
Abstract
The DNA region encoding the filipin gene cluster in Streptomyces avermitilis (pte) contains a PAS-LuxR regulatory gene, pteF, orthologue to pimM, the final pathway-specific positive regulatory protein of pimaricin biosynthesis in Streptomyces natalensis. Gene replacement of the gene from S. avermitilis chromosome resulted in a severe loss of filipin production and delayed spore formation in comparison to that of the wild-type strain, suggesting that it acts as a positive regulator of filipin biosynthesis and that it may also have a role in sporulation. Complementation of the mutant with a single copy of the gene integrated into the chromosome restored wild-type phenotypes. Heterologous complementation with the regulatory counterpart from S. natalensis also restored parental phenotypes. Gene expression analyses in S. avermitilis wild-type and the mutant by reverse transcription-quantitative polymerase chain reaction of the filipin gene cluster suggested the targets for the regulatory protein. Transcription start points of all the genes of the cluster were studied by 5′-rapid amplification of complementary DNA ends. Transcription start point analysis of the pteF gene revealed that the annotated sequence in the databases is incorrect. Confirmation of target promoters was performed by in silico search of binding sites among identified promoters and the binding of the orthologous regulator for pimaricin biosynthesis PimM to gene promoters by electrophoretic mobility shift assays. Precise binding regions were investigated by DNAse I protection studies. Our results indicate that PteF activates the transcription from two promoters of polyketide synthase genes directly, and indirectly of other genes of the cluster.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Streptomyces are soil-dwelling filamentous bacteria well-known for their ability to produce a variety of antibiotics and other secondary metabolites. Production of these compounds is regulated in response to nutritional status alteration, population density and a variety of environmental conditions and hence occurs in a growth-phase-dependent manner and usually accompanied by morphological differentiation (Martín et al. 2000; Bibb 2005; Liu et al. 2013).
Filipin is a 28-membered ring pentaene macrolide antifungal antibiotic produced by Streptomyces filipinensis, Streptomyces avermitilis and other Streptomyces strains (Whitfield et al. 1955; Ikeda et al. 2014; Kim et al. 2012). It represents the archetype of non-glycosylated polyenes, and as a polyene, it interacts with membrane sterols, thus causing the alteration of membrane structure and leading to the leakage of cellular materials (Aparicio et al. 2004). Contrary to most polyene macrolides which display higher affinity for ergosterol (the main sterol in fungal membranes) than for other sterols, filipin shows a similar affinity for cholesterol and ergosterol. This property makes it useless for its application in human therapy due to its toxic side effects but has permitted its application as a tool for the diagnosis of Niemann-Pick type C disease, a characteristic cholesterol overloaded lysosomal disorder of genetic origin (Kruth et al. 1986), and as a probe for the detection and quantification of cholesterol in cellular membranes (Gimpl and Gehrig-Burger 2011). As other macrocyclic polyketides, filipin is synthesized by the action of type I modular polyketide synthases. Its biosynthetic gene cluster (pte) has been identified in the avermectin-producing S. avermitilis NRRL 8165 upon sequencing of its genome and encodes 14 polyketide synthase modules within five multifunctional enzymes and eight additional proteins that presumably govern modification of the polyketide skeleton and regulation of gene expression (Fig. 1a) (Omura et al. 2001; Ikeda et al. 2003). In nature, filipin is produced as a mixture of related compounds known as the filipin complex (filipins I to IV) (Fig. 1b) (Bergy and Eble 1968), being filipin III the major component.

