
Abstract
The spatiotemporal distribution of microbial diversity, community composition, and their major drivers are fundamental issues in microbial ecology. In this study, the planktonic bacterial and microeukaryotic communities of the Jiulong River were investigated across both wet and dry seasons by using denaturing gradient gel electrophoresis (DGGE). We found evidence of temporal change between wet and dry seasons and distinct spatial patterns of bacterial and microeukaryotic communities. Both bacterial and microeukaryotic communities were strongly correlated with temperature, NH4-N, PO4-P, and chlorophyll a, and these environmental factors were significant but incomplete predictors of microbial community composition. Local environmental factors combined with spatial and temporal factors strongly controlled both bacterial and microeukaryotic communities in complex ways, whereas the direct influence of spatial and temporal factors appeared to be relatively small. Path analysis revealed that the microeukaryotic community played key roles in shaping bacterial community composition, perhaps through grazing effects and multiple interactions. Both Betaproteobacteria and Actinobacteria were the most dominant and diverse taxa in bacterial communities, while the microeukaryotic communities were dominated by Ciliophora (zooplankton) and Chlorophyta (phytoplankton). Our results demonstrated that both bacterial and microeukaryotic communities along the Jiulong River displayed a distinct spatiotemporal pattern; however, microeukaryotic communities exhibited a stronger distance-decay relationship than bacterial communities and their spatial patterns were mostly driven by local environmental variables rather than season or spatial processes of the river. Therefore, we have provided baseline data to support further research on river microbial food webs and integrating different microbial groups into river models.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
It is both ecologically and economically important to investigate the mechanisms that regulate the community composition and geographical distribution of microbes in freshwater ecosystems. This is because these microorganisms constitute the major part of Earth’s biomass and play an indispensable role in biogeochemical cycles and ecosystem functions (Fuhrman 2009; Newton and McMahon 2011; Freimann et al. 2013; Liu et al. 2013). Planktonic microorganisms exhibit complex responses to environmental disturbances and stresses that are manifested through variation at both the population and community levels (Beier et al. 2008; Fuhrman 2009; Yang et al. 2012; Chen et al. 2013). The composition of planktonic bacterial and microeukaryotic communities can be highly variable across running water systems since the spatiotemporal dynamics in microbial community composition are often related to changes in the environmental parameters (Hullar et al. 2006; Stomp et al. 2011; Portillo et al. 2012; Liu et al. 2013; Read et al. 2015). Certain development activities, including agriculture, industry, and urbanization, are often important drivers of changes in environmental conditions of lotic ecosystems (Chen and Hong 2012; Staley et al. 2014), and the functions and community dynamics of plankton ecosystem (e.g., nutrient recycling, organic matter content) can strongly influence water quality (Fuhrman 2009; Hanson et al. 2012). Indeed, a wide range of toxins and algae blooms triggered by plankton community changes can negatively affect the municipal water supply (Yang et al. 2012; Pantelić et al. 2013). Therefore, the plankton community is generally thought to be a good indicator of ecosystem status and water quality, as ecosystem variations are reflected in relatively quick changes in plankton community composition (Bianchi et al. 2003; Crump et al. 2007; Yang et al. 2012; Liu et al. 2013; Staley et al. 2014).
Research to date attempting to illustrate the geographical patterns and mechanisms involved in shaping natural metacommunities have largely focused on macroorganisms while comparatively less work has been conducted on microorganisms (Nekola and White 1999; Lear et al. 2013; Liu et al. 2015; Read et al. 2015). Further, relatively few studies have quantified spatial patterns of both bacterial and microeukaryotic communities at the same time (Liu et al. 2011; Soininen et al. 2011, 2013), although biogeographical patterns of specific taxonomic groups, such as diatoms, have been studied in more detail (Astorga et al. 2012; Goldenberg et al. 2014). The effects of geographical factors and microbial properties including body size, trophic position, and the rate and extent of disturbance are considered to mainly determine the composition and diversity of macroorganism communities and need further investigation in microbial communities (Green and Bohannan 2006; de Bie et al. 2012; Hanson et al. 2012). Further, most published molecular studies on the diversity and community composition of microorganisms have been performed in lentic water; however, the environmental conditions of rivers or streams (lotic systems) are more complex and dynamic, and it is difficult to obtain integrated samples (Crump and Hobbie 2005; Liu et al. 2013; Read et al. 2015). Human settlements extend mainly along the rivers, which might result in intensive human impacts on river ecosystems. Furthermore, the connectivity of lotic systems has been shown to be important in affecting dispersal of aquatic microbial species (Read et al. 2015). Therefore, the biogeographical patterns of lotic microbial communities can be quite different from that of lentic ecosystem.
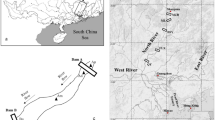
The Jiulong River, which is the second longest river in Fujian province in southeastern China, drains an area of approximately 14,700 km2 and is composed of two major tributaries (the North River and West River) and many small tributaries (Liu et al. 2011). The river is used for a wide range of agricultural, industrial, transportation, and recreational purposes and is an important drinking-water source used by many cities along its length. With rapid economic development in the Jiulong River Watershed in the past 30 years, it has been impacted by a variety of anthropogenic disturbances which contribute nutrients, chemicals, pharmaceuticals, and pathogenic bacteria (Liu et al. 2011; Zhang et al. 2012; Hu et al. 2014). In particular, the upstream city of Longyan is the main livestock-breeding area of Fujian Province, leading to a lot of nutrients and pharmaceuticals entering into the river (Liu et al. 2011; Zhang et al. 2012). The Jiulong River Watershed is ideally suited for us to study the spatiotemporal dynamics of plankton community at large scale. The dispersal capacity of river microbial communities makes them suitable for investigating how different communities are constrained by dispersal and how alternating wet and dry seasons affect dispersal.
