
Abstract
The accumulation of toxic inorganic nitrogen is one of the major water quality problems in intensive aquaculture systems, thus the N removal in aquaculture systems is an important issue for the sustainable development of aquaculture. To understand one of the major microbial N removal processes, anaerobic ammonium oxidation (anammox), phylogenetic diversity, and distribution of anammox bacteria in sediments of four different marine aquaculture zones in Hong Kong (HK) were investigated. The 16S rRNA genes analysis indicated that sequences detected from Cheung Sha Wan (CSW) and Sok Kwu Wan (SKW) were closely related to several clusters within the Scalindua genus of anammox bacteria, including a new habitat-specific group, while only several sequences related to Scalindua and Kuenenia were detected in Sham Wan (SW) and Yim Tin Tsai East (YTTE). Most of the sequences obtained in SW and YTTE with the same PCR primers showed a low similarity to the known anammox bacteria, forming several novel groups within the Planctomycetes. However, results from the hydrazine oxidoreductase (HZO) encoding gene showed that only sequences from SW were related to the genus of Kuenenia, and sequences from other three sites were closely related to the genus of Scalindua. The community analysis showed that CSW and SKW share similar anammox bacterial community structures while SW and YTTE contain a unique anammox bacterial community. Furthermore, correlations reflect that organic matter is positively correlated with Kuenenia-like anammox bacteria, while the redox potential is significantly correlated with Scalindua-like anammox bacteria in marine aquaculture zones. Our results extend the knowledge of anammox bacteria in marine aquaculture systems and highlight the importance of environmental factors in shaping the community structures of anammox bacteria.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Aquaculture, including the freshwater and marine aquaculture, is a rapidly growing primary production sector to feed the increasing human population. Since 1970, aquaculture has grown at an average rate of 8.9 % per year, and the percentage contribution of aquaculture to the total world fisheries has grown from 17.0 to 31.7 % from 1993 to 2003 (FAO 2004). Due to the worldwide decline of ocean fisheries and the continuous expansion of the human population, aquaculture will continue to grow greatly to satisfy the minimum protein requirement for human nutrition (FAO 2004). The intensive development of the marine aquaculture has been accompanied by an increase of environmental impacts on the marine ecosystems (FAO 2004). Large amounts of effluents (excessive feed and feces) would be generated during the aquaculture process. These effluents, containing nutrients, various organic and inorganic compounds such as nitrogen (N), phosphorus (P), and dissolved organic compounds (C), and organic matter, are discharged into coastal water. The excessive accumulation of nitrogenous compounds (e.g., ammonia and nitrite) not only leads to the deterioration of seawater quality but also causes poison of fish or shrimp (Crab et al. 2007). Previous studies have proven that the efficiency of N assimilation is one of the most important implications for water quality and profitability of aquaculture (Hargreaves 1998, Crab et al. 2007). The accumulation and subsequent toxicity of ammonia in the aquaculture system would strongly affect the production of aquaculture, and excessive nitrate and nitrite would also increase the possibility of harmful algal blooms in the aquaculture system, especially in the marine aquaculture zone (Avnimelech 1999, Crab et al. 2007). Thus, the N removal in aquaculture systems is always considered as an important priority issue for the sustainable development of aquaculture. It has been known that sediment represents a source of ammonia and a sink for nitrite and nitrate in marine aquaculture systems, and the large volume of oxygen-depleted reduced sediment also suggests that the potential for N removal by anaerobic processes, including denitrification and anaerobic ammonium oxidation (anammox). Previous studies have paid great efforts to understand the contribution of denitrification in marine aquaculture system, while only a few related researches have been reported on marine aquaculture zones (Crab et al. 2007, Martins et al. 2010, Holl et al. 2011).