Organizations of the filipin biosynthetic gene cluster and structure of filipins. a The pointed boxes indicate the direction of transcription. The pteF gene is indicated in black and the polyketide synthase genes are shown as stripped pointed boxes. The arrows indicate deduced transcriptional units. Square boxes are DNA fragments used in mobility shift experiments (sizes (bp) are indicated below). b Filipin I: R1 = H, R2 = H; filipin II: R1 = OH, R2 = H; filipin III: R1 = OH, R2 = OH. Filipin IV is a stereoisomer of filipin III
Control of secondary metabolite production is a complex process involving multiple signals and an intricate network of regulators that cross-talk with each other (Martín and Liras 2010). Generally, the lowest level of this network is played by regulatory genes that only affect a single antibiotic biosynthetic pathway. These pathway-specific regulatory genes are usually found within the respective antibiotic biosynthesis gene cluster. Two distinct regulators of filipin biosynthesis are encoded by genes located in the pte cluster, pteR and pteF (Ikeda et al. 2003).
PteR is orthologous to the transcriptional activator of pimaricin biosynthesis PimR, an archetype of a particular class of regulators that combine an N-terminal DNA-binding domain corresponding to the SARP family of transcriptional activators with a C-terminal half homologous to guanylate cyclases and large ATP-binding regulators of the LuxR family (LAL) (Antón et al. 2004). The C-terminal half includes the ATP/GTP-binding domain characteristic of these protein families but lacks the characteristic signature sequence at the N-terminus of guanylate cyclases or the LuxR-type helix-turn-helix (HTH) motif for DNA binding present at the C-terminus of LAL regulators. Members of this class also include the nikkomycin activator in Streptomyces ansochromogenes SanG (Liu et al. 2005) or the polyoxin regulator in Streptomyces cacaoi PolR (Li et al. 2009). Recently, we have characterized the PimR mode of action and determined that it binds an operator that contains three heptameric direct repeats of the consensus CGGCAAG with 4 bp spacers (Santos-Aberturas et al. 2012).
In turn, PteF is orthologous to PimM, the second activator of pimaricin biosynthesis (Antón et al. 2007). These PAS/LuxR regulators combine an N-terminal PAS sensory domain with a C-terminal HTH motif of the LuxR type. Recently, we have carried out the molecular characterization of PimM mode of action and determined the canonical binding site of this regulator as CTVGGGAWWTCCCBAG, just at the -35 hexamer of regulated promoters in Streptomyces natalensis (Santos-Aberturas et al. 2011b). Noteworthy, PimM homologous regulatory proteins have been found to be encoded in all known biosynthetic gene clusters of polyene polyketides (Santos-Aberturas et al. 2011b). PimR and PimM act in a hierarchical way, PimR binds pimM promoter and activates its transcription (Santos-Aberturas et al. 2012), and the gene product of the latter activates transcription from eight different promoters of pimaricin structural genes directly (Santos-Aberturas et al. 2011a).
In order to understand if there is a molecular mechanism of polyene control common to all actinomycetes, it was of great interest to study the transcriptional control of the filipin genes (pte) of S. avermitilis by the pathway-specific regulator PteF. In this work, we have generated mutants of the regulator and characterized its mode of action.
Materials and methods
Bacterial strains and cultivation
S. avermitilis NRRL 8165 was routinely grown in yeast extract-malt extract (YEME) medium (Kieser et al. 2000) without sucrose. Sporulation was generally achieved in TBO medium (Higgens et al. 1974) at 30 °C. Spore production was assessed in YEME, TBO and minimal medium (Kieser et al. 2000) by spreading 2.5 × 106 spores of the mutant and the parental strain and incubation at 30 °C for 5, 7 and 10 days. After making the spore suspension, spore counting was done by plating serially diluted spore suspension on TSA medium. Escherichia coli strain DH5α was used as a host for DNA manipulation. E. coli BL21 (DE3) was used for expression studies. E. coli BW25113 [pIJ790] was used for gene replacement experiments. E. coli ET12567 [pUZ8002] was used as donor in intergeneric conjugations. Candida utilis (syn. Pichia jadinii) CECT 1061 was used for bioassay experiments.