Two domains of microbial life (Bacteria and Eukarya) were studied through denaturing gradient gel electrophoresis (DGGE) and clone sequencing to investigate spatiotemporal changes of both bacterial and microeukaryotic communities and to assess the main drivers of the trends we observed. The two groups have fundamentally different characteristics (e.g., cell size) and ecological roles (Liu et al. 2013). Although it has been partly superseded by more recently developed approaches, the DGGE method remains a rapid and cost-effective approach to provide intuitive information on community composition changes for the analysis of a large number of samples (Yu et al. 2015). First, we examined whether temporal change or distance decay drives compositional dissimilarity. Next, we sought to examine the relationships between environmental factors and the microbial community composition. We further used variance partitioning to evaluate which spatial, temporal, or environmental factors were most associated with the variation in the composition of microbial communities. We also attempted to address the interactions of bacterial and microeukaryotic communities in response to spatiotemporal changes in the environment. Finally, we used clone sequencing to identify the dominant planktonic bacterial and microeukaryotic species in the Jiulong River.
Materials and methods
Study area and field sampling
Water samples were collected in both July 2012 and January 2013 (representing the wet and dry seasons, respectively) from 16 sites along the Jiulong River. Seventy-five percent of the annual precipitation occurs from April to November in this area (Chen et al. 2008). The sample locations and codes are presented in Fig. 1. Study sites were chosen to span the entire watershed and to capture unaffected communities as well as possible changes influenced by major urban and agricultural inputs and variations at river channel confluences. The latitude and longitude for the studied sites were determined by a portable global positioning system (GPS Jisibao G330, Bejing, China). For planktonic bacterial and microeukaryotic community analyses, surface water samples (<0.5 m depth) were pre-filtered through a 200-μm mesh in order to remove larger metazoan and debris, and then 300–500 ml water was subsequently filtered on a 0.2-μm-pore-size, 47-mm-diameter polycarbonate membrane (Millipore, Billerica, MA, USA). The membranes were immediately frozen at −20 °C in the field and subsequently stored at −80 °C in the laboratory until DNA extraction.

Distribution of the 16 sampling sites in the Jiulong River Watershed
Physicochemical analyses
The analysis of water temperature, dissolved oxygen (DO), electrical conductivity (EC), salinity, pH, ORP, chlorophyll a (Chl-a), and turbidity was carried out in situ using a multiparameter water-quality analyzer (Hydrolab DS5, Hach Company, Loveland, CO, USA). The current velocity was measured with a flowtracker (SonTek, FlowTracker Handheld-ADV® YSI, San Diego, CA, USA). Suspended solids concentrations were gravimetrically measured by filtering water samples of 100 ml through pre-weighed 0.45-μm-pore-size filters; these filters were reweighed again after evaporation at 105 °C. A TOC/TN-VCPH analyzer (Shimadzu, Kyoto, Japan) was employed to determine the concentrations of total nitrogen (TN), total organic carbon (TOC), and total carbon (TC). A spectrophotometric method was used to measure total phosphorus (TP) after digestion. The concentrations of ammonium nitrogen (NH4-N), nitrite and nitrate nitrogen (NO x -N), and phosphate phosphorus (PO4-P) were determined with a Lachat QC8500 Flow Injection Autoanalyzer (Lachat Instruments, Hach Company, Loveland, CO, USA). All physicochemical analyses were measured according to methods used in our previous work (Yang et al. 2012; Liu et al. 2013).
DNA extraction and PCR amplification
Total DNA was obtained directly from cells trapped in the filters using the methods described in Liu et al. (2013). The bacterial 16S rRNA genes were amplified with primers 341F-GC (5′-CGC CCG CCG CGC CCC GCG CCC GTC CCG CCG CCC CCG CCC GCC TAC GGG AGG CAG CAG-3′) and 907R (5′-CCG TCA ATT CMT TTG AGT TT−3′), which are specific for most bacteria (Schäfer and Muyzer 2001). For the microeukaryotic species, PCR primer pair Euk1A (5′-CTG GTT GAT CCT GCC AG-3′) and Euk516R-GC (5′-ACC AGA CTT GCC CTC CCG CCC GGG GCG CGC CCC GGG CGG GGC GGG GGC ACG GGG GG−3′) were used for amplification of 18S rRNA genes (Díez et al. 2001). The PCR reaction mixture (50 μl) contained 100 ng template DNA, 1 μl (0.25 pmol each) of primer set, 4 μl (2.5 mM each) of deoxyribonucleotide triphosphate mixture (Takara Bio Inc., Otsu, Shiga, Japan), 5 μl (20 mM MgCl2) of Ex Taq buffer, and 0.5 μl (1.25 U) of Ex Taq DNA polymerase (Takara Bio Inc., Otsu, Shiga, Japan). PCR conditions for bacterial communities were set as follows: initial denaturation for 5 min at 94 °C; followed by 30 cycles of 30 s at 94 °C, annealing for 30 s at 52 °C, and elongation for 1 min at 72 °C; a final elongation step for 10 min at 72 °C was performed. Similar cycle parameters and reaction composition were used for microeukaryotic species for 30 cycles (30 s at 94 °C, annealing for 30 s at 50 °C, and elongation for 1 min at 72 °C). The 1 % agarose gels were used to check the size of PCR products. The PCR products were then used for subsequent DGGE analysis.
Denaturing gradient gel electrophoresis
DGGE analysis was performed with a DCode system (Bio-Rad, Hercules, CA, USA). The gradient of denaturants increased from 30 to 60 % for 16S rRNA genes and from 20 to 50 % for 18S rRNA genes in a 6 % (w/v) polyacrylamide gel, respectively. The DGGE gel was pre-run at 60 °C and 200 V for 10 min, and then electrophoresis was run at 60 °C and 100 V for 16 h. The gel was rinsed in distilled water after SYBR Green I nucleic acid staining, and then inspected with a Gel Doc EQ imager (Bio-Rad, Hercules, CA, USA). A sterile scalpel was used to recover the DGGE bands for cloning and sequencing.
Sequencing and phylogenetic analysis
The recovered bands were washed twice by 1 × PCR buffer, suspended in 50 μl of Tris-EDTA buffer and deposited at 4 °C overnight. Two milliliters DNA supernatant was used as template in a PCR re-amplification with non-GC-clamped original primers. The product was gel-checked and purified with an Agarose Gel DNA Purification Kit Ver. 2.0 (Takara Bio Inc., Otsu, Shiga, Japan). Each purified product was sequenced, respectively. DNA fragments were individually cloned into T-vector pMD18-T (Takara Bio Inc., Otsu, Shiga, Japan) and transformed into the competent Escherichia coli DH5α cells (Takara Bio Inc., Otsu, Shiga, Japan) following the procedures of Liu et al. (2011). Successfully inserted plasmids were unidirectionally sequenced using an ABI3730 automated sequencer (Applied Biosystems, Foster City, CA, USA). Sequences of 16S rRNA and 18S rRNA genes were checked and manually modified with BioEdit software version 7.0.4.1 (IBIS Biosciences, Carlsbad, CA, USA) (Hall 1999), and then compared with bacterial and eukaryotic sequences recorded in the GenBank using BLASTN to obtain the closest relatives (Liu et al. 2011).
