
Abstract
The recent focus on the cystic fibrosis (CF) complex microbiome has led to the recognition that the microbes can interact between them and with the host immune system, affecting the disease progression and treatment routes. Although the main focus remains on the interactions between traditional pathogens, growing evidence supports the contribution and the role of emergent species. Understanding the mechanisms and the biological effects involved in polymicrobial interactions may be the key to improve effective therapies and also to define new strategies for disease control. This review focuses on the interactions between microbe–microbe and host–microbe, from an ecological point of view, discussing their impact on CF disease progression. There are increasing indications that these interactions impact the success of antimicrobial therapy. Consequently, a new approach where therapy is personalized to patients by taking into account their individual CF microbiome is suggested.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cystic fibrosis (CF) is a common lethal disease affecting nearly 70,000 people around the world. It is characterized by the build-up of thick mucus overlying lung epithelial cells, wherein persistent cycles of chronic infection and inflammation occur (Gibson et al. 2003; Goss and Burns 2007). The CF airways provide microorganisms with heterogeneous microenvironments containing varying levels of oxygen, pH, nutrients, and antibiotics. This heterogeneity contributes largely for the proliferation of a phylogenetically diverse ecosystem, influencing the consortia of microbes able to occupy it (Yang et al. 2011a).
A complex microbiome has been previously described in the context of CF (e.g., Lopes et al. (2014a)). This microbiome encompasses species that are believed to be clinically significant and species thought to be bystanders, i.e., microorganisms for which no direct evidence exists to support their impact in the disease. Pseudomonas aeruginosa is recognized as the most significant and the most commonly isolated pathogen in CF infections, worsening CF pulmonary status due to chronic infections and being responsible for higher fatality rates (Winstanley and Fothergill 2009). In addition, a small number of other pathogens, such as Staphylococcus aureus, Haemophilus influenzae, and the Burkholderia cepacia complex, have been also documented as having repercussions in disease progression (Alexander and Hudson 2001; Lambert 2002; Lyczak et al. 2002; Yang et al. 2006; Starner et al. 2006; Treggiari et al. 2007; Drevinek and Mahenthiralingam 2010; Kahl 2010; Hauser et al. 2011; Høiby et al. 2011; Huang and Lynch 2011). Novel molecular technologies have more recently detected and identified a diverse microbial community inhabiting CF lungs (Inquilinus limosus and Dolosigranulum pigrum, etc.) of unexplored relevance in CF disease (Coenye et al. 2002; Bittar et al. 2008).
The multispecies microbiome composition in CF is constantly shaped by selective pressures exerted by the niche characteristics at sites of infection. It is increasingly recognized that the properties of such communities may be distinct from those of their individual members, due in large part to interspecies interactions shaping behavior (Faust et al. 2012; Korgaonkar et al. 2013). This may, in part, explain the lack of response to conventional therapeutic regimens that primarily target single causative agents instead of all members in the community.
Nonetheless, the precise ways under which the many different organisms interact within the CF airways and how these interactions influence the behavior of the individual species, the activities of the polymicrobial communities, and the relationship between host and microbes are poorly understood questions. Some studies have highlighted the potentially important roles of such interspecies interactions in disease phenotype and clinical outcome of CF infections (e.g., Amin et al. 2010; Chattoraj et al. 2010; Bragonzi et al. 2012; Twomey et al. 2012; Lopes et al. 2012). Such studies suggest that both synergistic and antagonistic interactions in mixed-species infections can impact microbial virulence and antibiotic resistance, which most likely will have clinical effects on disease severity and responses to therapy. Consequently, it is pertinent that therapeutic/prophylactic strategies should not be limited, and/or focused only, on the major pathogenic members that are able to directly cause disease but also on polymicrobial communities that involve complex interactions among members of the CF microbiota.
In a recently published review (Lopes et al. 2014a). we described the diversity of the CF microbiome and provided a few examples of possible interactions between different microorganisms. In here, we consider the collective microbiome as a potential pathogenic entity in itself, describe in detail the social behavior within CF communities, highlighting the interactions established among microbes and between microbes and their host in the context of CF and analyzing whether a particular community causes or worsens disease, in a manner analogous to individual pathogens. We conclude that the relationship between a microbial community and disease is better understood from an ecological perspective and can improve clinical understanding, ultimately providing guidelines for an effective treatment and chronic infection suppression.
Relevant aspects of CF—pathogenesis, reduced-oxygen environment, and microbial colonization
CF is a human genetic disorder that results from mutations in the CF transmembrane conductance regulator (CFTR) gene. The most prevalent of those mutations (∆F508) is the deletion of three nucleotides at the position 508 of the CFTR protein sequence (Lopes et al. 2014a). The CFTR protein acts as a channel for the chloride and sodium ions transport across the cell membranes. Therefore, a dysfunctional CFTR protein leads to the absence or a decreased chloride secretion, resulting in an intracellular accumulation of those ions and ultimately to the depletion of chloride, sodium, and water from the airway lumen. This causes abnormal thick and viscous secretions and impairs mucociliary clearance in CF airways (Rowe et al. 2005; Davis 2006; Farrell et al. 2008).
The clinical manifestations of CF are quite variable, affecting individuals throughout their entire life. CF-affected individuals typically have a lifespan of approximately 30–40 years (Castellani et al. 2008). It is well established that the greatest contributor to the morbidity and/or mortality is the failure in lung function that generally occurs in older patients, caused by the build-up of mucus that clogs the airways and leads to persistent colonization by different microorganisms (frequently bacterial species). Hence, recurrent cycles of infections and inflammation lead to progressive airway and lung damage, respiratory failure and eventually death (Fig. 1) (Nixon et al. 2001; De Boeck et al. 2006; Boucher 2007).

Mechanism of the CF pulmonary disease. In the lungs, the defective chloride ion transport results in the decrease of the volume of the periciliary fluid, compromising the mucociliary clearance and triggering the overproduction of dehydrated and viscous mucus. This leads to the persistent colonization of bacteria in the lungs, and the physiologic consequences are persistent inflammatory responses, obstructive lung physiology, respiratory insufficiency, which ultimately results in death from chronic respiratory failure. Adapted from Kirkby et al. (2011)
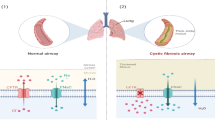
The existence of steep oxygen gradients within the CF airway mucus is well-known, with zones ranging from aerobic (in the top layers) to completely anaerobic (deeper mucus layers) (Fig. 2 steps 1–3) (Worlitzsch et al. 2002).

a Alterations in mucus of normal epithelial airway cells (Step 1 to 3): (Step 1) On a normal airway epithelia, a thin mucus layer (yellow) resides on top of the PCL (clear). The presence of the low-viscosity PCL facilitates efficient mucociliary clearance (denoted by black arrow). A normal rate of epithelial O2 consumption (Qo2; left) produces no O2 gradients within this thin ASL (denoted by the red bar). (Step 2) Excessive CF volume depletion (denoted by vertical arrows) removes the PCL, mucus becomes adherent to epithelial surfaces, and mucus transport slows/stops (bidirectional black arrow). The raised O2 consumption (left) associated with accelerated CF ion transport does not generate gradients in thin films of ASL. (Step 3) Persistent mucus hypersecretion (denoted as mucus secretory gland; gray) with time increases the height of luminal mucus masses/plugs. The raised CF epithelial Qo2 generates steep hypoxic gradients (blue color in bar) in thickened mucus masses. b Schematic model for P. aeruginosa biofilm in the CF mucus (Step 4 to 6): (Step 4) P. aeruginosa are deposited on the thickened mucus surfaces and can penetrate the mucus actively (e.g., by inhalation, flagellum- or pili-dependent motility) and/or passively (due to mucus turbulence) into the CF mucus. (Step 5) Afterward, P. aeruginosa start to develop bacterial aggregates (the biofilms), which are protected by an alginate capsule. In this step, the consumption of O2 is drastically increased by the bacterial cells, and hypoxic and/or anaerobic pockets are formed. (Step 6) In the final stages, where O2 is almost depleted, the bacterial aggregates become highly resistant to the neutrophils and antibiotics, setting the stage for persistent chronic infection (Worlitzsch et al. 2002; Hassett et al. 2002; Boucher 2004)
Typically, the airway epithelial cells have a thin and hydrated mucus layer, located on top of the periciliary liquid layer (PCL), which enables an efficient mucociliary clearance (Worlitzsch et al. 2002; Hassett et al. 2002; Boucher 2004). A normal rate of epithelial O2 consumption (Qo2) produces no O2 gradients within the thin airway surface liquid (ASL). In CF, the airway epithelium absorbs the sodium (Na+) and chloride (Cl−) ions and water from the lumen, depletes the PCL and slow down or even stop the mucus transport. The increased O2 consumption associated with accelerated CF ion transport does not generate gradients in the thin biofilm of ASL, but the persistent mucus hyper secretion leads to the production of luminal mucus plugs, hence increasing the mucus layer on the epithelial cells and generating steep oxygen gradients, with zones ranging from aerobic (generally located at the top) and microaerobic and/or even completely anaerobic (located in the deeper layers) (Worlitzsch et al. 2002; Hassett et al. 2002; Boucher 2004).
Patients suffering from CF are prone to develop severe biofilm-related infections that are thought to contribute greatly to the emergence and dissemination of antibiotic resistance (Høiby et al. 2010a). The biofilm formation represents a protective mode of growth that allows microorganisms to survive in hostile environments and disperse by seeding cells to colonize new niches under desirable conditions (Wei and Ma 2013). P. aeruginosa persists in the CF airways due to its ability to form biofilms, being considered the key CF pathogen (Hassett et al. 2010). P. aeruginosa presents a notorious ability to develop resilient biofilms in the form of “bacterial aggregates” within the CF mucus (Fig. 2, steps 4–6) (Worlitzsch et al. 2002; Hassett et al. 2002; Boucher 2004; Wei and Ma 2013). The persistence of chronic P. aeruginosa lung infections in CF patients is due to biofilm-growing mucoid strains, protected by alginate overproduction (Høiby et al. 2010b). The persistence of these biofilms into the CF airway mucus often leads to a high tolerance to many antibiotics (Borriello et al. 2004). Conventional resistance mechanisms, such as the presence of a chromosomal β-lactamase, upregulated efflux pumps, and mutations of antibiotic target molecules in the bacteria, have also contributed to the adaptation of P. aeruginosa biofilms to the CF environment (Høiby et al. 2010b).