Anammox, an important pathway of the N cycle, allows ammonium to be oxidized by nitrite under anoxic condition yielding the dinitrogen (N2) gas (Mulder et al. 1995, van de Graaf et al. 1995). Anammox is considered as one of the most important ways for N removal from the marine ecosystem, which might contribute more than 50 % of the total N loss in world oceans (Devol 2003). Since their discovery in 1990s, the anammox bacteria, affiliated to the Planctomycetes (Strous et al. 1999), have been detected in wastewater treatment plants and in various natural ecosystems, including marine (Thamdrup and Dalsgaard 2002, Dalsgaard et al. 2003, Kuypers et al. 2003, Kuypers et al. 2005a, Kuypers et al. 2005b, Hamersley et al. 2007, Lam et al. 2009, Ward et al. 2009, Li and Gu 2013, Castro-Gonzalez et al. 2014), freshwater (Penton et al. 2006, Schubert et al. 2006, Zhang et al. 2007, Lee et al. 2014, Smith et al. 2015), terrestrial (Humbert et al. 2010, Wang and Gu 2013), extreme (Byrne et al. 2009, Jaeschke et al. 2009, Li et al. 2010a, Zhu et al. 2015), intestinal tracts of fish (Chan et al. 2016), and polychaetes (Li and Gu 2016). Several studies have also reported the activity and biodiversity of anammox bacteria in aquaculture systems, such as the biofilters in freshwater recirculating aquaculture systems (Tal et al. 2006, Lahav et al. 2009, van Kessel et al. 2010, van Kessel et al. 2011, Castine et al. 2012), a denitrification reactor for a recirculating aquaculture system (Lahav et al. 2009), and sediments of the shrimp ponds (Amano et al. 2011) and marine aquaculture zones (Li et al. 2010b, Li et al. 2011b). However, researches about anammox bacteria diversity and distribution in marine aquaculture systems are still very limited, and more efforts should be taken in order to comprehensively describe this important N removal process in aquaculture systems. In the present study, we selected four long-term marine aquaculture zones of Hong Kong as our research areas to investigate the diversity and distribution of anammox bacteria in the sediments based on the analyses of 16S rRNA and hydrazine oxidoreductase (HZO)-encoding genes. Moreover, we also investigated the influence of environmental factors on the diversity distribution of anammox bacteria in these marine aquaculture zones.
Materials and methods
Sample collection and chemical analysis
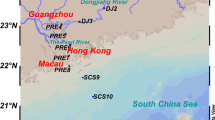
Four different marine fish aquaculture zones of Hong Kong were selected as research area, including Cheung Sha Wan (CSW, 22°14′28.1″N 114°00′24.6″E), Sok Kwu Wan (SKW, 22°12′35.6″N 114°08′00.6″E), Sham Wan (SW, 22°26′55.2″N 114°20′53.0″E), and Yim Tin Tsai East (YTTE, 22°26′54.0″N 114°13′06.2″E) (Fig. 1). The CSW and SKW are located at the southern part of Hong Kong, while SW and YTTE are near the northeastern part of Hong Kong. Triplicate surface sediments (1–2 cm) were collected from these marine aquaculture zones in July 2009. Samples were immediately transferred into a −20 °C freezer after being taken and kept frozen before analysis. The concentrations of phosphate, nitrate, nitrite, and ammonium in the pore water of the sediment samples were measured with an autoanalyzer (QuickChem, Milwaukee, WI) according to standard methods of the American Public Health Association (APHA 1995). Redox and pH were measured in situ using an IQ180G Bluetooth Multi-Parameter System (Hach Company, Loveland, CO). The organic matter content of each sediment sample was quantified by the loss on ignition based on the method reported by Heiri et al. (2001).

A map showing the selected sampling locations of the marine aquaculture zone around Hong Kong
DNA extraction and PCR amplification
The total genomic DNA of all the sediment samples was extracted using the Powersoil DNA Isolation Kit (MO BIO, Carlsbad, CA) according to the manual of the manufacturer. Triplicate DNA extracts of each sampling site were pooled together for further analysis. PCR amplifications of anammox bacterial 16S rRNA genes were made with primer sets (Broad541F-Amx820R, Amx368F-Amx820R) (Schmid et al. 2003, Schmid et al. 2005, Penton et al. 2006, Li et al. 2010b, Han and Gu 2013, Han et al. 2013) and the hzo genes were amplified by the primer set HZOF1-HZOR1 (H4) (Li et al. 2010b), and the details of the PCR conditions for 16S rRNA and hzo genes were described elsewhere (Li et al. 2010b). In brief, reaction mixtures contained 2 μl of DNA (30–50 ng μl−1), 1 μl of bovine serum albumin (100 mg ml−1, Roche), 5 μl of 10× GoTaq Flexi buffer (Promega, Hong Kong) and 4 μl of MgCl2 (25 mM, Promega), 1 μl of dNTPs (5 mM, Invitrogen, Hong Kong), 1 μl of each forward and reverse primer (20 μM), and 0.25 μl of GoTaq Flexi polymerase (5 U ml−1, Promega, Hong Kong), and the final volume for each reaction was 50 μl. The thermocycling regimes were set as follows: 95 °C for 3 min; 30 cycles of 95 °C for 45 s, 60 °C (for 16S rRNA gene), or 53 °C (for hzo gene) for 1 min, followed by 72 °C for 1 min; and finally 72 °C for 7 min. PCR products were electrophoresed on 1 % agarose gels and visualized by subsequent staining with ethidium bromide (0.5 mg ml−1).