Data analyses
The band profiles generated by DGGE were normalized and compared using the Quantity One 4.4.0 software (Bio-Rad, Hercules, CA, USA). A binary matrix was constructed based on band absence (0) and presence (1) patterns. This matrix was used for further statistical analysis and a Jaccard dissimilarity matrix was calculated (Wang et al. 2015). To investigate the possible differences in microbial communities between the wet and dry seasons, we used nonmetric multidimensional scaling ordination (NMDS) (Clarke and Gorley 2001). Further, we used an analysis of similarity (ANOSIM) to statistically test for significant difference in microbial communities between wet and dry seasons. R = 0 suggests no separation, whereas R = 1 indicates complete separation. Both NMDS and ANOSIM were performed using PRIMER 5.0 (Clarke and Gorley 2001).
Prior to analysis, all variables for statistical tests were ln(x + 1) transformed to improve their homoscedasticity and normality with the exception of pH (Legendre and Legendre 1998). The environmental distance matrix was calculated based on Euclidean distance using PRIMER 5.0. We then used Mantel tests to determine the correlation between geographical distance matrix and community dissimilarity or environmental distance matrices. We used a one-way ANOVA with Scheffe’s post hoc test to test statistically whether there was significant difference in both biological and physicochemical parameters between wet and dry seasons using SPSS 19.0 (SPSS Inc., Crawfordville, FL, USA). The longest gradient lengths of detrended correspondence analysis (DCA) on the bacterial and microeukaryotic communities data sets were 2.32 and 2.31, respectively, thereby indicating that redundancy analysis (RDA) was suitable to determine environmental factors which were significantly related to both communities (Lepš and Šmilauer 2003). RDAs with forward manual selection procedure and Monte Carlo tests (P < 0.05) were then conducted on the species data against environmental variables, respectively, to select predictors in each group that explained significant proportions of the variance in community composition data (Lepš and Šmilauer 2003). A set of spatial eigenvectors were derived though PCNM (principal coordinates of neighbor matrices) analysis based on the geographical distances using the R package Vegan (Borcard and Legendre 2002; Oksanen et al. 2013). A qualitative matrix composed of the binary data set provided a representation of the wet and dry seasons. The relative importance of the environmental, spatial, and temporal factors and their co-variations for explaining changes in community composition was distinguished using variation partitioning. DCA, RDA, and variation partitioning were performed using the CANOCO 4.5 software package. Path analysis with structural equation modeling further explored the specific effects of the environmental, spatial, and temporal factors on microbial community composition. The RV coefficient was used to determine the multivariate correlation between standardized data matrices with the FactoMineR package in R (Robert and Escoufier 1976). The chi-square test was performed to assess the adequacy between the original correlation matrix and the overall causal model, in which a high P value indicates that the data fit the model well. Both Bentler comparative fit index (CFI) and the goodness-of-fit index (GFI) were used to test how well the model fitted the data (Schermelleh-Engel et al. 2003). We used the AMOS 17.0 (SPSS Inc., Crawfordville, FL, USA) to calculate individual path coefficients which indicate the strength of the statistical relationships among variables and to evaluate the fit of the final path model.
Accession number
The 16S and 18S ribosomal RNA gene sequences from this study were deposited in the GenBank. Their accession numbers and closest known matches in GenBank are summarized in Table S2 and Table S3.
Results
Spatial and temporal variations in environmental parameters
The 19 environmental variables from both wet and dry seasons are summarized in Table S1. We detected the highest concentrations of TP, TN, PO4-P, and NH4-N in the Longyan City region (sites 1, 2, and 3), indicating the nutrient discharges from livestock and poultry breeding in the upstream regions. In general, electrical conductivity, salinity, and nutrient concentrations of TC, TOC NH4-N, TP, and TN were lower in the wet season than those of the dry season (Table S1). However, there were no significant differences in the NO x -N and PO4-P concentrations between wet and dry seasons. Water temperature in the wet season was higher than in the dry season. The highest chlorophyll a concentration occurred in the wet season at site 16 located downstream of Zhangzhou City. In general, however, the chlorophyll a concentration in the wet season was lower than in the dry season except for sites 6 and 16. According to Mantel tests, the correlation coefficient between geographical distance and environmental distance was 0.193 (P = 0.026) in dry season, while no significant correlation was found in wet season (r = 0.121, P = 0.089).
Spatial and temporal variations in microbial communities
Clear seasonal patterns of microbial communities were identified by the DGGE profiles of both 16S and 18S rRNA genes (Fig. S1 and Fig. S2). Nonmetric multidimensional scaling ordination suggested that the bacterial or microeukaryotic communities collected in the wet season clustered together, whereas those in the dry season gathered together (Fig. 2). The results of ANOSIM further corroborated that the bacterial or microeukaryotic communities could be significantly distinguished between wet and dry seasons (P = 0.001). According to a Mantel test, bacterial and microeukaryotic communities showed significant concordance in their spatiotemporal patterns (r = 0.351, P = 0.001).

Two-dimensional nonmetric multidimensional scaling ordination using Jaccard indices of a bacterial communities and b microeukaryotic communities based on the DGGE profiles. The samples were collected in both wet (July 2012) and dry (January 2013) seasons
Not surprisingly, there is no significant correlation (r = 0.092, P = 0.315) between geographical distance and bacterial community dissimilarity during the wet season (Fig. 3a). During the dry season, however, there was a statistically significant relationship between geographical distance and bacterial community dissimilarity (r = 0.209, P = 0.022) (Fig. 3b). Interestingly, the positive correlations between geographical distance and microeukaryotic community dissimilarity were statistically significant during both wet (r = 0.212, P = 0.020) and dry (r = 0.263, P = 0.004) seasons (Fig. 3c, d). Overall, the difference in microbial community composition between any two sampling sites increased with the geographical distance between the sample locations, except for the bacterial community from the wet season. Further, both bacterial and microeukaryotic communities exhibited a stronger distance-decay relationship in the dry season than in the wet season, indicating that seasonal monsoonal hydrology may affect distance-decay relationships. More importantly, bacterial community exhibited a weaker distance-decay relationship than microeukaryotic communities in both seasons.