Although P. aeruginosa prefers oxygen respiration as the highest energy-yielding process for growth, it can survive in the mucus anaerobic zones (Hassett et al. 2002). The ability of this bacterium to adapt to the oxygen-limited environments is associated with a drastic physiological change in P. aeruginosa (e.g., increased alginate production; alterations in the outer membrane; biofilm development), which contributes to an increased antibiotic tolerance (Schobert and Jahn 2010). The alginate produced by the biofilm bacteria in CF lung infections also provides a physical barrier to host defense systems (Worlitzsch et al. 2002; Hassett et al. 2002; Boucher 2004).
The complex CF microbiome
Traditional CF microbiology
As stated above, the infections in the CF airway are frequently polymicrobial (Rogers et al. 2004; Sibley et al. 2006; Bittar and Rolain 2010). The CF airways offer a favorable environment for the colonization and proliferation of a large variety of organisms, including as bacteria, fungi, and viruses, with bacterial species being the ones that are more frequently isolated (Guss et al. 2011).
Traditionally, the detection and identification of microbial species has relied on culture-based studies, using sputum or bronchial alveolar lavage samples for microbial detection and identification (Price et al. 2013). These techniques allow identification of several key microbial species that contribute to CF lung infection and disease progression, beginning early in life with S. aureus and H. influenzae and culminating in chronic infections caused by P. aeruginosa or B. cepacia complex species (Table 1) (Razvi et al. 2009; Price et al. 2013).
S. aureus, one of the first pathogens isolated from CF samples, is the most prevalent pathogen in children and adolescents; however, 40 % of adult patients still remain colonized (Kahl 2010). S. aureus has the ability to cause chronic infection (Alexander and Hudson 2001; Kahl 2010; Hauser et al. 2011). H. influenzae is also involved in chronic lung infections in CF pediatric patients, forming structures consistent with biofilms even before the onset of clinical signs or symptoms of lung disease (Starner et al. 2006). B. cepacia complex is a group of 18 Burkholderia species infecting 2 to 8 % of patients with CF, with some of them (B. cenocepacia, B. multivorans, B. cepacia, and B. dolosa) being highly transmissible, presenting pathogenic potential and very high resistance to antibiotic therapy (Yang et al. 2006; Lynch 2009; Drevinek and Mahenthiralingam 2010).
Approximately 50 % of CF patients are colonized with P. aeruginosa (Government 2013). which remains the most common pathogen isolated from CF sputum, being more prevalent in adults (Folkesson et al. 2012). The presence of P. aeruginosa in CF airways is highly associated with poor lung function, morbidity, and mortality of patients. After colonization with P. aeruginosa, consecutive episodes of recolonization frequently occur, resulting in a chronic infection that can persist for years or even never being eradicated in CF patient lungs (Sousa and Pereira 2014).
Emergent CF microbiology
In addition to the bacterial species documented as CF pathogens, recent molecular methodologies have documented complex microbial ecosystems in CF samples, with a wide array of uncommon microorganisms coexisting with traditional pathogens acting collectively to facilitate disease progression (Peters et al. 2012). Figure 3 discloses all the genera of microorganisms recovered so far from the respiratory tracts of patients with CF. The analysis of this figure highlight that almost 120 genera were recovered from CF airways, with a clear dominance of bacterial genera and only 7 genera of viruses detected. These microorganisms include bacteria (e.g., I. limosus, D. pigrum, Stenotrophomonas maltophilia), fungi (e.g., Aspergillus fumigatus, Candida albicans), and viruses (e.g., rhinovirus, adenovirus, influenza). In addition, Worlitzsch and colleagues (2009). identified in a cross-sectional study of 15 genera of obligate anaerobes in 91 % of patients suffering from CF. Tunney and colleagues (2008) also reported anaerobic species within the genera Prevotella, Veillonella, Propionibacterium, and Actinomyces, which were isolated in high numbers (>64 %) in sputum samples from patients in adulthood. The high numbers of anaerobic bacteria detected in the CF airways may be a result of oxygen consumption by aerobic pathogens (such as P. aeruginosa) that often colonize the airways, creating a favorable niche for the proliferation of anaerobes (Worlitzsch et al. 2002; Yoon et al. 2002). Using molecular methods (16S rRNA gene clone libraries and pyrosequencing), Guss and colleagues (2011) have identified, only in 4 CF sputum samples, more than 60 bacterial genera, including facultative and obligate anaerobes, oral bacteria, and opportunistic pathogens, many of which have never before been found in the CF lung. Bittar and colleagues (2008) identified 53 different bacterial species from 25 sputum samples. Additionally, standard microbiological culture and phenotypic identification of bacteria in sputum from CF patients have been compared to molecular methods by the use of 16S rDNA amplification, cloning, and sequencing. Twenty-five sputa from CF patients were cultured yielding 33 isolates (13 species) known to be pathogens during CF. For molecular cloning, 760 clones were sequenced (7.2 ± 3.9 species/sputum), and 53 different bacterial species were identified including 16 species of anaerobes (30 %). These results indicate that the traditional culture methods are insufficient to describe the polymicrobial populations actually present in the CF lung. A recent review provides a comprehensive understanding of the great complexity of the microbiome existing in CF, detected and/or identified employing recent molecular methodologies (Lopes et al. 2014a).

Genus of microorganisms identified in respiratory tracts of patients with CF (lung image adapted from http://lungdiseasenews.com)
Although the role of some of these emergent microorganisms in the pathogenesis of the disease and their clinical relevance remain unclear, there are already some hints about the implication of some unusual species in the pathophysiology of CF (Caraher et al. 2008; Ulrich et al. 2010; Costello et al. 2011; Sherrard et al. 2014; O’Neill et al. 2015; Pustelny et al. 2015; Benedyk et al. 2015). Further studies of this complex niche by, e.g., metagenomic analysis (Bittar et al. 2008; Price et al. 2013; Lim et al. 2013; Hauser et al. 2014; Lim et al. 2014) are currently needed to better understand these microbial communities, their implication in treatment and antibiotic resistance, their role in the development of chronic respiratory infections, and to identify their clinical significance in order to find new therapeutic targets.
Ecological perspective of the CF microbiome
Microbial interactions might exist within CF polymicrobial communities, so it is not surprising that these infections are increasingly viewed as complex communities of interacting organisms, with dynamic processes key to their pathogenicity. Similarly to the relative contribution to clinical status, disease progression and efficacy of antibiotic therapy by newly identified members of a polymicrobial community, which remain to be fully explored, the know-how on the consequences of the interplay among potential pathogens and/or between them and their eukaryotic host is also pivotal for understanding and treat CF-associated infections. These interactions can be mediated by a large number of mechanisms, which encompasses interspecies signaling, metabolite exchange, and cell–cell contact and are often implicated in the modulation of microbial behavior, ultimately contributing to disease progression and clinical outcome. In addition, many types of infections are caused by biofilm-associated microorganisms (Burmølle et al. 2010). which are harder to eradicate compared with planktonic exponentially growing cells, due to several factors operating concurrently (e.g., changed structure and reduced diffusion rates of the compounds in the biofilm matrix, changed gene expression pattern, and low growth rates of the biofilm-encased cells) (Sousa and Pereira 2014).This protective effect may be further enhanced if multiple species are present within the biofilm, where the dynamics between the resident species may potentially evolve and change the volume and function of the whole biofilm both qualitatively and quantitatively (Burmølle et al. 2014). In these consortia, microorganisms frequently communicate via quorum sensing (QS) complex systems, which play an important role in the social behavior, regulation of microbial population density, and expression of virulence factors (e.g., resistance genes and proteins) among members of a microbial community (Rutherford and Bassler 2012).
Although particular microbial communities may be associated with certain clinical outcomes, the heterogeneous nature of the airway environment (nutrients, as well as physiochemical characteristics, such as oxygen tension, temperature, and pH) will influence the mix of microbes that are able to occupy it, through exerting selective pressures. In addition, it is increasingly recognized that the microbes can alter the characteristics of the niche in which they grow, by influencing the behavior of other colonizing species (such as pathogenicity (Sibley et al. 2008)), or by directly interacting with the eukaryotic host (e.g., by damaging airway epithelia (King 2011) or triggering inflammation (Essilfie et al. 2012)), as well as the impact of changes in antibiotic treatment that follow clinical worsening, such as the type and intensity of antibiotic exposure (Rogers et al. 2013). For instance, Tunney et al. (2011) have reported that substantial shifts in bacterial abundance within the microbial community can be detected following antibiotic therapy. However, Stressmann and colleagues (2012) showed that antibiotic therapy can temporarily perturb these communities, which tend to return to their pretreatment configuration following cessation of antibiotics.
The pressures affecting microbiome composition are dynamic, and the comprehensive understanding of the drives of microbial community stability is fundamental for predicting the way in which a microbiome will respond to perturbation. Microbial activity will influence the processes that select for subsequent members of the microbiome; therefore, the infection by one species can indirectly dictate microbiome composition (Rogers et al. 2013).
Hence, it becomes imperative to understand the molecular basis and the biological effect of those interplay processes within multispecies communities to help improve clinical understanding and the in-use treatment regimens, devising new targets and disease control strategies.