Plasmids and DNA manipulation procedures
Standard genetic techniques with E. coli and in vitro DNA manipulations were as described by Sambrook and Russell (2001). Recombinant DNA techniques in Streptomyces species and isolation of Streptomyces total DNA were performed as previously described (Kieser et al. 2000). Southern hybridization was carried out with probes labelled with digoxigenin by using the DIG DNA labelling kit (Roche Biochemicals). Intergeneric conjugation between E. coli ET12567 [pUZ8002] and S. avermitilis was performed as described (Enríquez et al. 2006). pUC19 (New England Biolabs) was used as the routine cloning vector, pSETneo (AmR, KanR, pUC18 replicon, ΦC31 attP; Vicente et al. 2009) was used for intergeneric conjugation and pJMDBD was the vector used for protein expression (Santos-Aberturas et al. 2011a). Polymerase chain reactions were carried out using Phusion DNA Polymerase as described by the enzyme supplier (Finnzymes). DNA sequencing was accomplished by the dideoxynucleotide chain-termination method using the Perkin Elmer Amplitaq Gold Big Dye-terminator sequencing system with an Applied Biosystems ABI 3130 DNA genetic analyzer (Foster City, CA, USA).
Construction of ΔpteF mutant
Deletion of pteF from S. avermitilis chromosome was made by replacing the wild-type gene with a cassette containing an apramycin selective marker using a PCR-based system (Gust et al. 2003). The plasmid pIJ773 containing the apramycin resistance gene (aac(3)IV) and the oriT replication origin was used as a template. The mutant was constructed using the oligonucleotides 5′-ccgcgcgagcgggcgccgggagggcccgatgcccgctcaTGTAGGCTGGAGCTGCTTC-3′ and 5′-gttcaacatctggcgataccgctggtcccagggccgatgATTCCGGGGATCCGTCGACC-3′ as the forward and reverse primers, respectively (the sequence identical to the DNA segment upstream from the start codon of pteF is in lower case italics and the sequence identical to the segment downstream from the stop codon of pteF is underlined and in lower case). These two long PCR primers (59 and 58 nt) were designed to produce a deletion of pteF just after its start codon, leaving only its stop codon behind. The 3′ sequence of each primer matches the right or left end of the disruption cassette (the sequence is shown uppercase in both primers). The extended resistance cassette was amplified by PCR, and E. coli BW25113 [pIJ790] bearing cosmid CL240_D05 was electro-transformed with this cassette. The isolated mutant cosmid was introduced into non-methylating E. coli ET12567 containing the RP4 derivative pUZ8002. The mutant cosmid was then transferred to S. avermitilis by intergeneric conjugation. Double cross-over exconjugants were screened for their apramycin resistance followed by confirmation by both PCR and Southern blot analysis (Fig. S1).
Construction of plasmids for gene complementation
In order to complement pteF replacement mutant, a 1,122-bp DNA fragment containing the entire pteF gene plus its own promoter was amplified by PCR with primers pteF fw (5′-GAGGTGCTTCTCCCGTG-3′) and pteF rev (5′-CCATCGTGCTTGCGG-3′) using S. avermitilis chromosomal DNA as template. The PCR product was ligated into a EcoRV-cut pSETneo to yield pSETneo::pteF.
For the heterologous complementation of S. avermitilis ΔpteF, the entire pimM gene plus its own promoter region were amplified by PCR using the primers pimM + P forward (5′-GGTTGAATTCTTGCGGTCGGTGGTGCGGGC-3′) and pimM + P reverse (5′-GTCGTCCTCGGGTTCTTGCGG-3′) using S. natalensis chromosomal DNA as template. The PCR product was ligated into a EcoRV-digested pSETneo to obtain pSETneo::pimM.
Isolation of total RNA
For RNA extraction, 2 ml from liquid cultures in YEME medium without sucrose was harvested by centrifugation and immediately frozen by immersion in liquid nitrogen. Cells were resuspended in lysis solution [600 μl RLT buffer (RNeasy Mini Kit; Qiagen); 60 μl 2-mercaptoethanol] and disrupted using a sonicator (Ultrasonic processor XL; Misonix Inc.). RNeasy ® Mini kit (Qiagen) was used for RNA isolation using RNase-Free DNase Set (Qiagen) as specified by the manufacturer, followed by two consecutive digestions with TURBOTM DNase from Ambion® according to the manufacturer’s instructions.