Cloning, sequencing, and phylogenetic analysis
Clone libraries were constructed from the PCR products as described previously (Li et al. 2009). Briefly, the PCR-amplified products were purified using a Qiaex II Gel Extraction Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and cloned into the PMD18 T-vector (Takara, Japan). The insertion of an appropriate-sized DNA fragment was verified by PCR amplification using the primer set M13F and M13R. Clones were selected from each library for sequencing after RFLP analysis (Hong et al. 2011). Sequencing was performed with the Big Dye Terminator kit (Applied Sciences, Foster City, CA) and an ABI Prism 3730 DNA analyzer. Sequences were analyzed and edited by MEGA software (Tamura et al. 2007). Before alignment, sequences were inspected manually or with the CHIMERA_CHECK program available from RDP for the presence of chimeric sequences (Cole et al. 2005). Sequences were aligned and phylogenetic trees were constructed using MEGA, version 4.0 (Tamura et al. 2007), and subjected to phylogenetic inference using the neighbor-joining algorithm followed by 1000 cycles of bootstrap sampling.
Statistical analysis
Sequences were analyzed by the distance-based OTU and richness (DOTUR) program (Schloss and Handelsman 2005). Operational taxonomic units (OTUs) for community analysis were defined by a 1 % variation in nucleotide and amino acid sequences for 16S rRNA gene and hzo genes, respectively (Hong et al. 2011). The Shannon and Chaol were also generated by DOTUR for each clone library. The geographic distribution of phylogenetic structure anammox bacteria in four marine aquaculture zones were investigated by the online software UniFrac (http://bmf2.colorado.edu/unifrac/index.psp) using the principal coordinates analysis (PCoA) and Jackknife environmental clusters as suggested previously (Lozupone et al. 2006). Canonical correspondence analysis (CCA) was performed in CANOCO 4.5 for windows to identify the relationships between two bacterial community structures and environmental parameters (ter Braak and Smilauer 2002).
Nucleotide sequence accession numbers
The GenBank accession numbers for the 16S rRNA gene sequences reported here are KX092470–KX092642, and the GenBank accession numbers for the hzo gene sequences are KX146969–KX147061.
Results
Physicochemical characteristics of sediment samples
The redox potential and ammonium concentrations showed a reverse variable trend in the four marine aquaculture zone sediments (Table 1). Furthermore, sediment collected from SW and YTTE had significantly higher organic matter contents than that of CSW and SKW. It could also be found that the sediments collected from SW had the highest pH value and ammonium concentration but the lowest redox potential and phosphate concentration, indicating a location with specific habitat characteristics of these four samples (Table 1).
Anammox bacteria diversity by 16S rRNA gene
Anammox bacteria communities in surface sediments of marine aquaculture zones were detected successfully with 16S rRNA gene. From the four 16S rRNA gene clone libraries, a total of 173 clones were selected for sequencing after the RFLP analysis. Using 1 % sequence variation cut-off, 15 and 4 OTUs of anammox bacteria were obtained from CSW and SKW, respectively (Table 2). However, only eight sequences from YTTE were affiliated to the known anammox bacteria, representing two OTUs at the same sequence cutoff. More interestingly, all sequences obtained from SW with the same PCR primer sets were not related to any known anammox bacteria closely, showing a low similarity (83–86 %) with the available anammox bacterial 16S rRNA gene sequences (Table 2). Even though the sequencing clone numbers increased to more than two times than that of the other sites, we still could not detect any anammox bacterial-related 16S rRNA genes by selected PCR primers. Results indicated that CSW has the highest diversity of anammox bacteria, followed by SKW and YTTE; no anammox bacterial 16S rRNA genes were detected from SW sediments (Table 2).
Phylogenetic analysis showed that these obtained 16S rRNA sequences were clearly divided into two groups, one closely related to the known anammox bacteria and another affiliated with uncultured Planctomycetes (Fig. 2). Figure 2a showed the phylogenetic relationships of anammox bacteria obtained from sediments of the marine aquaculture zones in this study. All anammox bacterial 16S rRNA sequences were divided into six different clusters, including five Scalindua-related clusters and one Kuenenia-related cluster. Sequences obtained from CSW were widely distributed into five Scalindua-related clusters, representing Brade cluster, Arabica cluster, Zhenghei II cluster, Wagneri cluster, and one new cluster specific to the marine aquaculture zone of this study, but sequences of SKW were closely related to Brade cluster, Zhenghei II cluster, and the new cluster. For YTTE site, not only four sequences were detected to relate to the Zhenghei II cluster but also another four sequences were affiliated with Kuenenia cluster (Fig. 2A).