An extensive research in recent literature has identified studies reporting interactions among microbes and between microbes and their host in the context of CF, which is summarized in Table 2.
The interactions described within Table 2 are divided into two different categories, synergism and antagonism. Contrariwise to synergistic interactions, which represent mutual benefit to all species present, antagonistic interactions result in a negative effect for at least one species. As it is possible to observe, microorganisms can use simultaneously different mechanisms to interact with other species, which may be associated with the niche characteristics and selective pressures exerted that shaped the behavior and the way in which the species interact.
The majority of the studies found in the literature (Table 2) are carried out under in vitro conditions so that the effect of interaction in the host is only predictive. Although the predictive effects for most microbe–host interactions (most of them carried out in vivo) are considered negative, some mechanisms involve interactions that can have a predictable positive effect on the host and thereby be used as a therapeutic approach. Similarly, molecules that block key signal sensing or transduction steps in pathogens could represent lead compounds for new drugs.
In any polymicrobial infection, the combined effect of two or more microbes on the disease progression can be more dramatic than any of the individuals alone and can display enhanced pathogen persistence in the infection site, increased disease severity, and increased antimicrobial resistance in a phenomenon known as polymicrobial synergism (Dalton et al. 2011; Peters et al. 2012; Murray et al. 2014). Synergistic interactions between different bacterial species allow reaping benefits that would be unattainable to them as individual cells, such as increased antibiotic tolerance, biofilm development, defense against competitors, adaptation to changing environments, increased tissue damage, and declined pulmonary infection (Jacques et al. 1998; Duan et al. 2003; Dalton et al. 2011). As examples of synergistic interactions occur in the CF context, several authors (Pilkington et al. 2011; Bragonzi et al. 2012) have demonstrated that a higher number of cells in the biofilm can be produced, which may have a great impact in antibiotic tolerance.
However, in some cases, the antagonistic interactions between organisms within a community are unavoidable due to competition for finite resources, with effects on the growth or viability of competitors (Harrison 2007). In CF, these interactions were found, for example, between P. aeruginosa and the fungal species A. fumigatus and C. albicans, with the small diffusive molecules secreted by P. aeruginosa inhibiting the biofilm formation of those fungal populations (Hogan et al. 2004; Cugini et al. 2007; Mowat et al. 2010). and between B. cenocepacia and C. albicans with QS signal produced by B. cenocepacia inhibiting the filament formation by C. albicans (Boon et al. 2008). Bacteria produce many types of diffusible molecules that can interact with other bacteria during disease. The various chemical cell-to-cell signaling mechanisms that are used by bacteria are collectively known as QS systems (Fuqua et al. 1994). a bacterial cell-to-cell communication process that involves the production, detection, and response to extracellular signaling molecules called autoinducers (AIs) (Rutherford and Bassler 2012). Some signal molecules such as autoinducer-2 (AI-2), Pseudomonas Quinolone Signal (PQS), 2-heptyl-4-hydroxy quinoline N-oxide (HQNO), and signal molecules of the diffusible signal factor (DSF) have been found to be produced during the infection and to influence other bacteria. For example, the ability to stimulate S. aureus biofilm formation was strongly associated to the production of HQNO and PQS by P. aeruginosa isolates (Fugère et al. 2014).
While some studies have revealed the interplay among typical CF bacteria (Hoffman et al. 2006). only few have reported the role of emergent species on lung disease chronicity (Lopes et al. 2014a, 2014b) or the interactions between those atypical microorganisms with eminent pathogens (Lopes et al. 2012). mainly when the micorganisms are encased in biofilms. Recently, Lopes et al. (2012) showed that the CF atypical bacteria I. limosus and D. pigrum could interact synergistically with P. aeruginosa, developing dual-species consortia with increased tolerance to several antibiotics. This suggests that previously thought clinically insignificant species may influence the behavior of individual species or even the whole microbial community. Based on these ecological interactions, it is strongly suggested to have a focus shift from an individual species to a polymicrobial community management and that modeling such multispecies interactions will help to predict the effects of new therapeutic interventions, dismissing much of the current antibiotic therapy empiricism and increasing its effectiveness.
In addition to microbial–microbial interactions, microbial–host interactions also exist in CF, and the most significant features is the ability of the pathogens to deceive or modulate the multifaceted host response following colonization. The airway epithelium recognizes and responds to pathogens through the interaction between host pathogen recognition receptors and pathogen-associated membrane proteins (Callaghan and McClean 2012). The airway epithelium is one of several sources of chemokine interleukin-8 (IL-8) (Standiford et al. 1990) that acts as the first line of host defense against pathogens. In CF patients, the ΔF508-CFTR mutation results in increased levels of IL-8 and neutrophils, responsible for the development of chronic obstructive and inflammatory lung diseases (Bodas and Vij 2010). Furthermore, neutrophils resulting in DNA release and increased mucous viscosity worsen the problem of bacterial attachment (Callaghan and McClean 2012). Recent studies have demonstrated that the conventional pathogens P. aeruginosa and B. cenocepacia can trigger an excessive inflammatory response in the host (Kotrange et al. 2011; Pilkington et al. 2011; Lorè et al. 2012; Kim et al. 2014). Deregulation of matrix metalloproteases (MMPs) in CF is another contributor to CF lung disease and to bacterial colonization (Pilkington et al. 2011). So, while many of the modifications and adaptations serve to promote inflammation and to benefit the colonization, other strategies are used to avoid and minimize the host response (Patankar et al. 2013). Additionally, colonization by multiple pathogens may trigger unknown repercussions in the host, although it is suspected that for most cases, adverse effects can occur with greater impact in antibiotic therapy.
The majority of studies about interactions in the polymicrobial CF community focus on the traditional pathogen P. aeruginosa, due to its prevalence in CF lung, its ability to form biofilms that protect the organism to the host responses and to numerous antibiotics, and its potential do develop chronic infection. Therefore, more research is needed to provide a better mechanistic insight into the complex interplay between potential pathogenic agents, commensal organisms, and the host response in the polymicrobial infections.
Understanding polymicrobial interactions to better treat CF
The resistance to antimicrobial agents is currently one of the major problems in the healthcare setting worldwide (French 2010). Antimicrobial resistance is potentiated in CF patients due to the extensive use of antimicrobial agents from a young age, both for the prophylaxis and treatment of respiratory infection (Oliver 2010).
When the chronic infection is established, pathogens such as P. aeruginosa growing as biofilms in the CF lung can exhibit increased resistance to antibiotics (Hassett et al. 2009; Bjarnsholt et al. 2009; Høiby et al. 2011). In fact, bacteria in the form of biofilms show increased resistance to several antibiotics when compared to planktonic or free living counterparts (Sriramulu et al. 2005). The minimal inhibitory concentration (MIC) and minimal bactericidal concentration (MBC) of antibiotics to biofilm-growing bacteria may be up to 100–1000-fold higher compared with planktonic bacteria (Anwar et al. 1990; Moskowitz et al. 2004).
Apart from the conventional resistance mechanisms presented by bacteria (e.g., chromosomal beta-lactamase, upregulated efflux pumps, and mutations in antibiotic target molecules), biofilms also present an extracellular polymeric matrix. The reduced diffusion of antibiotics through the exopolysaccharide matrix (alginate, in the case of P. aeruginosa biofilm) retards the movement of antimicrobial agents (Costerton 2001; Bagge et al. 2004; Chan et al. 2005; Anderson and O’Toole 2008; Vettoretti et al. 2009) and contributes for the resistance and/or tolerance of biofilms to the antimicrobial agents (Høiby et al. 2010a; Sriramulu 2013).
When CF was first described in 1938 (Andersen 1938). the predicted survival age of patients was only 6 months (Cohen-Cymberknoh et al. 2011). For patients born in the 1990s, median survival is now predicted to exceed 40 years, due to the introduction of multiple therapies that treat the symptoms of CF (Wilschanski 2013).
Antibiotic therapy for CF patients is directed at preventing, eradicating, or controlling respiratory infections. The therapy generally starts with oral and inhaled therapies in an outpatient setting and the use of intravenous route for patients with severe exacerbations (McCaughey et al. 2012; Sriramulu 2013).
The fluoroquinolones (e.g., ciprofloxacin) are the most commonly used oral agents to treat acute exacerbations caused by P. aeruginosa infection (Sriramulu 2013). Other agents that have long been used by inhalation in CF patient for the treatment of P. aeruginosa lung infection are tobramycin, aztreonam, or colistin (Sriramulu 2013). Current standard care guidelines for antibiotic recommend in CF patients for most commonly bacterial species are described in Table 3.
Recently, new antibiotic combinations have been developed (MacLeod et al. 2009; McCaughey et al. 2012; McCaughey et al. 2013; Anderson et al. 2013). One example is the combination of fosfomycin/tobramycin (FTI), an inhaled antibiotic with broad-spectrum antibacterial activity for treatment of bacterial respiratory infections. FTI consists of fosfomycin (F) and tobramycin (T) in a 4:1 weight-to-weight ratio (w/w); this combination has promising activity against MRSA and P. aeruginosa with greater activity under aerobic and physiologically relevant anaerobic conditions, compared to F or T alone (MacLeod et al. 2009; McCaughey et al. 2012; McCaughey et al. 2013; Anderson et al. 2013). Lam and colleagues (2013) reported that tobramycin inhalation powder (TIP) represents the first dry powder inhaled antibiotic available for use in CF. TIP was approved in the USA in 2013 (Fiel 2014). Inhaled antibiotics have been probably the safest and most effective therapy for P. aeruginosa chronic lung infection in CF patients (Máiz et al. 2013). The use of inhaled antibiotics allows it to be delivered directly to the target area, with a lower dose than more conventional oral or intravenous delivery methods, with reduced systemic absorption and consequently reduced risk of toxic effects (Traini and Young 2009; Hoppentocht et al. 2014).