Relationship between community dissimilarity and geographical distance. a Bacterial community in wet season, b bacterial community in dry season, c microeukaryotic community in wet season, and d microeukaryotic community in dry season
Environmental factors influencing microbial community composition
Redundancy analysis (RDA) indicated that temperature, NH4-N, PO4-P, chlorophyll a, and DO were the factors that significantly affect the composition of the bacterial communities, while the microeukaryotic community was found to be significantly influenced by temperature, NH4-N, PO4-P, and chlorophyll a (Fig. 4). For both bacterial and eukaryotic communities, redundancy analysis revealed that the wet season samples formed one group, while the dry season samples formed the other group. Samples from Longyan City region (sites 1, 2, and 3) were quite different from the others due to their high concentrations of NH4-N and PO4-P in the upper Jiulong River.

RDA ordination based on the microbial community composition and selected environmental factors. All environmental factors were significantly related to the variation of microbial community (P < 0.05). a Bacterial community, b microeukaryotic community
Relative contribution of environmental and spatiotemporal factors on microbial communities
Variation partitioning using partial RDA showed that environmental, spatial, and temporal factors combined explained 41.3 % of the bacterioplankton community variation (Fig. 5b). The relative contribution of environmental factors (35.5 %) was considerably larger than that of spatial (12.1 %) or temporal (11.3 %) factors. For microeukaryotic communities, the variance explained by environmental factors (26.8 %) was also higher than that explained by spatial (13.6 %) or temporal (13.5 %) factors (Fig. 5c). In the case of the percentage change in microbial communities, clearly, the bacterial community variance explained by environmental factors was higher than that for the microeukaryotic community. However, the microeukaryotic community variance explained by spatial or temporal factors was slightly higher than that of the bacterial community.

Variation partitioning of microbial communities into the spatial, temporal, and environmental variables. a General outline, b bacterial community, c microeukaryotic community. The number stands for the percentage of community variation
Path analysis was performed to validate the causal relationships between spatial, temporal, and environmental factors with microbial community composition. We also examined the impacts of biological interactions by assessing the influence of microeukaryotic community on bacterial community. The initial model included all plausible pathways (Fig. S3) and we tried to find the optimal ecological model. Our final model was obtained by removing all non-significant pathways from the initial model (Fig. 6). Both bacterial and microeukaryotic communities were most directly influenced by the environmental factors. Noticeably, both bacterial and microeukaryotic communities were tightly coupled because bacterial community composition was most directly influenced by microeukaryotic community (path coefficient was 0.446). Environmental factors changed as the function of season (0.506) and space (0.400). Interestingly, both space and season had no direct effect on microbial community, but displayed indirect effects on microbial communities through their significant effects on environmental factors.

Path diagram obtained with the structural equation modeling. The chi-square test is performed to assess the adequacy between the original correlation matrix and the overall causal model. Path coefficients (numbers on each arrow) indicate the strength of each causal relationship
Taxonomic diversity of bacterial and microeukaryotic communities
The DGGE profiles of bacterial communities generated a total of 712 bands, averaging about 22 bands per sample, which were classified into 57 distinct bands. A DNA cloning approach was used to isolate 16S rDNA fragments from 27 dominant DGGE bands (Fig. S1, Table S2). In total, we phylogenetically characterized 493 bacterial bands clustered into 24 OTUs. The taxonomic distribution of bacterial communities among all samples was as follows: Betaproteobacteria, 45.1 %; Actinobacteria, 26.2 %; Bacteroidetes, 9.4 %; Alphaproteobacteria, 7.8 %; Firmicutes, 6.3 %; and Gammaproteobacteria, 5.2 % (Table S2). The majority of GenBank sequence matches were bacteria isolated from freshwater or water associated habitats. In the Jiulong River ecosystems, Betaproteobacteria was the most diverse and widely distributed taxon.
For microeukaryotic communities, a total of 398 bands were detected (ranging from 6 to 22 in samples, averaging about 12 bands per sample) (Fig. S2). Sequence data of 28 bands were retrieved (Table S3). Thus, we phylogenetically identified 357 microeukaryotic bands clustered into 26 OTUs. The Ciliophora (zooplankton) and Chlorophyta (phytoplankton) were the most common and dominant taxa. The taxonomic distribution of 18S rRNA gene sequences among all samples was as follows: Ciliophora, 27.2 %; Chlorophyta, 25.8 %; Fungi, 17.6 %; Rotifera, 10.9 %; Chrysophyta, 5.0 %; Dinophyceae, 4.8 %; Stramenopiles, 3.6 %; and Choanoflagellata, 2.5 %.
Discussion
There are a relatively small number of published studies addressing the identification, estimation, and biogeographical patterns of both bacterial and microeukaryotic species sampled at the same time in flowing waters (Liu et al. 2013; Read et al. 2015). We analyzed the spatiotemporal dynamics and diversity of bacterial and microeukaryotic communities in the Jiulong River Watershed (Southeast China) in order to better understand lotic microbial ecology. The composition of bacterial and microeukaryotic communities varied significantly in the Jiulong River between different seasons. More importantly, we found that the spatial patterns were closely related not only to seasonal monsoonal hydrology but also to organism characteristics. We further attempted to quantify and describe the role of spatial, temporal, and environmental variables on the dynamics of bacterial and microeukaryotic communities.
Spatiotemporal patterns of river ecosystem
The Jiulong River has two major tributaries (about 258 km in length), and more than 130 dams have been constructed along its length for hydropower stations (Chen et al. 2008; Liu et al. 2011). The changes of certain environmental variables between the wet and dry seasons at some sampling sites were found to be significant. Further, the dams might lead to decreases of flow rate, thereby promoting particle settling and water transparency increases and perhaps also enhancing primary production (Chen and Hong 2012). The dam effects are particular prominent in dry season, in which the shortage of rainfall and water can lead to the increased nutrient loading and decreased river connectivity.