Phylogenetic relationship between the of 16S rRNA gene sequences obtained from different marine aquaculture zones in Hong Kong and representatives of anammox bacteria (a) and other Planctomycetes bacteria (b). Bootstrap values represent 1000 replicates and only values above 50 % are shown. Branch lengths correspond to sequence differences as indicated by the scale bar. Numbers in parenthesis refer to the number of clones were analyzed
As mentioned above, many sequences detected from the two sites, SW (100 % of sequences) and YTTE (75 %), showed low similarities with any available anammox bacterial 16S rRNA gene sequences but are closely related to some uncultured Planctomycetes clones recovered from sediments of Jiaozhou Bay aquaculture zone (JN090949), Marmara sea (AM980569), tidal flat (JN010128) and mangrove (GQ331353), biofilms of biofilter (DQ664528), and seawater column of Black Sea (DQ368318), respectively (Fig. 2B). These sequences formed a subgroup between anammox and other representative Planctomycetes species, such as Pirellula staleyi (AF399914), Planctomyces maris (NR025327), and Isophaera sp. (FJ542905), representing new Planctomycetes subgroups (Fig. 2B).
Anammox bacteria diversity by hzo
In contrast to anammox bacterial 16S rRNA gene, all retrieved hzo gene sequences from the four marine aquaculture zones were closely related to anammox bacteria (Table 2). Using 1 % amino acid variation as cutoff, 5 to 12 OTUs could be obtained from each site. The SKW had the highest hzo gene diversity, while YTTE was the lowest (Table 2).
Phylogenetic analysis showed that all obtained hzo gene sequences from marine aquaculture zones were clearly divided into five subclusters, including four Scalindua-like subclusters and one Kuenenia-like subcluster (Fig. 3). From the phylogenetic tree, all hzo gene sequences recovered from CSW, SKW, and YTTE belonged to the four Scalindua-like subclusters, relating to the clones detected from Candidatus “Scalindua sp.” enrichments (CAQ57909), uncultured Planctomycetes (CAQ57913), and Mai Po costal wetland sediments (ACN61640 and ACN61646), respectively. However, sequences retrieved from site SW were more closely related to the hzo genes of Candidatus “Kuenenia stuttgartiensis” than of other described anammox bacteria or sequences from the other three marine aquaculture zones, forming a novel and habitat-specific group of SW (Fig. 3).

Phylogenetic relationship of anammox bacteria based on the deduced amino acids from hzo gene sequences in this study and representative database sequences. Bootstrap values represent 1000 replicates and only values above 50 % are shown. Branch lengths correspond to sequence differences as indicated by the scale bar. Numbers in parenthesis refer to the number of clones were analyzed
Classification of anammox bacteria
To understand the classification anammox bacterial community structures in the four marine aquaculture zones, UniFrac analysis was performed based on the 16S rRNA and hzo gene sequence phylogenetic contexts. From the results of the PCoA and Jackknife environmental clusters, sites CSW and SKW shared the similar microbial community structure while the community structure at YTTE and SW was relatively site specific, especially the community structure of SW was totally different from others (Fig. 4).

UniFrac PCoA (a) and environmental cluster (b) for anammox bacterial 16S rRNA (1) and hzo (2) genes
Spatial distribution of anammox bacteria and their correlations with environmental factors
The CCA of 16S rRNA and hzo genes were used to identify the influence of environmental factors, such as nutrient concentrations, pH, redox, and organic matter contents, on the anammox bacterial diversity distribution in four marine aquaculture zones. Similar with the community classification, the CCA axes of anammox bacterial 16S rRNA and hzo genes also clearly distinguished the anammox bacterial assemblage of SW from those of the three marine aquaculture zones (Fig. 5). In the 16S rRNA gene CCA plot, organic matter contents was the most important factor to influence the bacteria-environment relationship, while nitrate and nitrite played more important role in the bacteria-environment relationship (Fig. 5). Furthermore, CCA results indicated that the spatial distribution of most of Scalindua-related sequences of 16S rRNA and hzo genes were related to selective environmental factors of marine aquaculture zone. For example, redox potential and concentration of phosphate and nitrite were positively related to all Scalindua-related clusters in 16S rRNA and hzo genes except the Zhenghei cluster in 16S rRNA gene analysis, while the organic matter contents of the sediments were positively correlated with Kuenenia-related groups in both 16S rRNA and hzo genes.