With the increased antibiotic resistance in CF patients, the need for new strategies in the lifelong treatment of pulmonary infection has to be validated (van Westreenen and Tiddens 2010). In a cross-sectional study, the detection of 2,3-butanedione in the breath gases of CF patients indicated the presence of Streptococcus spp. (Whiteson et al. 2014). Linking together products that are unique to microbial metabolism with the genes detected by metagenomic sequences of microbial communities in sputum may enable development of biomarkers for early detection of exacerbations.
Lim and colleagues (2014) combined the use of metagenomic sequencing and clinical microbiology for monitoring polymicrobial infections in individual patients. Their findings highlighted that information on the predicted resistance of the whole microbial community is perhaps one of the most useful pieces of information extracted from metagenome sequencing, which will be important to understand how quickly antibiotic resistance might change in the microbial community. The implementation of metagenomic analysis as a clinical diagnostic tool can give rise to vital information for clinicians to prescribe the appropriate antibiotic therapy.
Because CF infection is no longer viewed as being caused by a single pathogen, antibiotics used to target a small group of species recognized as key CF pathogens are generally ineffective when other atypical species are present (Lopes et al. 2012, 2014b) or fail in many cases (Leekha et al. 2011). This problem is compounded by the huge polymicrobial CF community and the bacterial interactions occurring in lung. Due to the complex interactions that result between traditional and emergent CF pathogens—for instance, a study by Lopes and colleagues (2012) demonstrated that the association among atypical and conventional CF bacteria could result in the impact of the antibiotic resistance—a new approach where antibiotic therapy is personalized to each patient, based on comprehensive microbiological analyses, could be development for treating lung infections (Short et al. 2014). There is an increasing appreciation of the polymicrobial nature of many bacterial infections such as those associated with CF and of the potentially important role for interspecies interactions in influencing both bacterial virulence and response to therapy, as previously discussed in the earlier section. Twomey and colleagues (2012) demonstrated that antibiotic resistance of P. aeruginosa biofilms was enhanced in the presence of diffusible signal molecules, produced by Stenotrophomas maltophilia and B. cenocepacia. On the other hand, a recent study showed that P. aeruginosa might be responsible for the protection of S. maltophilia against tobramycin (Pompilio et al. 2015).
Antivirulence drugs are a new type of therapeutic drug that target virulence factors, without killing or inhibiting bacterial growth. Many antivirulence strategies are being explored, including inhibiting bacterial adhesion to the host cell (inhibiting biofilm formation), inhibiting cell-to-cell signaling (known as quorum quenchers by inhibiting QS systems), and interfering with gene regulation of virulence traits (Rasko and Sperandio 2010; Rutherford and Bassler 2012; Allen et al. 2014). Other innovative therapeutic approach is the development of CFTR-modulating drug as potential treatment for cystic fibrosis. Ivacaftor is the first licensed CFTR modulator drug and, although only targets ∼5 % of CF patients, may indeed be one of many therapeutic agents that point to the emergence of a new era of personalized medicine (Ramsey et al. 2011). These drugs will allow treatment of the basic defect in CF disease and open the door for therapy according to gene sequencing—true personalized medicine (Wilschanski 2013). Moreover, every person with CF is unique and requires personalized diagnosis and therapy.
In addition to recognizing the polymicrobial nature of CF community, understanding the molecular mechanisms and biological effects from the microbe–microbe and host–microbe interactions is also crucial to improve therapy regimens and also define new antimicrobial agents, new targets and strategies for CF disease control. We are facing a postantibiotic era with limited capability to combat polymicrobial infections.
Conclusions
In the past decades, technological advances in diagnostic tools have led to the recognition that many infections, including CF, are far more complex than originally believed. It has become apparent that, although therapies are focused on the treatment of the dominant microbial species of an infection, other microbes may have a profound significance on both the response to antibiotic therapy and virulence. While microbes–microbes and microbes–host interactions are not fully understood, it is suspected that the consequences of such interplay carry out synergistic and antagonistic effects either for CF treatment or for antibiotic resistance, ultimately contributing to disease progress and clinical outcome.
Accordingly, the challenge is now to explore multispecies biofilms in further detail, by examining their physiology, function, and underlying mechanisms but specifically enhancing the focus for microbial–microbial and/or microbial–host interactions in these communities. Understanding the physical and chemical interactions between microorganisms in these polymicrobial communities will help to define potential new targets for disrupting biofilm-community development and, in cystic fibrosis, affect the ecology of biofilms in the airways of patients. Since several pathogens employ QS regulation to express a specific broad range of virulence factors, this has spurred interest in QS inhibitors as antivirulence drugs.
The medical community is now starting to recognize the significance of polymicrobial diseases and the major types of microbial community interactions associated with human health and disease. Many traditional therapies are just starting to take into account the polymicrobial cause of diseases and the repercussions of treatment and prevention. By taking into account the complexity of infecting organisms and their interplay, it is intended to develop a new approach where therapy is personalized to each patient or to a group of patients. If successful, the approach may pave the way for more effective therapeutic regimens and ultimately contribute to personalized treatment for these diseases, based on unique microbial profile of a given patient, and further extrapolate for analogous polymicrobial infections.
References
Alexander EH, Hudson MC (2001) Factors influencing the internalization of Staphylococcus aureus and impacts on the course of infections in humans. Appl Microbiol Biotechnol 56:361–366
Allen RC, Popat R, Diggle SP, Brown SP (2014) Targeting virulence: can we make evolution-proof drugs? Nat Rev Microbiol 12:300–308. doi:10.1038/nrmicro3232
Amin R, Dupuis A, Aaron SD, Ratjen F (2010) The effect of chronic infection with Aspergillus fumigatus on lung function and hospitalization in patients with cystic fibrosis. Chest 137:171–176. doi:10.1378/chest.09-1103
Andersen DH (1938) Cystic fibrosis of the pancreas and its relation to celiac disease. Am J Dis Child 56:344. doi:10.1001/archpedi.1938.01980140114013
Anderson GG, Kenney TF, Macleod DL, Henig NR, O’Toole GA (2013) Eradication of Pseudomonas aeruginosa biofilms on cultured airway cells by a fosfomycin/tobramycin antibiotic combination. Pathog Dis 67:39–45. doi:10.1111/2049-632X.12015
Anderson GG, O’Toole GA (2008) Innate and induced resistance mechanisms of bacterial biofilms. Curr Top Microbiol Immunol 322:85–105
Anwar H, Dasgupta MK, Costerton JW (1990) Testing the susceptibility of bacteria in biofilms to antibacterial agents. Antimicrob Agents Chemother 34:2043–2046
Bagge N, Schuster M, Hentzer M, Ciofu O, Givskov M, Greenberg EP, Høiby N (2004) Pseudomonas aeruginosa biofilms exposed to imipenem exhibit changes in global gene expression and beta-lactamase and alginate production. Antimicrob Agents Chemother 48:1175–1187
Bakare N, Rickerts V, Bargon J, Just-Nübling G (2003) Prevalence of Aspergillus fumigatus and other fungal species in the sputum of adult patients with cystic fibrosis. Mycoses 46:19–23
Baldan R, Cigana C, Testa F, Bianconi I, De Simone M, Pellin D, Di Serio C, Bragonzi A, Cirillo DM (2014) Adaptation of Pseudomonas aeruginosa in cystic fibrosis airways influences virulence of Staphylococcus aureus in vitro and murine models of co-infection. PLoS One. doi:10.1371/journal.pone.0089614
Barton RC, Borman AM, Johnson EM, Houbraken J, Hobson RP, Denton M, Conway SP, Brownlee KG, Peckham D, Lee TWR (2010) Isolation of the fungus Geosmithia argillacea in sputum of people with cystic fibrosis. J Clin Microbiol 48:2615–2617. doi:10.1128/JCM.00184-10
Ben Dekhil SM, Peel MM, Lennox VA, Stackebrandt E, Sly LI (1997) Isolation of Lautropia mirabilis from sputa of a cystic fibrosis patient. J Clin Microbiol 35:1024–1026
Benedyk M, Byrne DP, Glowczyk I, Potempa J, Olczak M, Olczak T, Smalley JW (2015) Pyocycanin, a contributory factor in haem acquisition and virulence enhancement of Porphyromonas gingivalis in the lung. PLoS One 10:e0118319. doi:10.1371/journal.pone.0118319