Our results revealed that bacterial and microeukaryotic communities exhibited distinct spatial patterns. Geographical distance affected microbial community composition—presumably due to dispersal limitation (Yang et al. 2010). However, bacteriaplankton did not show a significant distance-decay pattern during the wet season, perhaps indicating that they were best at dispersing across the river systems during periods of high water flows. Bacterial, or microeukaryotic, community dissimilarity increased more slowly with geographical distance during the wet season than the dry season. Therefore, the distance-decay patterns exhibited by the microbial community were more evident during the dry season in the Jiulong River. This was not a surprising finding since wet season rainfall could result in significant increase in habitat homogeneity and river connectivity. In dry season, the translocation of microbial taxa from the upstream to the downstream might decrease due to strong barrier effect of the ubiquitous dams on water flow (Chen and Hong 2012). There was a reduction on river connectivity in dry season and thereby promoting the heterogenicity in microhabitats and microbial communities.
More interestingly, the correlation between geographical distance and microeukaryotic community (with a larger size, 5–200 μm) dissimilarity was stronger than that observed for bacterial community (with a smaller size, 0.5–5 μm) dissimilarity during both wet and dry seasons. That is to say, bacterial community dissimilarity increased more slowly with geographical distance than microeukaryotic community dissimilarity. Our findings concur with Soininen et al. (2011) where within 100 small lakes, the dispersal ability of bacterioplankton was higher than that of phytoplankton and zooplankton. Niche plasticity might be greater among smaller organisms and is possible that short-lived and smaller organisms are not as tenacious as larger organisms in a dynamic colonization-extinction equilibrium. This finding is consistent with Soininen et al. (2013), who found that the predictability of phytoplankton or zooplankton distributions was stronger than that of bacteria. We also found that microeukaryotic communities were much less similar to each other overall than bacterial communities in both wet and dry seasons. The observed spatial patterns of each microbial community could be influenced by potential factors related to its own ecological characteristics and requirements (Soininen et al. 2011). Although bacterial and microeukaryotic communities responded to the same environmental conditions, the different population abundance, generation time, and organism size of each community have led to different speciation, extinction, or dispersal rates (Mazaris et al. 2010; Yang et al. 2010; Hanson et al. 2012).
Environmental factors regulating the microbial community
Clearly, variations in both bacterial and microeukaryotic communities responded to the same environmental factors with an exception of DO in this study. This synchrony was also reflected in their significant cross-taxon concordance. Temperature has been previously proven to have a significant effect on the dynamics and composition of plankton communities in rivers (Crump and Hobbie 2005; Chen et al. 2013; Liu et al. 2013) and in other natural environments such as lakes (Ju et al. 2014), thermal springs (Miller et al. 2009), and marine environments (Fuhrman et al. 2008). The composition of the bacterial and microeukaryotic communities may be both directly and indirectly affected by water temperature which is one of the main seasonal factors. As the range of optimal growth temperature for each phylotype is different, water temperature changes can result in variation of microbial community composition (Pomeroy and Wiebe 2001).
Further, our findings are consistent with reports of microbial communities’ responses to nutrient concentrations (Newton and McMahon 2011; Muscarella et al. 2014). Nutrient availability plays a critical role in driving the activities and large-scale distributions of freshwater bacteria and phytoplankton communities (Yang et al. 2012; Liu et al. 2014). Higher nutrient concentrations of TN, NH4-N, TP, and PO4-P occurred at upper river sites (sites 1, 2, and 3) that drain livestock farming areas (Liu et al. 2011). Animal feeding is the most significant source of NH4-N and PO4-P in existing inventories (Goebes et al. 2003). More recently, Li et al. (2015) reported that livestock production was the dominant influencing factor for the NH4-N loadings in agricultural watersheds. Although NH4-N is an essential nutrient in aquatic ecosystems, it is toxic to aquatic creatures in high concentrations (Ip et al. 2001). The higher NH4-N concentrations indicate a greater environmental risk to the aquatic ecosystem. Therefore, regulations and restrictions on agricultural activities are needed to restore and maintain water quality in Longyan City region. In addition, chlorophyll a was significantly related to bacterial community composition. It is well known that chlorophyll a can be used as a measure of algal biomass. During the dry season, the low current velocity and the high nutrient concentration were favorable to the growth of algae (phytoplankton) (Yang et al. 2012). Correlations between phytoplankton dynamics and bacterial composition have been reported (Pinhassi et al. 2004; Liu et al. 2014). It is possible that bacterial communities may be more strongly influenced by phytoplankton communities during the dry season.
Determinants of bacterial and microeukaryotic communities
The bacterial and microeukaryotic communities were significantly influenced by environmental, spatial, and temporal factors. For both bacterial and microeukaryotic communities, the variance explained by environmental factors was higher than that explained by either spatial or temporal factors. This suggests that the composition of microbial communities may be more related to site-specific conditions rather than watershed scale or seasonal factors. Similar results have been observed by other authors showing that differences in bacterial community were more closely related to variability in environmental factors that to spatial factors (Langenheder and Ragnarsson 2007; Liu et al. 2013, 2015). This suggests that the composition of microbial communities is largely dependent on niche-based processes (species distributions due to environmental factors) rather than on neutral processes (community dissimilarity caused by geographically limited dispersal). Fierer et al. (2007) have argued that environmental factors, not spatial factors, appear to have significant effects on bacterial community composition. It has to be emphasized that some of the variation was shared between the explanatory variables in both environmental and spatial factors. One possible explanation is that the spatial and temporal factors affecting community composition are closely tied to the changes in environmental factors.
The percentage of bacterial community variance explained by environmental factors was higher than for the microeukaryotic community. It is therefore presumed that niche-based processes played a larger role for bacterial communities. In the study by de Bie et al. (2012), the microorganisms were more strongly determined by the environment and showed weaker spatial patterns. Conversely, the microeukaryotic community variance explained by spatial or temporal factors was higher than that of the bacterial community, respectively. Our findings suggest that microeukaryotic community composition responds more to the changes in spatiotemporal factors than bacterial community, which better reflects the changes in environmental factors. This is because bacteria (smaller taxa) perceive the world at smaller spatial and temporal scales (Azovsky 2002). Recently, Soininen and Luoto (2014) found that trophic positions and body size of microorganisms are closely related to the predictability of aquatic species distributions. However, it should be noted that about 60 % of the variation in compositions of bacterial and microeukaryotic communities cannot be explained. The unexplained variation in the compositions of microbial communities might be related to unmeasured environmental factors, other biotic interactions (such as fish predation), and historical processes (Hanson et al. 2012). In fact, our species accumulation curves showed that the expected number of OTUs leveled off for both bacteria and microeukaryotes (Fig. S4); the detection limitation in DGGE analyses (estimated to be 1 % of the total abundance) might be a reason of the high percentage of unexplained variance (Muyzer et al. 1993).