Reverse transcriptase-PCR experiments
Transcription was studied by using the SuperScriptTM One-Step reverse transcriptase-PCR (RT-PCR) system with Platinum® Taq DNA polymerase (Invitrogen), using 150 ng of total RNA as template. Conditions were as follows: first strand complementary DNA (cDNA) synthesis, 45 °C for 40 min followed by heating at 94 °C for 2 min; amplification, 30 cycles of 94 °C for 40 s, 63–67 °C (depending of the set of primers used) for 30 s, and 72 °C for 1 min. Primers (18–26 mers; Table S1) were designed to cover intergenic regions, generating PCR products of approximately 300–800 bp. Negative controls were carried out with each set of primers and Platinum® Taq DNA polymerase in order to confirm the absence of contaminating DNA in the RNA preparations. The identity of each amplified product was corroborated by direct sequencing of the PCR product.
Rapid amplification of cDNA ends
The 5′ ends of transcripts were identified by using a 5′ rapid amplification of cDNA ends (RACE) system for rapid amplification of cDNA ends kit (Invitrogen) following the manufacturer’s instructions (version 2.0). Briefly, first strand cDNA synthesis was carried out using 5 μg of total RNA, reverse transcriptase and the gene-specific primer (numbers 1 in Table S2). The cDNA was purified using the SNAP columns provided in the kit, and poly(dC) tails were added to the 3′ ends using terminal deoxynucleotidyl transferase. PCR amplification of the tailed cDNA was carried out using the 5′ RACE abridged anchor primer with the first nested primer (numbers 2 in Table S2). A dilution of the PCR mixture was then subjected to reamplification using the abridged universal amplification primer with the second nested primer (numbers 3 in Table S2). The PCR products were gel-purified and sequenced. When cDNA tailing with poly(dC) did not permit the identification of the transcription start point, poly(dA) tails were added to the 3′ ends of cDNA. In these cases, second strand cDNA synthesis was necessary prior to nested amplifications and was carried out using the 3′ RACE adapter primer (Invitrogen). PCR amplification of the cDNA was then carried out using the abridged universal amplification primer with the first nested primer (numbers 2 in Table S2). Final nested amplification was carried out as before.
Reverse transcription-quantitative PCR
Reverse transcription of total RNA was performed on selected samples with 5 μg of total RNA and 12.5 ng/μl of random hexamer primer (Invitrogen) using SuperScript™ III reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. All RNA samples were analyzed with the Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA) and only those with RNA integrity number (RIN) values (Schroeder et al. 2006) raging from 8.0 to 9.0 were selected. Each reaction was performed in 20 μl with SYBR® Premix Ex TaqTM (TaKaRa), and 100–350 nM of each primer and the template cDNA 1:2 diluted and run on a StepOnePlus Real-Time PCR system (Applied Biosystems). Reactions were carried out on three biological replicates with three technical replicates each and appropriate controls were included to verify the absence of gDNA contamination in RNA and primer-dimer formation. Primers (see Table S3) were designed to generate PCR products between 96 and 153 bp, near the 5′-end of messenger RNA (mRNA) using the Primer3 software (Rozen and Skaletsky 2000). The PCR reactions were initiated by incubating the sample at 95 °C for 10 min followed by 40 cycles at 95 °C for 15 s, 62–70 °C (depending of the set of primers used) for 34 s. To check the specificity of real-time PCR reactions, a DNA melting curve analysis was performed by holding the sample at 60 °C for 60 s followed by slow ramping of the temperature to 95 °C. SYBR fluorescence was normalized by ROX fluorescence. Baseline and threshold values were determined by the StepOnePlus software. C t values were normalized with respect to rrnA1 mRNA (encoding 16S ribosomal RNA (rRNA)). Relative changes in gene expression were quantified using the Pfaffl (2001) method and the REST© software (Pfaffl et al. 2002). The corresponding real-time PCR efficiency (E) of one cycle in the exponential phase was calculated according to the equation E = 10[−1/slope] (Rasmussen 2000) using fivefold dilutions of genomic DNA raging from 0.013 to 40 ng (n = 5 or 6 with three replicates for each dilution) with a coefficient of determination R 2 > 0.98 (Fig. S2).
