z, Daria SP, Marlena S, Ryszard S, Daniel L (2015) 1,3-propanediol production by new recombinant Escherichia coli containing genes from pathogenic bacteria. Microbiol Res 171:1–7
Holm AK, Blank LM, Marco O, Andreas S, Christian S, Jensen PR (2010) Metabolic and transcriptional response to cofactor perturbations in Escherichia coli. J Biol Chem 285:17498–17506
Jeyakanthan J, Thamotharan S, Panjikar S, Kitamura Y, Nakagawa N, Shinkai A, Kuramitsu S, Yokoyama S (2010) Expression, purification and X-ray analysis of 1,3-propanediol dehydrogenase (Aq_1145) from Aquifex aeolicus VF5. Acta Crystallogr F 66:184–186
Jiang GR, Nikolova S, Clark DP (2001) Regulation of the ldhA gene, encoding the fermentative lactate dehydrogenase of Escherichia coli. Microbiol 147:2437–2446
Jin P, Li S, Lu SG, Zhu JG, Huang H (2011) Improved 1,3-propanediol production with hemicellulosic hydrolysates (corn straw) as cosubstrate: impact of degradation products on Klebsiella pneumoniae growth and 1,3-propanediol fermentation. Bioresour Technol 102:1815–1821
Kaeding T, Daluz J, Kube J, Zeng AP (2015) Integrated study of fermentation and downstream processing in a miniplant significantly improved the microbial 1,3-propanediol production from raw glycerol. Bioproc Biosyst Eng 38:575–586
Kaur G, Srivastava AK, Chand S (2012) Advances in biotechnological production of 1,3-propanediol. Biochem Eng J 64:106–118
Kumar V, Sankaranarayanan M, Durgapal M, Zhou S, Ko Y, Ashok S, Sarkar R, Park S (2012) Simultaneous production of 3-hydroxypropionic acid and 1,3-propanediol from glycerol using resting cells of the lactate dehydrogenase-deficient recombinant Klebsiella pneumoniae over-expressing an aldehyde dehydrogenase. Bioresour Technol 135:555–563
Lee CS, Aroua MK, Daud WMAW, Cognet P, Peres-Lucchese Y, Fabre PL, Reynes O, Latapie L (2015) A review: conversion of bioglycerol into 1,3-propanediol via biological and chemical method. Renew Sust Energ Rev 42:963–972
Luhe AL, Tan L, Wu J, Zhao H (2011) Increase of ethanol tolerance of Saccharomyces cerevisiae by error-prone whole genome amplification. Biotechnol Lett 33:1007–1011
Marcal D, Rego AT, Carrondo MA, Enguita FJ (2009) 1,3-propanediol dehydrogenase from Klebsiella pneumoniae: decameric quaternary structure and possible subunit cooperativity. J Bacteriol 191:1143–1115
Mu Y, Xiu ZL, Zhang DJ (2008) A combined bioprocess of biodiesel production by lipase with microbial production of 1,3-propanediol by Klebsiella pneumoniae. Biochem Eng J 40:537–541
Nakamura CE, Whited GM (2003) Metabolic engineering for the microbial production of 1,3-propanediol. Curr Opin Biotech 14:454–459
Nemeth A, Sevella B (2008) Development of a new bioprocess for production of 1,3-propanediol.I.: modeling of glycerol bioconversion to 1,3-propanediol with Klebsiella pneumoniae enzymes. Appl Biochem Biotech 144:47–58
Saxena RK, Pinki A, Saurabh S, Jasmine I (2009) Microbial production of 1,3-propanediol: recent developments and emerging opportunities. Biotechnol Adv 27:895–913
Stefan P, Hans M, Diethard M, Michael S (2014) Heading for an econo-mic industrial upgrading of crude glycerol from biodiesel production to 1,3-propanediol by Lactobacillus diolivorans. Bioresour Technol 152:499–504
Tarmy EM, Kaplan NO (1968) Kinetics of Escherichia coli B D-lactate dehydrogenase and evidence for pyruvate-controlled change in conformation. J Biol Chem 243:2587–2596
Vieira PB, Kilikian BV, Bastos RV, Perpetuo EA, Nascimento AAO (2015) Process strategies for enhanced production of 1,3-propandiol by Lactobacillus reuteri using glycerol as a co-substrate. Biochemical Eng J 94:30–38