Bittar F, Richet H, Dubus J-C, Reynaud-Gaubert M, Stremler N, Sarles J, Raoult D, Rolain J-M (2008) Molecular detection of multiple emerging pathogens in sputa from cystic fibrosis patients. PLoS One 3:e2908. doi:10.1371/journal.pone.0002908
Bittar F, Rolain J-M (2010) Detection and accurate identification of new or emerging bacteria in cystic fibrosis patients. Clin Microbiol Infect 16:809–820. doi:10.1111/j.1469-0691.2010.03236.x
Bjarnsholt T, Jensen PØ, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, Pressler T, Givskov M, Høiby N (2009) Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr Pulmonol 44:547–558. doi:10.1002/ppul.21011
Bodas M, Vij N (2010) The NF-kappaB signaling in cystic fibrosis lung disease: pathophysiology and therapeutic potential. Discov Med 9:346–356
Boon C, Deng Y, Wang L-H, He Y, Xu J-L, Fan Y, Pan SQ, Zhang L-H (2008) A novel DSF-like signal from Burkholderia cenocepacia interferes with Candida albicans morphological transition. Isme j 2:27–36. doi:10.1038/ismej.2007.76
Borriello G, Werner E, Roe F, Kim AM, Ehrlich GD, Stewart PS (2004) Oxygen limitation contributes to antibiotic tolerance of Pseudomonas aeruginosa in biofilms. Antimicrob Agents Chemother 48:2659–2664. doi:10.1128/AAC.48.7.2659-2664.2004
Boucher RC (2007) Evidence for airway surface dehydration as the initiating event in CF airway disease. J Intern Med 261:5–16. doi:10.1111/j.1365-2796.2006.01744.x
Boucher RC (2004) New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J 23:146–158
Bragonzi A, Farulla I, Paroni M, Twomey KB, Pirone L, Lorè NI, Bianconi I, Dalmastri C, Ryan RP, Bevivino A (2012) Modelling co-infection of the cystic fibrosis lung by Pseudomonas aeruginosa and Burkholderia cenocepacia reveals influences on biofilm formation and host response. PLoS One. doi:10.1371/journal.pone.0052330
Burmølle M, Ren D, Bjarnsholt T, Sørensen SJ (2014) Interactions in multispecies biofilms: do they actually matter? Trends Microbiol 22:84–91. doi:10.1016/j.tim.2013.12.004
Burmølle M, Thomsen TR, Fazli M, Dige I, Christensen L, Homøe P, Tvede M, Nyvad B, Tolker-Nielsen T, Givskov M, Moser C, Kirketerp-Møller K, Johansen HK, Høiby N, Jensen PØ, Sørensen SJ, Bjarnsholt T (2010) Biofilms in chronic infections—a matter of opportunity—monospecies biofilms in multispecies infections. FEMS Immunol Med Microbiol 59:324–336. doi:10.1111/j.1574-695X.2010.00714.x
Burns JL, Emerson J, Stapp JR, Yim DL, Krzewinski J, Louden L, Ramsey BW, Clausen CR (1998) Microbiology of sputum from patients at cystic fibrosis centers in the United States. Clin Infect Dis 27:158–163
Butler WR, Sheils CA, Brown-Elliott BA, Charles N, Colin AA, Gant MJ, Goodill J, Hindman D, Toney SR, Wallace RJ, Yakrus MA (2007) First isolations of Segniliparus rugosus from patients with cystic fibrosis. J Clin Microbiol 45:3449–3452. doi:10.1128/JCM.00765-07
Callaghan M, McClean S (2012) Bacterial host interactions in cystic fibrosis. Curr Opin Microbiol 15:71–77. doi:10.1016/j.mib.2011.11.001
Caraher E, Collins J, Herbert G, Murphy PG, Gallagher CG, Crowe MJ, Callaghan M, McClean S (2008) Evaluation of in vitro virulence characteristics of the genus Pandoraea in lung epithelial cells. J Med Microbiol 57:15–20. doi:10.1099/jmm.0.47544-0
Castellani C, Cuppens H, Macek M, Cassiman JJ, Kerem E, Durie P, Tullis E, Assael BM, Bombieri C, Brown A, Casals T, Claustres M, Cutting GR, Dequeker E, Dodge J, Doull I, Farrell P, Ferec C, Girodon E, Johannesson M, Kerem B, Knowles M, Munck A, Pignatti PF, Radojkovic D, Rizzotti P, Schwarz M, Stuhrmann M, Tzetis M, Zielenski J, Elborn JS (2008) Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cyst Fibros 7:179–196. doi:10.1016/j.jcf.2008.03.009
Chan C, Burrows LL, Deber CM (2005) Alginate as an auxiliary bacterial membrane: binding of membrane-active peptides by polysaccharides. J Pept Res 65:343–351. doi:10.1111/j.1399-3011.2005.00217.x
Chattoraj SS, Murthy R, Ganesan S, Goldberg JB, Zhao Y, Hershenson MB, Sajjan US (2010) Pseudomonas aeruginosa alginate promotes Burkholderia cenocepacia persistence in cystic fibrosis transmembrane conductance regulator knockout mice. Infect Immun 78:984–993. doi:10.1128/IAI.01192-09
Cimon B, Carrere J, Chazalette JP, Vinatier JF, Chabasse D, Bouchara JP (1999) Chronic airway colonization by Penicillium emersonii in a patient with cystic fibrosis. Med Mycol 37:291–293
Cimon B, Challier S, Béguin H, Carrère J, Chabasse D, Bouchara J-P (2005) Airway colonization by Acrophialophora fusispora in patients with cystic fibrosis. J Clin Microbiol 43:1484–1487. doi:10.1128/JCM.43.3.1484-1487.2005
Coenye T, Goris J, Spilker T, Vandamme P, LiPuma JJ (2002) Characterization of unusual bacteria isolated from respiratory secretions of cystic fibrosis patients and description of Inquilinus limosus gen. nov., sp. nov. J Clin Microbiol 40:2062–2069
Cohen-Cymberknoh M, Shoseyov D, Kerem E (2011) Managing cystic fibrosis: strategies that increase life expectancy and improve quality of life. Am J Respir Crit Care Med 183:1463–1471. doi:10.1164/rccm.201009-1478CI
Costello A, Herbert G, Fabunmi L, Schaffer K, Kavanagh KA, Caraher EM, Callaghan M, McClean S (2011) Virulence of an emerging respiratory pathogen, genus Pandoraea, in vivo and its interactions with lung epithelial cells. J Med Microbiol 60:289–299. doi:10.1099/jmm.0.022657-0
Costerton JW (2001) Cystic fibrosis pathogenesis and the role of biofilms in persistent infection. Trends Microbiol 9:50–52
Cugini C, Calfee MW, Farrow JM, Morales DK, Pesci EC, Hogan DA (2007) Farnesol, a common sesquiterpene, inhibits PQS production in Pseudomonas aeruginosa. Mol Microbiol 65:896–906. doi:10.1111/j.1365-2958.2007.05840.x
Dalton T, Dowd SE, Wolcott RD, Sun Y, Watters C, Griswold JA, Rumbaugh KP (2011) An in vivo polymicrobial biofilm wound infection model to study interspecies interactions. PLoS One 6:e27317. doi:10.1371/journal.pone.0027317
Davis PB (2006) Cystic fibrosis since 1938. Am J Respir Crit Care Med 173:475–482. doi:10.1164/rccm.200505-840OE
De Boeck K, Wilschanski M, Castellani C, Taylor C, Cuppens H, Dodge J, Sinaasappel M (2006) Cystic fibrosis: terminology and diagnostic algorithms. Thorax 61:627–635. doi:10.1136/thx.2005.043539
Defontaine A, Zouhair R, Cimon B, Carrère J, Bailly E, Symoens F, Diouri M, Hallet J-N, Bouchara J-P (2002) Genotyping study of Scedosporium apiospermum isolates from patients with cystic fibrosis. J Clin Microbiol 40:2108–2114
Delhaes L, Monchy S, Fréalle E, Hubans C, Salleron J, Leroy S, Prevotat A, Wallet F, Wallaert B, Dei-Cas E, Sime-Ngando T, Chabé M, Viscogliosi E (2012) The airway microbiota in cystic fibrosis: a complex fungal and bacterial community–implications for therapeutic management. PLoS One 7:e36313. doi:10.1371/journal.pone.0036313
Döring G, Flume P, Heijerman H, Elborn JS (2012) Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J Cyst Fibros 11:461–479. doi:10.1016/j.jcf.2012.10.004
Drevinek P, Mahenthiralingam E (2010) Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin Microbiol Infect 16:821–830. doi:10.1111/j.1469-0691.2010.03237.x
Duan K, Dammel C, Stein J, Rabin H, Surette MG (2003) Modulation of Pseudomonas aeruginosa gene expression by host microflora through interspecies communication. Mol Microbiol 50:1477–1491. doi:10.1046/j.1365-2958.2003.03803.x
Essilfie A-T, Simpson JL, Dunkley ML, Morgan LC, Oliver BG, Gibson PG, Foster PS, Hansbro PM (2012) Combined Haemophilus influenzae respiratory infection and allergic airways disease drives chronic infection and features of neutrophilic asthma. Thorax 67:588–599. doi:10.1136/thoraxjnl-2011-200160
Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, Durie PR, Legrys VA, Massie J, Parad RB, Rock MJ, Campbell PW (2008) Guidelines for diagnosis of cystic fibrosis in newborns through older adults: cystic fibrosis foundation consensus report. J Pediatr 153:S4–S14. doi:10.1016/j.jpeds.2008.05.005
Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C (2012) Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 8:e1002606. doi:10.1371/journal.pcbi.1002606
Fiel SB (2014) Aerosolized antibiotics in cystic fibrosis: an update. Expert Rev Respir Med 8:305–314. doi:10.1586/17476348.2014.896205
Folkesson A, Jelsbak L, Yang L, Johansen HK, Ciofu O, Høiby N, Molin S (2012) Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat Rev Microbiol 10:841–851. doi:10.1038/nrmicro2907
French GL (2010) The continuing crisis in antibiotic resistance. Int J Antimicrob Agents 36(Suppl 3):S3–S7. doi:10.1016/S0924-8579(10)70003-0
Friman VP, Ghoul M, Molin S, Johansen HK, Buckling A (2013) Pseudomonas aeruginosa adaptation to lungs of cystic fibrosis patients leads to lowered resistance to phage and protist enemies. PLoS One 8:1–9. doi:10.1371/journal.pone.0075380
Fugère A, Séguin DL, Mitchell G, Déziel E, Dekimpe V, Cantin AM, Frost E, Malouin F (2014) Interspecific small molecule interactions between clinical isolates of Pseudomonas aeruginosa and Staphylococcus aureus from adult cystic fibrosis patients. PLoS One. doi:10.1371/journal.pone.0086705
Fuqua WC, Winans SC, Greenberg EP (1994) Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol 176:269–275
Gibson RL, Burns JL, Ramsey BW (2003) Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168:918–951. doi:10.1164/rccm.200304-505SO
Goss CH, Burns JL (2007) Exacerbations in cystic fibrosis. 1: epidemiology and pathogenesis. Thorax 62:360–367. doi:10.1136/thx.2006.060889
Government US a (2013) Annual Data Report.