CCA ordination plot for the physicochemical parameters, anammox bacteria groups (triangle) representing by 16S rRNA (a) and hzo (b) sequences and their sampling locations (small circle). Correlations between environmental variables and CCA axes are represented by the length and angle of arrows (environmental factors)
Discussion
In the current study, the diversity of anammox bacteria was comprehensively described by 16S rRNA and hzo genes in four different marine aquaculture zones in Hong Kong. Except the SW site, both 16S rRNA and hzo gene diversity analyses indicate that the major anammox bacteria in the sediments of marine aquaculture zones were Scalindua-related species although Kuenenia-like anammox bacteria could also be detected from one of the four selected sampling sites. The results of the current study showed quite different anammox bacteria community structures as carried out in previous studies at both marine and freshwater recirculating aquaculture systems where the detected anammox bacteria were closely related to Brocadia (Tal et al. 2006) and Kuenenia (van Kessel et al. 2010). The differences of anammox bacteria community structures of the present study and the previous ones are possibly due to the difference in aquaculture systems, where previous reports were related to the recirculating aquaculture of freshwater and marine systems (Tal et al. 2006, van Kessel et al. 2010, van Kessel et al. 2011, Castine et al. 2012). However, some results of the current ones were quite similar to those of previous studies (Egli et al. 2001, Tal et al. 2006, Lahav et al. 2009, van Kessel et al. 2010), like the novel Planctomycete-like bacterial 16S rRNA gene sequences that were detected in two marine aquaculture zones with relatively high diversity, indicating that these new Planctomycete-like bacteria are very common in aquaculture systems. These unknown bacterial sequences share 83–86 % sequence identity to known anammox members, but their activity and ecophysiology still need to be confirmed in future studies. Furthermore, from the results of 16S rRNA and hzo genes in the four marine aquaculture zone sediments, we obtained a very consistent phylogenetic relationship of anammox bacteria from sites CSW and SKW, indicating that the detected anammox bacteria were Scalindua-related groups. However, inconsistent phylogenetic relationships were also found at sites SW and YTTE, which was similar to our previous studies (Li et al. 2010b, Li et al. 2011b), indicating the different converge and specificity of two molecular biomarkers for anammox bacteria detection from environmental samples.
Both the UniFrac environmental clustering and PCoA analyses revealed considerable heterogeneity of the sediment anammox bacterial assemblages in four marine aquaculture zones of Hong Kong (Fig. 4). The SCW and SKW sites share a similar anammox bacterial community structure, while the SW and YTTE have distinct anammox bacterial assemblages, especially the SW site. The analyses of both biomarkers 16S rRNA and hzo genes indicate that the anammox bacterial community of site SW was distinctively different from those of the others. CCA analyses indicate that the environmental factors including ammonium and pH might contribute significantly to the uniqueness of the anammox bacterial assemblage at site SW, where the sediments have the highest ammonium concentration and the lowest pH value. Furthermore, the CCA analyses also indicate that some environmental factors might contribute significantly to the diversity distribution of anammox bacteria in marine aquaculture zones. For example, the organic matter contents of the sediments positively correlate to the Kuenenia-like anammox bacteria, while redox, nitrite, and phosphate also positively relate to Scalindua-like anammox bacteria. In previous studies, the organic matter contents, redox, and nitrite concentration of the sediments were identified as the key environmental factors to regulate the sediment anammox bacterial composition, community structure, abundance, distribution, or potential activity in the coastal and marine sediments, such as Jiaozhou Bay (Dang et al. 2010), mangrove (Li et al. 2011c), Pearl River estuary (Cao et al. 2011, Li et al. 2011a), and South China Sea deep-sea subsurface area (Hong et al. 2011). Finally, we found that the phosphate might also play an important role to shape the community structure of anammox bacteria. Although the exact metabolism for these environmental factors influencing the dynamics of anammox bacterial assemblages is still not very clear, the results of the present study provide a clear description of anammox bacteria diversity and distribution in the marine aquaculture zone, and the results of this study also further confirm the previous hypothesis that anthropogenic activities may have an impact on the coastal anammox microbiota and activities (Dang et al. 2010, Li et al. 2010b, Cao et al. 2011, Li et al. 2011a).