DNA-protein binding assays
DNA binding tests were performed by electrophoretic mobility shift assay (EMSA). The DNA fragments used for EMSA were amplified by PCR using the primers listed in Table S4, and either directly labelled at both ends with digoxigenin with DIG Oligonucleotide 3′-End Labeling Kit, 2nd Generation (Roche Applied Science) or first cloned into pUC19, and then amplified by PCR using the universal and reverse primers, one of them labelled with 6-carboxyfluorescein. Binding assays were performed with GST-PimMDBD, protein as described previously (Santos-Aberturas et al. 2011b, 2012).
Footprinting assays
DNase I footprinting assays were performed by the fluorescent labelling procedure, using the GST-PimMDBD protein as described in Santos-Aberturas et al. (2011a). The DNA fragment used was the same as that used for EMSA experiments labelled with 6-carboxyfluorecein as described above. In each case, the same unlabelled oligonucleotide served to prime the sequencing reaction. The PCR products were purified after agarose-gel electrophoresis and DNA concentrations were determined with a NanoDrop ND-1000 spectrophotometer (Thermo Scientific).
DNase I footprinting was performed by incubating 0.28 pmol of the DNA probe and GST-PimMDBD protein for 10 min at 30 °C. Lyophilized bovine pancreas DNase I (Roche grade I) was reconstituted in 20 mM Tris-HCl pH 7.0, 50 mM NaCl, 100 μg/ml BSA, 1 mM DTT and 10 % glycerol to a final concentration of 2.5 × 10−3 units/μl. Nuclease digestions were carried out with 0.01 units (4 μl) at 30 °C for 1 min and stopped with 120 μl of 40 mM EDTA in 9 mM Tris-HCl pH 8.0. After phenol-chloroform purification and ethanol precipitation, samples were loaded in an Applied Biosystems ABI 3130 DNA genetic analyzer (Foster City, CA, USA). Results were analysed with the Peak Scanner program (Applied Biosystems).
Assay of filipin production
To assay filipin in culture broths, 8 ml of culture was extracted with 16 ml of ethyl acetate and the organic phase was vacuum-dried and resuspended in 200 μl of pure methanol. Solutions of pure filipin III (Sigma) were used as control. Quantitative determination of filipin III was assessed by reverse-phase high performance liquid chromatography (HPLC) using a Waters 600 unit coupled to a PDA 996 detector equipped with a Mediterranea Sea C18 column (4.6 × 150 mm, particle size 3 μm) (Teknokroma). Elution was performed with a gradient (0.8 ml/min) of methanol-0.1 % formic acid according to the following program: 50:50 v/v 0–3 min, up to 90:10 v/v 3–12 min, 90:10 12–20 min, up to 100:0 v/v 20–21 min, 100:0 v/v 21–23 min, down to 0:100 v/v 23–24 min, 0:100 v/v 24–26 min, up to 50:50 v/v 26–27 min, 50:50 v/v 27–32 min. Retention time for filipin III was 18.2 min. When required, the fungicidal activity of filipin was tested by bioassay using C. utilis CECT 1061 as test organism.
Bioinformatic analysis
Candidate sequences to contain promoters were analyzed using the Patser algorithm (Hertz and Stormo 1999), implemented in the web resource Regulatory Sequence Analysis Tools (van Helden 2003). The pseudocount value was set to 10, and the alphabet parameter was adjusted to the GC content of Streptomyces genome: AT, 0.15; CG, 0.35. The matrices used to search for regions -35 and -10 were those derived from the alignments of class C and class A promoters of Bourn and Babb (1995). To search for a combination of ‘class C–n nucleotides of separation–class A’, we included n columns of null values in the combined matrix. To calculate the information content (R i value) of individual sequences (Schneider 1997) and to obtain the logo of the binding site of the regulator PimM, we used the BiPad Server (Bi and Rogan 2006).