Variation partitioning and path analysis are both ecological modeling methods for testing the effects of specific factors. While the explanatory and response variables are defined a priori in variation partitioning, path analysis determines the most likely causal modeling scenarios between different variables (Legendre and Legendre 1998). It seems that the combination of these two approaches should provide complementary ecological insights (Bienhold et al. 2011). Our path analysis revealed that both bacterial and microeukaryotic communities were most directly affected by the environmental parameters. The effect of space and time on microbial community composition was probably mediated by environmental parameters (Fig. 6). Further, the microeukaryotic community played key roles in shaping bacterial community composition. This phenomenon can be explained by several mechanisms. We found Ciliophora were the most dominant and diverse zooplankton taxa. According to the food web theory, the predators play a key role in controlling aquatic microbial communities (van Hannen et al. 1999). The nutritional mode of most groups of ciliates is bacterivore; thus, they are important consumers of bacteria (Mostajir et al. 2015). A rotifer species (Brachionus plicatilis) detected in our study can also feed on microbial bacteria and protists (Arndt 1993). Additionally, the predation of bacteria by ciliates and rotifers allows the recycling of various nutrients that are mobilized for bacterial growth (Sherr and Sherr 1987; Mostajir et al. 2015). One genus (Salpingoeca sp.) of heterotrophic nanoflagellates (HNFs) belonging to the choanoflagellates was recorded in our study. HNFs are one of the most important bacterivorous groups in aquatic ecosystems, and their abundance varies greatly in different pelagic habitats (Barcina et al. 1991; Pernthaler 2005). Perhaps, riverine conditions (turbulence, turbidity, and nutrient level) and PCR primers bias could result in a relatively low number of HNFs taxa detected in our study (Weitere and Arndt 2003; Pernthaler 2005; Yu et al. 2015). In both mesocosm experiments and natural systems, phytoplankton species are known to play a key role in shaping bacterial community composition through complex interactions with nutrients and signaling molecules (Pinhassi et al. 2004; Kent et al. 2007; Liu et al. 2014; Amin et al. 2015). Dissolved organic matter released from phytoplankton has been reported as a key good-quality carbon source to bacteria and it can be consumed and remineralized by bacteria (Chrost and Faust 1983; Obernosterer and Herndl 1995). In this study, three important phytoplankton taxa (Chlorophyta, Chrysophyta, and Dinophyceae) accounted for 35.6 % of the microeukaryotic sequences. It has been reported that both Chlorophyta and Chrysophyta were significantly related with bacterial community composition (van Hannen et al. 1999; Liu et al. 2014). Further, our RDA analysis showed that chlorophyll a was one of the factors that most influence the variations in the bacterial communities. This relationship, however, did not account for other unmeasured parameters which might simultaneously affect bacterial and microeukaryotic communities. Our results identified that the microeukaryotic community played a key role in shaping bacterial community—a result relevant to the understanding of ecosystem functioning and the microbial loop in river ecosystem, although the interaction mechanism between microeukaryotic and bacterial communities needs further study (Amin et al. 2015).
In conclusion, bacterial and microeukaryotic communities showed similar spatiotemporal variability within the Jiulong River ecosystem. Both communities were overall strongly spatially structured in dry season, and the microeukaryotic communities exhibited a stronger distance-decay relationship than bacterial communities—thereby indicating dispersal limitation of microorganisms can matter at organism characteristics and seasonal hydrology change. The bacterial and microeukaryotic communities were controlled by similar environmental factors, and variations in both communities were explained more by environmental factors than spatial and seasonal factors. However, the spatial or seasonal factors showed an indirect but significant relationship with both microbial communities. In addition, this study suggests that microeukaryotic community may strongly impact bacterial community through top down effects and complex interactions, and should be taken into account in aquatic microbial community analyses. Due to the existence of a large number of rare microbial species in the natural ecosystems, next-generation sequencing approaches are needed for relatively detailed analyses of the rare biosphere community (Portillo et al. 2012; Liu et al. 2015). To prove the generality of our conclusions, complete studies of more microbial taxa at different spatial scales will be needed. Further research on river microbial plankton should be helpful in establishing a relationship between the structure and function of the microbial food webs and developing better microbe-based monitoring methods in river ecosystems.