References
Amano T, Yoshinaga I, Yamagishi T, Thuoc CV, Thu PT, Ueda S, Kato K, Sako Y, Suwa Y (2011) Contribution of anammox bacteria to benthic nitrogen cycling in a mangrove forest and shrimp ponds, Haiphong, Vietnam. Microbes Environ 26:1–6
APHA (1995) Standard methods for the examination of water and wastewater. APHA, Washington D.C
Avnimelech Y (1999) Carbon nitrogen ratio as a control element in aquaculture systems. Aquaculture 176:227–235
Byrne N, Strous M, Crepeau V, Kartal B, Birrien JL, Schmid M, Lesongeur F, Schouten S, Jaeschke A, Jetten M, Prieur D, Godfroy A (2009) Presence and activity of anaerobic ammonium-oxidizing bacteria at deep-sea hydrothermal vents. ISME J 3:117–123
Cao H, Li M, Dang H, Gu JD (2011) Responses of aerobic and anaerobic ammonia/ammonium-oxidizing microorganisms to anthropogenic pollution in coastal marine environments. Methods Enzymol 496:35–62
Castine SA, Erler DV, Trott LA, Paul NA, de Nys R, Eyre BD (2012) Denitrification and anammox in tropical aquaculture settlement ponds: an isotope tracer approach for evaluating N2 production. PLoS One 7:e42810
Castro-Gonzalez M, Molina V, Rodriguez-Rubio E, Ulloa O (2014) The first report of a microdiverse anammox bacteria community in waters of Colombian Pacific, a transition area between prominent oxygen minimum zones of the eastern tropical Pacific. Environ Microbiol Rep 6:595–604
Chan, H.W., H. Meng, and J.-D. Gu (2016) Anamox bacteria detected in fish intestinal tract systems. Appl Environ Biotechnol 1. doi:10.18063/AEB.2016.01.010
Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM (2005) The ribosomal database project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res 33:D294–D296
Crab R, Avnimelech Y, Defoirdt T, Bossier P, Verstraete W (2007) Nitrogen removal techniques in aquaculture for a sustainable production. Aquaculture 270:1–14
Dalsgaard T, Canfield DE, Petersen J, Thamdrup B, Acuna-Gonzalez J (2003) N2 production by the anammox reaction in the anoxic water column of Golfo Dulce, Costa Rica. Nature 422:606–608
Dang H, Chen R, Wang L, Guo L, Chen P, Tang Z, Tian F, Li S, Klotz MG (2010) Environmental factors shape sediment anammox bacterial communities in hypernutrified Jiaozhou Bay, China. Appl Environ Microbiol 76:7036–7047
Devol AH (2003) Nitrogen cycle: solution to a marine mystery. Nature 422:575–576
Egli K, Fanger U, Alvarez PJ, Siegrist H, van der Meer JR, Zehnder AJ (2001) Enrichment and characterization of an anammox bacterium from a rotating biological contactor treating ammonium-rich leachate. Arch Microbiol 175:198–207
FAO (2004) State of world fisheries and aquaculture. FAO, Rome
Hamersley MR, Lavik G, Woebken D, Rattray JE, Lam P, Hopmans EC, Damste JSS, Kruger S, Graco M, Gutierrez D, Kuypers MMM (2007) Anaerobic ammonium oxidation in the Peruvian oxygen minimum zone. Limnol Oceanogr 52:923–933
Han P, Gu JD (2013) A newly designed degenerate PCR primer based on pmoA gene for detection of nitrite-dependent anaerobic methane-oxidizing bacteria from different ecological niches. Appl Microbiol Biotechnol 97:10155–10162
Han P, Huang YT, Lin JG, Gu JD (2013) A comparison of two 16S rRNA gene-based PCR primer sets in unraveling anammox bacteria from different environmental samples. Appl Microbiol Biotechnol 97:10521–10529
Hargreaves JA (1998) Nitrogen biogeochemistry of aquaculture ponds. Aquaculture 166:181–212
Heiri O, Lotter AF, Lemcke G (2001) Loss on ignition as a method for estimating organic and carbonate content in sediments: reproducibility and comparability of results. J Paleolimnol 25:101–110
Holl CM, Glazer CT, Moss SM (2011) Nitrogen stable isotopes in recirculating aquaculture for super-intensive shrimp production: tracing the effects of water filtration on microbial nitrogen cycling. Aquaculture 311:146–154
Hong YG, Li M, Cao H, Gu JD (2011) Residence of habitat-specific anammox bacteria in the deep-sea subsurface sediments of the South China Sea: analyses of marker gene abundance with physical chemical parameters. Microb Ecol 62:36–47