Guss AM, Roeselers G, Newton ILG, Young CR, Klepac-Ceraj V, Lory S, Cavanaugh CM (2011) Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis. Isme j 5:20–29. doi:10.1038/ismej.2010.88
Harris JK, De Groote MA, Sagel SD, Zemanick ET, Kapsner R, Penvari C, Kaess H, Deterding RR, Accurso FJ, Pace NR (2007) Molecular identification of bacteria in bronchoalveolar lavage fluid from children with cystic fibrosis. Proc Natl Acad Sci U S A 104:20529–20533. doi:10.1073/pnas.0709804104
Harrison F (2007) Microbial ecology of the cystic fibrosis lung. Microbiology 153:917–923. doi:10.1099/mic.0.2006/004077-0
Hassett DJ, Cuppoletti J, Trapnell B, Lymar SV, Rowe JJ, Yoon SS, Hilliard GM, Parvatiyar K, Kamani MC, Wozniak DJ, Hwang SH, McDermott TR, Ochsner UA (2002) Anaerobic metabolism and quorum sensing by Pseudomonas aeruginosa biofilms in chronically infected cystic fibrosis airways: rethinking antibiotic treatment strategies and drug targets. Adv Drug Deliv Rev 54:1425–1443
Hassett DJ, Korfhagen TR, Irvin RT, Schurr MJ, Sauer K, Lau GW, Sutton MD, Yu H, Hoiby N (2010) Pseudomonas aeruginosa biofilm infections in cystic fibrosis: insights into pathogenic processes and treatment strategies. Expert Opin Ther Targets 14:117–130. doi:10.1517/14728220903454988
Hassett DJ, Sutton MD, Schurr MJ, Herr AB, Caldwell CC, Matu JO (2009) Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol 17:130–138. doi:10.1016/j.tim.2008.12.003
Hauser AR, Jain M, Bar-Meir M, McColley SA (2011) Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin Microbiol Rev 24:29–70. doi:10.1128/CMR.00036-10
Hauser PM, Bernard T, Greub G, Jaton K, Pagni M, Hafen GM (2014) Microbiota present in cystic fibrosis lungs as revealed by whole genome sequencing. PLoS One 9:e90934. doi:10.1371/journal.pone.0090934
Hickey PW, Sutton DA, Fothergill AW, Rinaldi MG, Wickes BL, Schmidt HJ, Walsh TJ (2009) Trichosporon mycotoxinivorans, a novel respiratory pathogen in patients with cystic fibrosis. J Clin Microbiol 47:3091–3097. doi:10.1128/JCM.00460-09
Hoffman LR, Déziel E, D’Argenio DA, Lépine F, Emerson J, McNamara S, Gibson RL, Ramsey BW, Miller SI (2006) Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 103:19890–19895. doi:10.1073/pnas.0606756104
Hogan DA, Vik Å, Kolter R (2004) A Pseudomonas aeruginosa quorum-sensing molecule influences Candida albicans morphology. Mol Microbiol 54:1212–1223. doi:10.1111/j.1365-2958.2004.04349.x
Høiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O (2010a) Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents 35:322–332. doi:10.1016/j.ijantimicag.2009.12.011
Høiby N, Ciofu O, Bjarnsholt T (2010b) Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol 5:1663–1674. doi:10.2217/fmb.10.125
Høiby N, Ciofu O, Johansen HK, Song Z, Moser C, Jensen PØ, Molin S, Givskov M, Tolker-Nielsen T, Bjarnsholt T (2011) The clinical impact of bacterial biofilms. Int J Oral Sci 3:55–65. doi:10.4248/IJOS11026
Holcombe LJ, McAlester G, Munro CA, Enjalbert B, AJP B, N. A. R G, Ding C, Butler G, O’Gara F, Morrissey JP (2010) Pseudomonas aeruginosa secreted factors impair biofilm development in Candida albicans. Microbiology 156:1476–1485. doi:10.1099/mic.0.037549-0
Hoppentocht M, Hagedoorn P, Frijlink HW, de Boer AH (2014) Developments and strategies for inhaled antibiotic drugs in tuberculosis therapy: a critical evaluation. Eur J Pharm Biopharm 86:23–30. doi:10.1016/j.ejpb.2013.10.019
Huang YJ, Lynch SV (2011) The emerging relationship between the airway microbiota and chronic respiratory disease: clinical implications. Expert Rev Respir Med 5:809–821. doi:10.1586/ers.11.76
Iaria M, Caccuri F, Apostoli P, Giagulli C, Pelucchi F, Padoan RF, Caruso A, Fiorentini S (2015) Detection of KI WU and Merkel cell polyomavirus in respiratory tract of cystic fibrosis patients. Clin Microbiol Infect 21:603.e9–603.e15. doi:10.1016/j.cmi.2015.01.025
Jacques I, Derelle J, Weber M, Vidailhet M (1998) Pulmonary evolution of cystic fibrosis patients colonized by Pseudomonas aeruginosa and/or Burkholderia cepacia. Eur J Pediatr 157:427–431
Kahl BC (2010) Impact of Staphylococcus aureus on the pathogenesis of chronic cystic fibrosis lung disease. Int J Med Microbiol 300:514–519. doi:10.1016/j.ijmm.2010.08.002
Kenney DMC, Brown KE, Allison DG (1995) Influence of Pseudomonas aeruginosa exoproducts on virulence factor production in Burkholderia cepacia : evidence of interspecies communication. Influ Pseudomonas aeruginosa Exoproducts Virulence Factor Prod Burkholderia Cepacia : Evid 177:6989–6992
Kidd TJ, Ramsay KA, Hu H, Bye PTP, Elkins MR, Grimwood K, Harbour C, Marks GB, Nissen MD, Robinson PJ, Rose BR, Sloots TP, Wainwright CE, Bell SC (2009) Low rates of Pseudomonas aeruginosa misidentification in isolates from cystic fibrosis patients. J Clin Microbiol 47:1503–1509. doi:10.1128/JCM.00014-09
Kim YJ, Paek SH, Jin S, Park BS, Ha UH (2014) A novel Pseudomonas aeruginosa-derived effector cooperates with flagella to mediate the upregulation of interleukin 8 in human epithelial cells. Microb Pathog 66:24–28. doi:10.1016/j.micpath.2013.12.001
King P (2011) Pathogenesis of bronchiectasis. Paediatr Respir Rev 12:104–110. doi:10.1016/j.prrv.2010.10.011
Kirkby S, Novak K, McCoy K (2011) Aztreonam (for inhalation solution) for the treatment of chronic lung infections in patients with cystic fibrosis: an evidence-based review. Core Evid 6:59–66. doi:10.2147/CE.S11181
Kolak M, Karpati F, Monstein H-J, Jonasson J (2003) Molecular typing of the bacterial flora in sputum of cystic fibrosis patients. Int J Med Microbiol 293:309–317. doi:10.1078/1438-4221-00265
Korgaonkar A, Trivedi U, Rumbaugh KP, Whiteley M (2013) Community surveillance enhances Pseudomonas aeruginosa virulence during polymicrobial infection. Proc Natl Acad Sci U S A 110:1059–1064. doi:10.1073/pnas.1214550110
Kotrange S, Kopp B, Akhter A, Abdelaziz D, Abu Khweek A, Caution K, Abdulrahman B, Wewers MD, McCoy K, Marsh C, Loutet SA, Ortega X, Valvano MA, Amer AO (2011) Burkholderia cenocepacia O polysaccharide chain contributes to caspase-1-dependent IL-1beta production in macrophages. J Leukoc Biol 89:481–488. doi:10.1189/jlb.0910513
Kusenbach G, Skopnik H, Haase G, Friedrichs F, Döhmen H (1992) Exophiala dermatitidis pneumonia in cystic fibrosis. Eur J Pediatr 151:344–346
Lam J, Vaughan S, Parkins MD (2013) Tobramycin inhalation powder (TIP): an efficient treatment strategy for the management of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Clin Med Insights Circ Respir Pulm Med 7:61–77. doi:10.4137/CCRPM.S10592
Lambert PA (2002) Mechanisms of antibiotic resistance in Pseudomonas aeruginosa. J R Soc Med 95(Suppl 4):22–26
Leão RS, Pereira RHV, Ferreira AG, Lima AN, Albano RM, Marques EA (2010) First report of Paenibacillus cineris from a patient with cystic fibrosis. Diagn Microbiol Infect Dis 66:101–103. doi:10.1016/j.diagmicrobio.2009.06.011
Leekha S, Terrell CL, Edson RS (2011) General principles of antimicrobial therapy. Mayo Clin Proc 86:156–167. doi:10.4065/mcp.2010.0639
Lim YW, Evangelista JS, Schmieder R, Bailey B, Haynes M, Furlan M, Maughan H, Edwards R, Rohwer F, Conrad D, Forbes BA (2014) Clinical insights from metagenomic analysis of sputum samples from patients with cystic fibrosis. J Clin Microbiol 52:425–437. doi:10.1128/JCM.02204-13
Lim YW, Schmieder R, Haynes M, Willner D, Furlan M, Youle M, Abbott K, Edwards R, Evangelista J, Conrad D, Rohwer F (2013) Metagenomics and metatranscriptomics: windows on CF-associated viral and microbial communities. J Cyst Fibros 12:154–164. doi:10.1016/j.jcf.2012.07.009
Lopes SP, Azevedo NF, Pereira MO (2014a) Microbiome in cystic fibrosis: shaping polymicrobial interactions for advances in antibiotic therapy. Crit Rev Microbiol. doi:10.3109/1040841X.2013.847898
Lopes SP, Azevedo NF, Pereira MO (2014b) Emergent bacteria in cystic fibrosis: in vitro biofilm formation and resilience under variable oxygen conditions. Biomed Res Int 2014:678301. doi:10.1155/2014/678301
Lopes SP, Ceri H, Azevedo NF, Pereira MO (2012) Antibiotic resistance of mixed biofilms in cystic fibrosis: impact of emerging microorganisms on treatment of infection. Int J Antimicrob Agents 40:260–263. doi:10.1016/j.ijantimicag.2012.04.020