References
Amin SA, Hmelo LR, van Tol HM, Durham BP, Carlson LT, Heal KR, Morales RL, Berthiaume CT, Parker MS, Djunaedi B, Ingalls AE, Parsek MR, Moran MA, Armbrust EV (2015) Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522:98–101
Arndt H (1993) Rotifers as predators on components of the microbial web (bacteria, heterotrophic flagellates, ciliates)—a review. Hydrobiologia 255:231–246
Astorga A, Oksanen J, Luoto M, Soininen J, Virtanen R, Muotka T (2012) Distance decay of similarity in freshwater communities: do macro- and microorganisms follow the same rules? Glob Ecol Biogeogr 21:365–375
Azovsky AI (2002) Size-dependent species-area relationships in benthos: is the world more diverse for microbes? Ecography 25:273–282
Barcina I, Ayo B, Muela A, Egea L, Iriberri J (1991) Predation rates of flagellate and ciliated protozoa on bacterioplankton in a river. FEMS Microbiol Lett 85:141–149
Beier S, Witzel KP, Marxsen J (2008) Bacterial community composition in Central European running waters examined by temperature gradient gel electrophoresis and sequence analysis of 16S rRNA genes. Appl Environ Microbiol 74:188–199
Bianchi F, Acri F, Aubry FB, Berton A, Boldrin A, Camatti E, Cassin D, Comaschi A (2003) Can plankton communities be considered as bio-indicators of water quality in the lagoon of Venice? Mar Pollut Bull 46:964–971
Bienhold C, Boetius A, Ramette A (2011) The energy–diversity relationship of complex bacterial communities in Arctic deep-sea sediments. ISME J 6:724–732
Borcard D, Legendre P (2002) All-scale spatial analysis of ecological data by means of principal coordinates of neighbour matrices. Ecol Model 153:51–68
Chen NW, Hong HS (2012) Integrated management of nutrients from the watershed to coast in the subtropical region. Curr Opin Environ Sustain 4:233–242
Chen NW, Hong HS, Zhang LP, Cao WZ (2008) Nitrogen sources and exports in an agricultural watershed in Southeast China. Biogeochemistry 87:169–179
Chen ZB, Zhou ZY, Peng X, Xiang H, Xiang SN, Jiang ZX (2013) Effects of wet and dry seasons on the aquatic bacterial community structure of the three Gorges Reservoir. World J Microbiol Biotechnol 29:841–853
Chrost RH, Faust MA (1983) Organic carbon release by phytoplankton: its composition and utilization by bacterioplankton. J Plankton Res 5:477–493
Clarke KR, Gorley RN (2001) PRIMER v5: user manual/tutorial. PRIMER-E, Plymouth
Crump BC, Hobbie JE (2005) Synchrony and seasonality in bacterioplankton communities of two temperate rivers. Limnol Oceanogr 50:1718–1729
Crump BC, Adams HE, Hobbie JE, Kling GW (2007) Biogeography of bacterioplankton in lakes and streams of an arctic tundra catchment. Ecology 88:1365–1378
de Bie T, De Meester L, Brendonck L, Martens K, Goddeeris B, Ercken D, Hampel H, Denys L, Vanhecke L, Van der Gucht K, Van Wichelen J, Vyverman W, Declerck SAJ (2012) Body size and dispersal mode as key traits determining metacommunity structure of aquatic organisms. Ecol Lett 15:740–747
Díez B, Pedrós-Alió C, Marsh TL, Massana R (2001) Application of denaturing gradient gel electrophoresis (DGGE) to study the diversity of marine picoeukaryotic assemblages and comparison of DGGE with other molecular techniques. Appl Environ Microbiol 67:2942–2951
Fierer N, Morse JL, Berthrong ST, Bernhardt ES, Jackson RB (2007) Environmental controls on the landscape-scale biogeography of stream bacterial communities. Ecology 88:2162–2173
Freimann R, Bürgmann H, Findlay SE, Robinson CT (2013) Bacterial structures and ecosystem functions in glaciated floodplains: contemporary states and potential future shifts. ISME J 7:2361–2373
Fuhrman JA (2009) Microbial community structure and its functional implications. Nature 459:193–199
Fuhrman JA, Steele JA, Hewson I, Schwalbach MS, Brown MV, Green JL, Brown JH (2008) A latitudinal diversity gradient in planktonic marine bacteria. Proc Natl Acad Sci U S A 105:7774–7778
Goebes MD, Strader R, Davidson C (2003) An ammonia emission inventory for fertilizer application in the United States. Atmos Environ 37:2539–2550
Goldenberg A, Dam H, Loon EE, Vonk JA, Der HG, Admiraal W (2014) Eutrophication decreases distance decay of similarity in diatom communities. Freshw Biol 59:1522–1531
Green J, Bohannan BJ (2006) Spatial scaling of microbial biodiversity. Trends Ecol Evol 21:501–507
Hall T (1999) Biological sequence alignment editor for Win95/98/NT/2K/XP. IBIS Biosciences, Carlsbad
Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JBH (2012) Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Microbiol 10:497–506
Hu AY, Yang XY, Chen NW, Hou LY, Ma Y, Yu CP (2014) Response of bacterial communities to environmental changes in a mesoscale subtropical watershed, Southeast China. Sci Total Environ 472:746–756
Hullar MA, Kaplan LA, Stahl DA (2006) Recurring seasonal dynamics of microbial communities in stream habitats. Appl Environ Microbiol 72:713–722
Ip YK, Chew SF, Randall DJ (2001) Ammonia toxicity, tolerance, and excretion. In: Wright PA, Anderson PM (eds) Fish physiology, vol. 19 nitrogen excretion. Academic Press, New York, pp. 109–148
Ju LH, Yang J, Liu LM, Wilkinson DM (2014) Diversity and distribution of freshwater testate amoebae (Protozoa) along latitudinal and trophic gradients in China. Microb Ecol 68:657–670
Kent AD, Yannarell AC, Rusak JA, Triplett EW, McMahon KD (2007) Synchrony in aquatic microbial community dynamics. ISME J 1:38–47
Langenheder S, Ragnarsson H (2007) The role of environmental and spatial factors for the composition of aquatic bacterial communities. Ecology 88:2154–2161
Lear G, Washington V, Neale M, Case B, Buckley H, Lewis G (2013) The biogeography of stream bacteria. Glob Ecol Biogeogr 22:544–554
Legendre P, Legendre L (1998) Numerical ecology. Elsevier Science BV, Amsterdam
Lepš J, Šmilauer P (2003) Multivariate analysis of ecological data using CANOCO. Cambridge University Press, Cambridge
Li YY, Jiao JX, Wang Y, Yang W, Meng C, Li BZ, Li Y, Wu JS (2015) Characteristics of nitrogen loading and its influencing factors in several typical agricultural watersheds of subtropical China. Environ Sci Pollut Res 22:1831–1840
Liu LM, Yang J, Zhang YY (2011) Genetic diversity patterns of microbial communities in a subtropical riverine ecosystem (Jiulong River, southeast China). Hydrobiologia 678:113–125
Liu LM, Yang J, Yu XQ, Chen GJ, Yu Z (2013) Patterns in the composition of microbial communities from a subtropical river: effects of environmental, spatial and temporal factors. PLoS One 8:e81232
Liu LM, Yang J, Lv H, Yu Z (2014) Synchronous dynamics and correlations between bacteria and phytoplankton in a subtropical drinking water reservoir. FEMS Microbiol Ecol 90:126–138