Results
Organization of pte cluster transcriptional units
In order to define an overall picture of the transcriptional arrangement of the pte genes, an accurate identification of operons was needed. We thus decided to analyze the possible co-transcription of neighbouring genes by reverse transcriptase-polymerase chain reaction experiments. Total RNA was prepared from S. avermitilis after growth for 36 h (when filipin is actively produced). Primers were designed to obtain cDNAs corresponding to unabated transcription between two genes (Table S1). Transcripts were analyzed after 40 PCR cycles to ensure that even low-level transcribed genes were detected. A primer pair designed to amplify a cDNA of the rrnA1 gene was used as an internal control. These analyses were carried out at least three times for each primer pair. Following this strategy, pteA3, pteA4, pteA5, pteB, pteC, pteD and pteE could be co-transcribed since unabated transcription was observed between the upstream and the downstream gene, though pteD has its own promoter (see below). Similarly, pteA1 and pteA2 and pteF and pteG could form bicistronic transcripts, though pteA2 and pteG have their own promoters (see below). No transcripts were detected connecting pteA2 and pteA3 or pteG and pteH, thus suggesting that pteA3 and pteH should have their own promoters. The genes pteR and pteF must also have their own promoters as can be deduced from their chromosomal arrangement in a divergent manner, and the same applies to pteA1 which is arranged divergently from its upstream gene. Figure 1 shows the deduced organization of transcriptional units.
Inactivation of pteF reduces filipin production, and gene complementation restored antifungal production
In order to determine the function of pteF, we inactivated it by using the REDIRECT gene replacement technology as indicated in “Materials and methods”. Double-crossover mutants were screened by apramycin resistance. These were verified by both PCR and Southern blot analysis (Fig. S1).
The new mutant strain S. avermitilis ∆pteF showed delayed sporulation when compared with S. avermitilis wild-type when grown on solid YEME, MM or TBO media (at least 1 day later) (Fig. 2), whereas in submerged cultures, its growth was slightly higher (Fig. 3). Regardless of sporulation delay, the spore counts of both strains were similar after growth for 14 days at 30 °C on the three solid media.

The effect of pteF deletion on sporulation. Spores of the parental and the mutant strains were plated on YEME, TBO and MM agar followed by incubation at 30 °C for 5 or 7 days

Inactivation of pteF impairs filipin production, and gene complementation restores antifungal production. a Left: time course quantification of the filipin III production in S. avermitilis (black) and S. avermitilis ∆pteF (grey). Right: growth curves. b Quantification of the filipin III production attained by S. avermitilis ∆pteF complemented with pteF (dark grey) and pimM (white) genes after 48 and 72 h of growth. S. avermitilis::pSETneo was used as control (black). Data are the average of three flasks. Vertical bars indicate the standard deviation values. The three strains showed similar growth profiles
The fermentation broth, produced by the new mutant strain, was extracted with ethyl acetate and analyzed for the presence of filipin III (the major component of the filipin complex). HPLC assays indicated that filipin production by the mutant strain was severely reduced when compared with the parental strain. S. avermitilis ∆pteF produced only about 38 % of the filipin III accumulated by the wild-type strain at 48 h (51.1 μg/l) (Fig. 3). These results suggested that PteF was a positive modulator of filipin biosynthesis.