Lorè NI, Cigana C, De Fino I, Riva C, Juhas M, Schwager S, Eberl L, Bragonzi A (2012) Cystic fibrosis-niche adaptation of Pseudomonas aeruginosa reduces virulence in multiple infection hosts. PLoS One 7:e35648. doi:10.1371/journal.pone.0035648
Lumb R, Greville H, Martin J, Sangster N, Holmes M (2002) Nocardia asteroides isolated from three patients with cystic fibrosis. Eur J Clin Microbiol Infect Dis 21:230–233. doi:10.1007/s10096-001-0687-8
Lyczak JB, Cannon CL, Pier GB (2002) Lung infections associated with cystic fibrosis. Clin Microbiol Rev 15:194–222
Lynch JP (2009) Burkholderia cepacia complex: impact on the cystic fibrosis lung lesion. Semin Respir Crit Care Med 30:596–610. doi:10.1055/s-0029-1238918
MacLeod DL, Barker LM, Sutherland JL, Moss SC, Gurgel JL, Kenney TF, Burns JL, Baker WR (2009) Antibacterial activities of a fosfomycin/tobramycin combination: a novel inhaled antibiotic for bronchiectasis. J Antimicrob Chemother 64:829–836. doi:10.1093/jac/dkp282
Máiz L, Girón RM, Olveira C, Quintana E, Lamas A, Pastor D, Cantón R, Mensa J (2013) Inhaled antibiotics for the treatment of chronic bronchopulmonary Pseudomonas aeruginosa infection in cystic fibrosis: systematic review of randomised controlled trials. Expert Opin Pharmacother 14:1135–1149. doi:10.1517/14656566.2013.790366
Matos T, Cerar T, Praprotnik M, Krivec U, Pirš M (2015) First recovery of Rasamsonia argillacea species complex isolated in adolescent patient with cystic fibrosis in Slovenia—case report and review of literature. Mycoses 58:506–510. doi:10.1111/myc.12340
McCaughey G, Diamond P, Elborn JS, McKevitt M, Tunney MM (2013) Resistance development of cystic fibrosis respiratory pathogens when exposed to fosfomycin and tobramycin alone and in combination under aerobic and anaerobic conditions. PLoS One 8:e69763. doi:10.1371/journal.pone.0069763
McCaughey G, McKevitt M, Elborn JS, Tunney MM (2012) Antimicrobial activity of fosfomycin and tobramycin in combination against cystic fibrosis pathogens under aerobic and anaerobic conditions. J Cyst Fibros 11:163–172. doi:10.1016/j.jcf.2011.11.003
Menuet M, Bittar F, Stremler N, Dubus J-C, Sarles J, Raoult D, Rolain J-M (2008) First isolation of two colistin-resistant emerging pathogens, Brevundimonas diminuta and Ochrobactrum anthropi, in a woman with cystic fibrosis: a case report. J Med Case Rep 2:373. doi:10.1186/1752-1947-2-373
Moree WJ, Phelan VV, Wu C-H, Bandeira N, Cornett DS, Duggan BM, Dorrestein PC (2012) Interkingdom metabolic transformations captured by microbial imaging mass spectrometry. Proc Natl Acad Sci 109:13811–13816. doi:10.1073/pnas.1206855109
Moskowitz SM, Foster JM, Emerson J, Burns JL (2004) Clinically feasible biofilm susceptibility assay for isolates of Pseudomonas aeruginosa from patients with cystic fibrosis. J Clin Microbiol 42:1915–1922
Mowat E, Rajendran R, Williams C, McCulloch E, Jones B, Lang S, Ramage G (2010) Pseudomonas aeruginosa and their small diffusible extracellular molecules inhibit Aspergillus fumigatus biofilm formation. FEMS Microbiol Lett 313:96–102. doi:10.1111/j.1574-6968.2010.02130.x
Murray JL, Connell JL, Stacy A, Turner KH, Whiteley M (2014) Mechanisms of synergy in polymicrobial infections. J Microbiol 52:188–199. doi:10.1007/s12275-014-4067-3
Nagano Y, Millar BC, Goldsmith CE, Elborn JS, Rendall J, Moore JE (2007) Emergence of Scedosporium apiospermum in patients with cystic fibrosis. Arch Dis Child 92:607–607. doi:10.1136/adc.2007.119503
Nixon GM, Armstrong DS, Carzino R, Carlin JB, Olinsky A, Robertson CF, Grimwood K (2001) Clinical outcome after early Pseudomonas aeruginosa infection in cystic fibrosis. J Pediatr 138:699–704. doi:10.1067/mpd.2001.112897
O’Neill K, Bradley JM, Johnston E, McGrath S, McIlreavey L, Rowan S, Reid A, Bradbury I, Einarsson G, Elborn JS, Tunney MM (2015) Reduced bacterial colony count of anaerobic bacteria is associated with a worsening in lung clearance index and inflammation in cystic fibrosis. PLoS One 10:e0126980. doi:10.1371/journal.pone.0126980
Olesen HV, Nielsen LP, Schiotz PO (2006) Viral and atypical bacterial infections in the outpatient pediatric cystic fibrosis clinic. Pediatr Pulmonol 41:1197–1204. doi:10.1002/ppul.20517
Oliver A (2010) Mutators in cystic fibrosis chronic lung infection: prevalence, mechanisms, and consequences for antimicrobial therapy. Int J Med Microbiol 300:563–572. doi:10.1016/j.ijmm.2010.08.009
Patankar YR, Lovewell RR, Poynter ME, Jyot J, Kazmierczak BI, Berwin B (2013) Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa. Infect Immun 81:2043–2052. doi:10.1128/IAI.00054-13
Peters BM, Jabra-Rizk MA, O’May GA, Costerton JW, Shirtliff ME, William Costerton J, Shirtliff ME (2012) Polymicrobial interactions: impact on pathogenesis and human disease. Clin Microbiol Rev 25:193–213. doi:10.1128/CMR.00013-11
Pilkington R, Callaghan M, McClean S (2011) Activation of MMP-9 by human lung epithelial cells in response to the cystic fibrosis-associated pathogen Burkholderia cenocepacia reduced wound healing in. Am J Physiol - Lung Cell Mol Physiol 301:L575–L586. doi:10.1152/ajplung.00226.2010
Pompilio A, Crocetta V, De Nicola S, Verginelli F, Fiscarelli E, Di Bonaventura G (2015) Cooperative pathogenicity in cystic fibrosis: Stenotrophomonas maltophilia modulates Pseudomonas aeruginosa virulence in mixed biofilm. Front Microbiol 6:951. doi:10.3389/fmicb.2015.00951
Price KE, Hampton TH, Gifford AH, Dolben EL, Hogan DA, Morrison HG, Sogin ML, O’Toole GA (2013) Unique microbial communities persist in individual cystic fibrosis patients throughout a clinical exacerbation. Microbiome 1:27. doi:10.1186/2049-2618-1-27
Pustelny C, Komor U, Pawar V, Lorenz A, Bielecka A, Moter A, Gocht B, Eckweiler D, Müsken M, Grothe C, Lünsdorf H, Weiss S, Häussler S (2015) Contribution of veillonella parvula to Pseudomonas aeruginosa-mediated pathogenicity in a murine tumor model system. Infect Immun 83:417–429. doi:10.1128/IAI.02234-14
Ramirez IA, Caverly LL, Kalikin LM, Goldsmith AM, Lewis TC, Burke DT, JJ LP, Sajjan US, Hershenson MB (2014) Differential responses to rhinovirus- and influenza-associated pulmonary exacerbations in patients with cystic fibrosis. Ann Am Thorac Soc 11:554–561. doi:10.1513/AnnalsATS.201310-346OC
Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordoñez C, Elborn JS (2011) A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 365:1663–1672. doi:10.1056/NEJMoa1105185
Rasko DA, Sperandio V (2010) Anti-virulence strategies to combat bacteria-mediated disease. Nat Rev Drug Discov 9:117–128. doi:10.1038/nrd3013
Razvi S, Quittell L, Sewall A, Quinton H, Marshall B, Saiman L (2009) Respiratory microbiology of patients with cystic fibrosis in the United States, 1995 to 2005. Chest 136:1554–1560. doi:10.1378/chest.09-0132
Rogers GB, Carroll MP, Serisier DJ, Hockey PM, Jones G, Bruce KD (2004) Characterization of bacterial community diversity in cystic fibrosis lung infections by use of 16s ribosomal DNA terminal restriction fragment length polymorphism profiling. J Clin Microbiol 42:5176–5183. doi:10.1128/JCM.42.11.5176-5183.2004
Rogers GB, Hart CA, Mason JR, Hughes M, Walshaw MJ, Bruce KD (2003) Bacterial diversity in cases of lung infection in cystic fibrosis patients: 16S ribosomal DNA (rDNA) length heterogeneity PCR and 16S rDNA terminal restriction fragment length polymorphism profiling. J Clin Microbiol 41:3548–3558
Rogers GB, Hoffman LR, Carroll MP, Bruce KD (2013) Interpreting infective microbiota: the importance of an ecological perspective. Trends Microbiol 21:271–276. doi:10.1016/j.tim.2013.03.004
Rowe SM, Miller S, Sorscher EJ (2005) Cystic fibrosis. N Engl J Med 352:1992–2001. doi:10.1056/NEJMra043184
Rüger M, Ackermann M, Reichl U (2014) Species-specific viability analysis of Pseudomonas aeruginosa, Burkholderia cepacia and Staphylococcus aureus in mixed culture by flow cytometry. BMC Microbiol 14:56. doi:10.1186/1471-2180-14-56
Rutherford ST, Bassler BL (2012) Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med. doi:10.1101/cshperspect.a012427
Schobert M, Jahn D (2010) Anaerobic physiology of Pseudomonas aeruginosa in the cystic fibrosis lung. Int J Med Microbiol 300:549–556. doi:10.1016/j.ijmm.2010.08.007