Liu LM, Yang J, Yu Z, Wilkinson DM (2015) The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. doi:10.1038/ismej.2015.29
Mazaris AD, Moustaka-Gouni M, Michaloudi E, Bobori DC (2010) Biogeographical patterns of freshwater micro- and macroorganisms: a comparison between phytoplankton, zooplankton and fish in the eastern Mediterranean. J Biogeogr 37:1341–1351
Miller SR, Strong AL, Jones KL, Ungerer MC (2009) Bar-coded pyrosequencing reveals shared bacterial community properties along the temperature gradients of two alkaline hot springs in Yellowstone National Park. Appl Environ Microbiol 75:4565–4572
Mostajir B, Amblard C, Buffan-Dubau E, De Wit R, Lensi R, Sime-Ngando T (2015) Microbial food webs in aquatic and terrestrial ecosystems. In: Bertrand JC, Caumette P, Lebaron P, Matheron R, Normand P, Sime-Ngando T (eds) Environmental microbiology: fundamentals and applications. Springer, The Netherlands, pp. 485–509
Muscarella ME, Bird KC, Larsen ML, Placella SA, Lennon JT (2014) Phosphorus resource heterogeneity in microbial food webs. Aquat Microb Ecol 73:259–272
Muyzer G, De Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Nekola JC, White PS (1999) The distance decay of similarity in biogeography and ecology. J Biogeogr 26:867–878
Newton RJ, McMahon KD (2011) Seasonal differences in bacterial community composition following nutrient additions in a eutrophic lake. Environ Microbiol 13:887–899
Obernosterer I, Herndl GJ (1995) Phytoplankton extracellular release and bacterial growth: dependence on the inorganic N: P ratio. Mar Ecol Prog Ser 116:247–257
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H (2013) Vegan: community ecology package. R package version 2.0-6
Pantelić D, Svirčev Z, Simeunović J, Vidović M, Trajković I (2013) Cyanotoxins: characteristics, production and degradation routes in drinking water treatment with reference to the situation in Serbia. Chemosphere 91:421–441
Pernthaler J (2005) Predation on prokaryotes in the water column and its ecological implications. Nat Rev Microbiol 3:537–546
Pinhassi J, Sala MM, Havskum H, Peters F, Guadayol O, Malits A, Marrasé C (2004) Changes in bacterioplankton composition under different phytoplankton regimens. Appl Environ Microbiol 70:6753–6766
Pomeroy LR, Wiebe WJ (2001) Temperature and substrates as interactive limiting factors for marine heterotrophic bacteria. Aquat Microb Ecol 23:187–204
Portillo MC, Anderson SP, Fierer N (2012) Temporal variability in the diversity and composition of stream bacterioplankton communities. Environ Microbiol 14:2417–2428
Read DS, Gweon HS, Bowes MJ, Newbold LK, Field D, Bailey MJ, Griffiths RI (2015) Catchment-scale biogeography of riverine bacterioplankton. ISME J 9:516–526
Robert P, Escoufier Y (1976) A unifying tool for linear multivariate statistical methods: the RV-coefficient. Appl Stat 25:257–265
Schäfer H, Muyzer G (2001) Denaturing gradient gel electrophoresis in marine microbial ecology. Method Microbiol 30:425–468
Schermelleh-Engel K, Moosbrugger H, Müller H (2003) Evaluating the fit of structural equation models: tests of significance and descriptive goodness-of-fit measures. Method Psychol Res 8:23–74
Sherr EB, Sherr BF (1987) High rates of consumption of bacteria by pelagic ciliates. Nature 325:710–711
Soininen J, Luoto M (2014) Predictability in species distributions: a global analysis across organisms and ecosystems. Glob Ecol Biogeogr 23:1264–1274
Soininen J, Korhonen JJ, Karhu J, Vetterli A (2011) Disentangling the spatial patterns in community composition of prokaryotic and eukaryotic lake plankton. Limnol Oceanogr 56:508–520
Soininen J, Korhonen JJ, Luoto M (2013) Stochastic species distributions are driven by organism size. Ecology 94:660–670
Staley C, Gould TJ, Wang P, Phillips J, Cotner JB, Sadowsky MJ (2014) Bacterial community structure is indicative of chemical inputs in the Upper Mississippi River. Front Microbiol 5:524
Stomp M, Huisman J, Mittelbach GG, Litchman E, Klausmeier CA (2011) Large-scale biodiversity patterns in freshwater phytoplankton. Ecology 92:2096–2107
van Hannen EJ, Mooij W, van Agterveld MP, Gons HJ, Laanbroek HJ (1999) Detritus-dependent development of the microbial community in an experimental system: qualitative analysis by denaturing gradient gel electrophoresis. Appl Environ Microbiol 65:2478–2484
Wang YM, Yang J, Liu LM, Yu Z (2015) Quantifying the effects of geographical and environmental factors on distribution of stream bacterioplankton within nature reserves of Fujian, China. Environ Sci Pollut Res 22:11010–11021
Weitere M, Arndt H (2003) Structure of the heterotrophic flagellate community in the water column of the River Rhine (Germany). Eur J Protistol 39:287–300
Yang J, Smith HG, Sherratt TN, Wilkinson DM (2010) Is there a size limit for cosmopolitan distribution in free-living microorganisms? A biogeographical analysis of testate amoebae from polar areas. Microb Ecol 59:635–645
Yang J, Yu XQ, Liu LM, Zhang WJ, Guo PY (2012) Algae community and trophic state of subtropical reservoirs in southeast Fujian, China. Environ Sci Pollut Res 19:1432–1442
Yu LY, Zhang WJ, Liu LM, Yang J (2015) Determining microeukaryotic plankton community around Xiamen Island, southeast China, using Illumina MiSeq and PCR-DGGE techniques. PLoS One 10:e0127721
Zhang X, Zhang DD, Zhang H, Lou ZX, Yan CZ (2012) Occurrence, distribution, and seasonal variation of estrogenic compounds and antibiotic residues in Jiulongjiang River, South China. Environ Sci Pollut Res 19:1392–1404
Acknowledgments
We would like to thank Dr. Xian Zhang for the assistance in field sampling and Dr. David M. Wilkinson for the comments on an earlier version of this paper. This work was supported by the National Natural Science Foundation of China (31370471 and U1133601) and the Natural Science Foundation for Distinguished Young Scholars of Fujian Province (2012J06009).
Compliance with ethical standards
The authors have declared that no competing interests exist. No specific permissions were required for these activities. Informed consent was obtained from all participants and this article did not contain any studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 444 kb)
Rights and permissions
About this article

Cite this article
Wang, Y., Liu, L., Chen, H. et al. Spatiotemporal dynamics and determinants of planktonic bacterial and microeukaryotic communities in a Chinese subtropical river. Appl Microbiol Biotechnol 99, 9255–9266 (2015). https://doi.org/10.1007/s00253-015-6773-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6773-0