Wang HH, Isaacs FJ, Carr PA (2009) Programming cells by multiplex genome engineering and accelerated evolution. Nature 460:894–898
Wang M, Hu L, Fan L, Tan T (2015a) Enhanced 1-butanol production in engineered Klebsiella pneumoniae by NADH regeneration. Energ Fuel 29:1823–1829
Wang Y, Cai D, Chen C, Wang Z, Qin P, Tan T (2015b) Efficient magnesium lactate production with in situ product removal by crystallization. Bioresour Technol 198:658–663
Wilkens E, Ringel AK, Hortig D, Willke T, Vorlop KD (2012) High-level production of 1,3-propanediol from crude glycerol by Clostridium butyricum akr102a. Appl Microbiol Bio 93:1057–1063
Wu Z, Wang Z, Wang GQ, Tan TW (2013) Improved 1,3-propanediol production by engineering the 2,3-butanediol and formic acid pathways in integrative recombinant Klebsiella pneumoniae. J Biotechnol 168:194–200
Xu YZ, Guo NN, Zheng ZM, Ou XJ, Liu HJ, Liu DH (2009) Metabolism in 1,3-propanediol fed-batch fermentation by a D-lactate deficient mutant of Klebsiella pneumoniae. Biotechnol Bioeng 104:965–972
Yang G, Tian J, Li J (2007) Fermentation of 1,3-propanediol by a lactate deficient mutant of Klebsiella oxytoca under microaerobic conditions. Appl Microbiol Biot 73:1017–1024
Ye L, Zhao H, Li Z, Wu JC (2013) Improved acid tolerance of Lactobacillus pentosus by error-prone whole genome amplification. Bioresour Technol 135:459–463
Zeng AP, Biebl H (2002) Bulk chemicals from biotechnology: the case of 1,3-propanediol production and the new trends. Adv Biochem Eng Biot 74:239–259
Zhang G, Yang G, Wang X, Guo Q, Li Y, Li J (2012) Influence of blocking of 2,3-butanediol pathway on glycerol metabolism for 1,3-propanediol production by Klebsiella oxytoca. Appl Biochem Biotech 168:116–128
Zhang X, Zhang R, Bao T, Rao Z, Yang T, Xu M, Xu Z, Li H, Yang S (2014) The rebalanced pathway significantly enhances acetoin production by disruption of acetoin reductase gene and moderate-expression of a new water-forming NADH oxidase in Bacillus subtilis. Metab Eng 23:34–41
Zhang T, Wang Z, Li D, Tan T, Fang W, Yan Y (2015) Pull-in urea cycle for the production of fumaric acid in Escherichia coli. Appl Microbiol Biot 99:1–12
Zhao L, Ma X, Zheng Y, Zhang J, Wei G, Wei D (2009a) Over-expression of glycerol dehydrogenase and 1,3-propanediol oxidoreductase in Klebsiella pneumoniae and their effects on conversion of glycerol into 1,3-propanediol in resting cell system. J Chem Technol Biot 84:626–632
Zhao L, Zheng Y, Ma X, Wei D (2009b) Effects of over-expression of glycerol dehydrogenase and 1,3-propanediol oxidoreductase on bioconversion of glycerol into 1,3-propandediol by Klebsiella pneumoniae under micro-aerobic conditions. Bioproc Biosyst Eng 32:313–320
Zheng ZM, Xu YZ, Liu HJ, Guo NN, Cai ZZ, Liu DH (2008) Physiologicalmechanism of sequential products synthesis in 1,3-propanediol fed-batch fermentation by Klebsiella pneumoniae. Biotechnol Bioeng 100:923–932
Acknowledgments
This work was supported by the National Basic Research Program of China (2013CB733600), the National Nature Science Foundation of China (21390202), and National High Technology Research and Development Program of China (2014AA020522).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants performed by any of the authors.
Conflict of interest
The authors have declared no conflict of interest.
Electronic supplementary material
ESM 1
(PDF 412 kb).
Rights and permissions
About this article

Cite this article
Wang, M., Wang, G., Zhang, T. et al. Multi-modular engineering of 1,3-propanediol biosynthesis system in Klebsiella pneumoniae from co-substrate. Appl Microbiol Biotechnol 101, 647–657 (2017). https://doi.org/10.1007/s00253-016-7919-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7919-4