To confirm that the gene deletion was directly responsible for the impairment on filipin production, we complemented the mutant with pteF. A DNA fragment containing pteF plus its putative promoter region was inserted into the integrative vector pSETneo, giving rise to pSETneo::pteF (see “Materials and methods”). The plasmid was then transferred from E. coli ET12567 [pUZ8002] to S. avermitilis ∆pteF by conjugation. pSETneo was also introduced into S. avermitilis wild-type as control. Introduction of pSETneo::pteF restored filipin biosynthesis to the control levels (Fig. 3) and also its sporulation pace, thus suggesting that the mutant phenotype was due to the regulator-encoding gene knockout.
Positive heterologous complementation suggests functional conservation of PAS-LuxR regulators
Previous studies with PteF orthologous regulator PimM had shown that gene complementation of S. natalensis ∆pimM mutants with single copies of other PAS/LuxR counterparts restored pimaricin production, thus suggesting the functional conservation of these regulators (Santos-Aberturas et al. 2011b). To further corroborate this finding, we studied the effect of introducing one copy of pimM into the genome of S. avermitilis ∆pteF. For that purpose, we transferred the integrative plasmid pSETneo::pimM (see “Materials and methods”) by conjugation from E. coli ET12567 [pUZ8002] to the above said strain as described (Enríquez et al. 2006). pSETneo::pteF was also introduced as a control. Interestingly, introduction of the construct restored completely the ability of S. avermitilis ∆pteF to produce filipins (Fig. 3).
Quantitative analysis of expression of pte genes in the mutant
Total RNA was prepared from S. avermitilis wild-type and the mutant after growth for 36 h and used as template for gene expression analysis by reverse transcription-quantitative polymerase chain reaction (RT-qPCR). The expression levels of all pte genes in the mutant in relation to those of the wild-type strain (assigned a relative value of 1) are shown in Fig. 4. These analyses were carried out at least three times for each primer pair.

Gene expression analysis of the pte cluster in the mutant by RT-qPCR. Gene expression was assessed using RT-qPCR with the primers indicated in Table S3. The relative values are referred to 1, the assigned relative value for the expression of each gene in S. avermitilis NRRL 8165. The expression of rrnA1 (encoding 16S rRNA) was used as control. Error bars were calculated by measuring the standard deviation of the ratio value among three biological and three technical replicates of each sample. The RNA templates were from 36-h cultures grown in YEME medium without sucrose. Fold change values are indicated below
As expected, all structural biosynthetic genes (namely pteA1 to pteA5) showed reduced transcription in S. avermitilis ∆pteF, indicating that the promoters controlling the expression of these genes were likely targets, directly or indirectly, for regulatory control. According to our results, these promoters were pteA1p, pteA2p and pteA3p. Control on the transcription from any of these promoters, which drive the transcription/formation of polyketide synthases in charge of building the polyketide backbone of filipin (Ikeda et al. 2003), is sufficient to explain the reduced filipin production in the mutant. Interestingly, expression values among pteA3, pteA4 and pteA5 varied considerably (Fig. 4). Given that they belong to the same operon, it is possible that the multicistronic transcript could be processed and subject to different rates of RNA degradation.
Similarly, the expression of the 2-octenoyl-CoA carboxylase/reductase encoding gene pteB (Yoo et al. 2011) and the cytochrome P450 encoding genes pteC and pteD (Xu et al. 2010) was also reduced. The reduced expression of pteB and pteC is in agreement with the reduced expression of pteA3, pteA4 and pteA5 given that they are thought to be co-transcribed from pteA3p. The reduced transcription of pteD, however, could not have been predicted since this gene has its own promoter and suggests that such promoter (pteDp) is also a target for regulatory control.
Strikingly, pteR expression was dramatically triggered in the mutant, showing some 25-fold more expression than in the parental strain. This clearly indicates that pteR promoter is a target of PteF control, although this control turned out to be indirect (see below). This result is in clear contrast to what occurs in S. natalensis, where pimR expression is not affected in S. natalensis ∆pimM mutant (Santos-Aberturas et al. 2012). Given that filipin production is reduced in the mutant when compared with the parental strain, this result could indicate that PteF either acts directly as a negative regulator on pteR promoter or it activates a negative regulator controlling pteR expression.