Sherrard LJ, Schaible B, Graham KA, McGrath SJ, McIlreavey L, Hatch J, Wolfgang MC, Muhlebach MS, Gilpin DF, Schneiders T, Elborn JS, Tunney MM (2014) Mechanisms of reduced susceptibility and genotypic prediction of antibiotic resistance in Prevotella isolated from cystic fibrosis (CF) and non-CF patients. J Antimicrob Chemother 69:2690–2698. doi:10.1093/jac/dku192
Short FL, Murdoch SL, Ryan RP (2014) Polybacterial human disease: the ills of social networking. Trends Microbiol 22:508–516. doi:10.1016/j.tim.2014.05.007
Sibley CD, Duan K, Fischer C, Parkins MD, Storey DG, Rabin HR, Surette MG (2008) Discerning the complexity of community interactions using a Drosophila model of polymicrobial infections. PLoS Pathog 4:e1000184. doi:10.1371/journal.ppat.1000184
Sibley CD, Rabin H, Surette MG (2006) Cystic fibrosis: a polymicrobial infectious disease. Future Microbiol 1:53–61. doi:10.2217/17460913.1.1.53
Smyth AR, Smyth RL, Tong CY, Hart CA, Heaf DP (1995) Effect of respiratory virus infections including rhinovirus on clinical status in cystic fibrosis. Arch Dis Child 73:117–120
Sousa AM, Pereira MO (2014) Pseudomonas aeruginosa diversification during infection development in cystic fibrosis lungs—a review. Pathog (Basel, Switzerland) 3:680–703. doi: 10.3390/pathogens3030680
Sriramulu D (2013) Evolution and impact of bacterial drug resistance in the context of cystic fibrosis disease and nosocomial settings. Microbiol Insights 6:29–36. doi:10.4137/MBI.S10792
Sriramulu DD, Lünsdorf H, Lam JS, Römling U (2005) Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J Med Microbiol 54:667–676. doi:10.1099/jmm.0.45969-0
Standiford TJ, Kunkel SL, Basha MA, Chensue SW, Lynch JP, Toews GB, Westwick J, Strieter RM (1990) Interleukin-8 gene expression by a pulmonary epithelial cell line. A Model for Cytokine Networks in the Lung J Clin Invest 86:1945–1953. doi:10.1172/JCI114928
Starner TD, Zhang N, Kim G, Apicella MA, McCray PB (2006) Haemophilus influenzae forms biofilms on airway epithelia: implications in cystic fibrosis. Am J Respir Crit Care Med 174:213–220. doi:10.1164/rccm.200509-1459OC
Stressmann FA, Rogers GB, van der Gast CJ, Marsh P, Vermeer LS, Carroll MP, Hoffman L, Daniels TWV, Patel N, Forbes B, Bruce KD (2012) Long-term cultivation-independent microbial diversity analysis demonstrates that bacterial communities infecting the adult cystic fibrosis lung show stability and resilience. Thorax 67:867–873. doi:10.1136/thoraxjnl-2011-200932
Traini D, Young PM (2009) Delivery of antibiotics to the respiratory tract: an update. Expert Opin Drug Deliv 6:897–905. doi:10.1517/17425240903110710
Treggiari MM, Rosenfeld M, Retsch-Bogart G, Gibson R, Ramsey B (2007) Approach to eradication of initial Pseudomonas aeruginosa infection in children with cystic fibrosis. Pediatr Pulmonol 42:751–756. doi:10.1002/ppul.20665
Tunney MM, Field TR, Moriarty TF, Patrick S, Doering G, Muhlebach MS, Wolfgang MC, Boucher R, Gilpin DF, McDowell A, Elborn JS (2008) Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med 177:995–1001. doi:10.1164/rccm.200708-1151OC
Tunney MM, Klem ER, Fodor AA, Gilpin DF, Moriarty TF, Mcgrath SJ, Muhlebach MS, Boucher RC, Cardwell C, Doering G, Elborn JS, Wolfgang MC (2011) Use of culture and molecular analysis to determine the effect of antibiotic treatment on microbial community diversity and abundance during exacerbation in patients with cystic fibrosis. Thorax 66:579–584. doi:10.1136/thx.2010.137281
Twomey KB, O’Connell OJ, McCarthy Y, Dow JM, O’Toole GA, Plant BJ, Ryan RP (2012) Bacterial cis-2-unsaturated fatty acids found in the cystic fibrosis airway modulate virulence and persistence of Pseudomonas aeruginosa. Isme j 6:939–950. doi:10.1038/ismej.2011.167
Ulrich M, Beer I, Braitmaier P, Dierkes M, Kummer F, Krismer B, Schumacher U, Gräpler-Mainka U, Riethmüller J, Jensen PØ, Bjarnsholt T, Høiby N, Bellon G, Döring G (2010) Relative contribution of Prevotella intermedia and Pseudomonas aeruginosa to lung pathology in airways of patients with cystic fibrosis. Thorax 65:978–984. doi:10.1136/thx.2010.137745
van Westreenen M, Tiddens HAWM (2010) New antimicrobial strategies in cystic fibrosis. Paediatr Drugs 12:343–352. doi:10.2165/11316240-000000000-00000
Vettoretti L, Plésiat P, Muller C, El Garch F, Phan G, Attrée I, Ducruix A, Llanes C (2009) Efflux unbalance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob Agents Chemother 53:1987–1997. doi:10.1128/AAC.01024-08
Wei Q, Ma LZ (2013) Biofilm matrix and its regulation in Pseudomonas aeruginosa. Int J Mol Sci 14:20983–21005. doi:10.3390/ijms141020983
Wellinghausen N, Wirths B, Poppert S (2006) Fluorescence in situ hybridization for rapid identification of Achromobacter xylosoxidans and Alcaligenes faecalis recovered from cystic fibrosis patients. J Clin Microbiol 44:3415–3417. doi:10.1128/JCM.00508-06
Whiteson KL, Meinardi S, Lim YW, Schmieder R, Maughan H, Quinn R, Blake DR, Conrad D, Rohwer F (2014) Breath gas metabolites and bacterial metagenomes from cystic fibrosis airways indicate active pH neutral 2,3-butanedione fermentation. Isme j 8:1247–1258. doi:10.1038/ismej.2013.229
Wilschanski M (2013) Novel therapeutic approaches for cystic fibrosis. Discov Med 15:127–133
Winstanley C, Fothergill JL (2009) The role of quorum sensing in chronic cystic fibrosis Pseudomonas aeruginosa infections. FEMS Microbiol Lett 290:1–9. doi:10.1111/j.1574-6968.2008.01394.x
Worlitzsch D, Rintelen C, Böhm K, Wollschläger B, Merkel N, Borneff-Lipp M, Döring G (2009) Antibiotic-resistant obligate anaerobes during exacerbations of cystic fibrosis patients. Clin Microbiol Infect 15:454–460. doi:10.1111/j.1469-0691.2008.02659.x
Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiss T, Botzenhart K, Yankaskas JR, Randell S, Boucher RC, Döring G (2002) Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 109:317–325. doi:10.1172/JCI13870
Yang JH, Spilker T, LiPuma JJ (2006) Simultaneous coinfection by multiple strains during Burkholderia cepacia complex infection in cystic fibrosis. Diagn Microbiol Infect Dis 54:95–98. doi:10.1016/j.diagmicrobio.2005.08.020
Yang L, Jelsbak L, Molin S (2011a) Microbial ecology and adaptation in cystic fibrosis airways. Environ Microbiol 13:1682–1689. doi:10.1111/j.1462-2920.2011.02459.x
Yang L, Liu Y, Markussen T, Høiby N, Tolker-Nielsen T, Molin S (2011b) Pattern differentiation in co-culture biofilms formed by Staphylococcus aureus and Pseudomonas aeruginosa. FEMS Immunol Med Microbiol 62:339–347. doi:10.1111/j.1574-695X.2011.00820.x
Yoon SS, Hennigan RF, Hilliard GM, Ochsner UA, Parvatiyar K, Kamani MC, Allen HL, DeKievit TR, Gardner PR, Schwab U, Rowe JJ, Iglewski BH, McDermott TR, Mason RP, Wozniak DJ, Hancock REW, Parsek MR, Noah TL, Boucher RC, Hassett DJ (2002) Pseudomonas aeruginosa anaerobic respiration in biofilms: relationships to cystic fibrosis pathogenesis. Dev Cell 3:593–603
Acknowledgments
The authors acknowledge the Portuguese Foundation for Science and Technology (FCT), the strategic funding of UID/BIO/04469/2013-CEB and UID/EQU/00511/2013-LEPABE units. This study was also supported by FCT and the European Community fund FEDER, through Program COMPETE, under the scope of the Projects “DNA mimics” PIC/IC/82815/2007, RECI/BBB-EBI/0179/2012 (FCOMP-01-0124-FEDER-027462), “BioHealth—Biotechnology and Bioengineering approaches to improve health quality”, Ref. NORTE-07-0124-FEDER-000027 and NORTE-07-0124-FEDER-000025—RL2_ Environment and Health, co-funded by the Programa Operacional Regional do Norte (ON.2 – O Novo Norte), QREN, FEDER. The authors also acknowledge the grant of Susana P. Lopes (SFRH/BPD/95616/2013) and of the COST-Action TD1004: Theragnostics for imaging and therapy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Declaration of interest
The authors report no declarations of interest.
Rights and permissions
About this article

Cite this article
Magalhães, A.P., Azevedo, N.F., Pereira, M.O. et al. The cystic fibrosis microbiome in an ecological perspective and its impact in antibiotic therapy. Appl Microbiol Biotechnol 100, 1163–1181 (2016). https://doi.org/10.1007/s00253-015-7177-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-7177-x