Humbert S, Tarnawski S, Fromin N, Mallet MP, Aragno M, Zopfi J (2010) Molecular detection of anammox bacteria in terrestrial ecosystems: distribution and diversity. ISME J 4:450–454
Jaeschke A, Op den Camp HJ, Harhangi H, Klimiuk A, Hopmans EC, Jetten MS, Schouten S, Sinninghe Damste JS (2009) 16S rRNA gene and lipid biomarker evidence for anaerobic ammonium-oxidizing bacteria (anammox) in California and Nevada hot springs. FEMS Microbiol Ecol 67:343–350
Kuypers MM, Lavik G, Thamdrup B (2005a) Anaerobic ammonium oxidation in the marine environment. In: Neretin LN (ed) Past and present water column anoxia. Springer, Netherlands
Kuypers MM, Lavik G, Woebken D, Schmid M, Fuchs BM, Amann R, Jorgensen BB, Jetten MS (2005b) Massive nitrogen loss from the Benguela upwelling system through anaerobic ammonium oxidation. Proc Natl Acad Sci U S A 102:6478–6483
Kuypers MM, Sliekers AO, Lavik G, Schmid M, Jorgensen BB, Kuenen JG, Sinninghe Damste JS, Strous M, Jetten MS (2003) Anaerobic ammonium oxidation by anammox bacteria in the Black Sea. Nature 422:608–611
Lahav O, Bar Massada I, Yackoubov D, Zelikson R, Mozes N, Tal Y, Tarre S (2009) Quantification of anammox activity in a denitrification reactor for a recirculating aquaculture system. Aquaculture 288:76–82
Lam P, Lavik G, Jensen MM, van de Vossenberg J, Schmid M, Woebken D, Gutierrez D, Amann R, Jetten MS, Kuypers MM (2009) Revising the nitrogen cycle in the Peruvian oxygen minimum zone. Proc Natl Acad Sci U S A 106:4752–4757
Lee KH, Wang YF, Zhang GX, Gu JD (2014) Distribution patterns of ammonia-oxidizing bacteria and anammox bacteria in the freshwater marsh of Honghe wetland in Northeast China. Ecotoxicology 23:1930–1942
Li H, Chen S, Mu BZ, Gu JD (2010a) Molecular detection of anaerobic ammonium-oxidizing (anammox) bacteria in high-temperature petroleum reservoirs. Microb Ecol 60:771–783
Li M, Cao H, Hong YG, Gu JD (2011a) Seasonal dynamics of anammox bacteria in estuarial sediment of the Mai Po nature reserve revealed by analyzing the 16S rRNA and hydrazine oxidoreductase (hzo) genes. Microbes Environ 26:15–22
Li M, Ford T, Li X, Gu JD (2011b) Cytochrome cd1-containing nitrite Reductase encoding Gene nirS as a new functional biomarker for detection of anaerobic ammonium oxidizing (anammox) bacteria. Environ Sci Technol 45:3547–3553
Li M, Gu JD (2013) Community structure and transcript responses of anammox bacteria, AOA, and AOB in mangrove sediment microcosms amended with ammonium and nitrite. Appl Microbiol Biotechnol 97:9859–9874
Li, M., and J.-D. Gu (2016) Molecular evidence of the existence of anaerobic ammonia oxidation bacteria in the gut of Polychaete (Neanthes glandicincta). Appl Environ Biotechnol 1. doi:10.18063/AEB.2016.01.011
Li M, Hong Y, Klotz MG, Gu JD (2010b) A comparison of primer sets for detecting 16S rRNA and hydrazine oxidoreductase genes of anaerobic ammonium-oxidizing bacteria in marine sediments. Appl Microbiol Biotechnol 86:781–790
Li M, Hong YG, Cao H, Gu JD (2011c) Mangrove trees affect the community structure and distribution of anammox bacteria at an anthropogenic-polluted mangrove in the Pearl River Delta reflected by 16S rRNA and hzo genes analyses. Ecotoxicology 20:1780–1790
Li M, Yang H, Gu JD (2009) Phylogenetic diversity and axial distribution of microbes in the intestinal tract of the polychaete Neanthes glandicincta. Microb Ecol 58:892–902
Lozupone C, Hamady M, Knight R (2006) UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinf 7:371
Martins CIM, Eding EH, Verdegem MCJ, Heinsbroek LTN, Schneider O, Blancheton JP, d’Orbcastel ER, Verreth JAJ (2010) New developments in recirculating aquaculture systems in Europe: a perspective on environmental sustainability. Aquac Eng 43:83–93
Mulder A, Vandegraaf AA, Robertson LA, Kuenen JG (1995) Anaerobic ammonium oxidation discovered in a denitrifying fluidized-bed reactor. FEMS Microbiol Ecol 16:177–183
Penton CR, Devol AH, Tiedje JM (2006) Molecular evidence for the broad distribution of anaerobic ammonium-oxidizing bacteria in freshwater and marine sediments. Appl Environ Microbiol 72:6829–6832
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Schmid M, Walsh K, Webb R, Rijpstra WI, van de Pas-Schoonen K, Verbruggen MJ, Hill T, Moffett B, Fuerst J, Schouten S, Damste JS, Harris J, Shaw P, Jetten M, Strous M (2003) Candidatus “Scalindua brodae”, sp. nov., Candidatus “Scalindua wagneri”, sp. nov., two new species of anaerobic ammonium oxidizing bacteria. Syst Appl Microbiol 26:529–538
Schmid MC, Maas B, Dapena A, van de Pas-Schoonen K, van de Vossenberg J, Kartal B, van Niftrik L, Schmidt I, Cirpus I, Kuenen JG, Wagner M, Sinninghe Damste JS, Kuypers M, Revsbech NP, Mendez R, Jetten MS, Strous M (2005) Biomarkers for in situ detection of anaerobic ammonium-oxidizing (anammox) bacteria. Appl Environ Microbiol 71:1677–1684
Schubert CJ, Durisch-Kaiser E, Wehrli B, Thamdrup B, Lam P, Kuypers MM (2006) Anaerobic ammonium oxidation in a tropical freshwater system (Lake Tanganyika). Environ Microbiol 8:1857–1863
Smith RL, Bohlke JK, Song B, Tobias CR (2015) Role of anaerobic ammonium oxidation (anammox) in nitrogen removal from a freshwater aquifer. Environ Sci Technol 49:12169–12177
Strous M, Kuenen JG, Jetten MS (1999) Key physiology of anaerobic ammonium oxidation. Appl Environ Microbiol 65:3248–3250
Tal Y, Watts JE, Schreier HJ (2006) Anaerobic ammonium-oxidizing (anammox) bacteria and associated activity in fixed-film biofilters of a marine recirculating aquaculture system. Appl Environ Microbiol 72:2896–2904
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
ter Braak, C., and P. Smilauer. 2002. CANOCO Reference Manual and CanoDraw for Windows User’s Guide: Software for Canonical Community Ordination (Version 4.5). Microcomputer Power, Ithaca, NY, USA.
Thamdrup B, Dalsgaard T (2002) Production of N(2) through anaerobic ammonium oxidation coupled to nitrate reduction in marine sediments. Appl Environ Microbiol 68:1312–1318
van de Graaf AA, Mulder A, de Bruijn P, Jetten MS, Robertson LA, Kuenen JG (1995) Anaerobic oxidation of ammonium is a biologically mediated process. Appl Environ Microbiol 61:1246–1251
van Kessel MA, Harhangi HR, Flik G, Jetten MS, Klaren PH, Op den Camp HJ (2011) Anammox bacteria in different compartments of recirculating aquaculture systems. Biochem Soc Trans 39:1817–1821
van Kessel MAHJ, Harhangi HR, van de Pas-Schoonen K, van de Vossenberg J, Flik G, Jetten MSM, Klaren PHM, den Camp HJMO (2010) Biodiversity of N-cycle bacteria in nitrogen removing moving bed biofilters for freshwater recirculating aquaculture systems. Aquaculture 306:177–184
Wang J, Gu JD (2013) Dominance of Candidatus Scalindua species in anammox community revealed in soils with different duration of rice paddy cultivation in Northeast China. Appl Microbiol Biotechnol 97:1785–1798
Ward BB, Devol AH, Rich JJ, Chang BX, Bulow SE, Naik H, Pratihary A, Jayakumar A (2009) Denitrification as the dominant nitrogen loss process in the Arabian Sea. Nature 461:78–81
Zhang Y, Ruan XH, Op den Camp HJ, Smits TJ, Jetten MS, Schmid MC (2007) Diversity and abundance of aerobic and anaerobic ammonium-oxidizing bacteria in freshwater sediments of the Xinyi River (China). Environ Microbiol 9:2375–2382
Zhu G, Xia C, Shanyun W, Zhou L, Liu L, Zhao S (2015) Occurrence, activity and contribution of anammox in some freshwater extreme environments. Environ Microbiol Rep 7:961–969
Acknowledgments
We thank Dr. Zhenye Zhao, Mr. Kuwan and Ms. Jessie Lai for sample collection and laboratory assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This project was supported by the National Natural Science Foundation of China (grant no. 41506163), Natural Science Foundation of Guangdong Province (grant no. 2014A030310056), Shenzhen City (grant nos. JCY20140828163633985 and KQCX2015032416053646) and SZU (grant no. 000066) (M. L), and RGC GRF grant no. 701913 (J-D. G).
Conflict of interests
The authors declare that they have no conflict of interest.
Human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Li, M., Gu, JD. The diversity and distribution of anammox bacteria in the marine aquaculture zones. Appl Microbiol Biotechnol 100, 8943–8953 (2016). https://doi.org/10.1007/s00253-016-7690-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7690-6