Interestingly, pteG and pteH expression were also triggered in the mutant. This is also in contrast with what occurs in S. natalensis where expression of both counterparts (pimI and pimE) is reduced in S. natalensis ∆pimM mutant.
In silico analysis of PimM targets within the pte cluster reveals the binding sites of PteF
Given the high degree of conservation between PteF and PimM regulators (94 % identity, Santos-Aberturas et al. 2011b), we searched for the presence of sequences similar to PimM binding site throughout the filipin cluster from S. avermitilis. We thus found four matching sequences, two in the intergenic region between pteA1 and its upstream divergent gene (SAV420) and another two upstream the intergenic region between pteA1 and pteA2, i.e. within pteA1 coding sequence. According to PimM mode of action, these operators should lie in regulated promoters; thus, the first two would lie within the pteA1 promoter region and the remaining two in the pteA2 promoter. Table 1 shows the sequences and R i values of the binding sites found. The sequences found in pteA1p coincide with the binding sites already identified by Santos-Aberturas et al. (2011b) and have been corroborated experimentally, whereas the sequences found in pteA2p have not been described previously. This is because according to the results presented here, pteA2 promoter lies within the coding sequence of the upstream gene pteA1, and in this position, some 180 nucleotides upstream from pteA1 stop codon was unexpected (see below). It is noteworthy that no operator was identified in the promoter region of pteA3, thus suggesting that the control exerted by PteF on pteA3A4A5BC transcription must be indirect. Similarly, no operators were found in the promoters of pteD, pteG, pteH or the bidirectional promoter region pteFR, thus suggesting an indirect control of PteF on the expression of these genes.
PimMDBD binds two targets within the filipin cluster
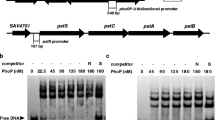
Incubation of GST-PimMDBD with each labelled DNA fragment from the putative promoter regions selected was assessed (Fig. 5) using an electrophoretic mobility shift assay (EMSA). For each experiment, two negative control reactions were performed: absence of protein and use of GST (isolated separately). The appearance of retarded band(s) was observed upon incubation of GST-PimMDBD with all the promoter regions selected, whereas it was not with the upstream regions of genes which did not contain sequences that fit the consensus. The intensity of the retarded band(s) was diminished by the addition of the same unlabeled DNA and increased when a higher concentration of protein was used (Fig. 5).

Electrophoretic mobility analyses (EMSA). a Electrophoretic mobility analysis of GST-PimM binding to different promoter regions. Promoter names are indicated above the pictures. All experiments were carried out with 10-ng labelled DNA probe and 20 nM of GST-PimMDBD protein. b Left panel: binding to pteA1 and pteA2 promoter regions with increasing concentrations of GST-PimMDBD. Central panel: competition experiment between labelled pteA2p and unlabeled pteA2p and between pteA2p and pteA3p. Note that 150-fold-higher concentrations of pteA3p competitor DNA failed to decrease the intensity of the pteA2p retardation bands. Both experiments were performed with 11 nM of GST-PimMDBD protein. Right panel: control reactions with 60 μM of pure GST protein. In all cases, lane C indicates control without protein
As expected, the regions retarded by GST-PimMDBD were the pteA1 promoter (two retardation bands) and the pteA2 promoter (two shifted bands) (Fig. 5b). Upstream regions of other genes such as pteA3, pteA4, pteR, pteH or pteG were not retarded, indicating that PimM does not interact with them. In all cases, control reactions made with pure GST protein were negative, excluding a possible binding of this protein to the promoters. The specificity of binding of GST-PimMDBD to target promoters was previously demonstrated by competition EMSA assays (Santos-Aberturas et al. 2011a).
Characterization of promoters
To determine the transcriptional start sites of promoters, 5′-RACE experiments were carried out. Once the +1 sites were known, the corresponding -10 and -35 boxes of each promoter were established by comparison with the matrices reported by Bourn and Babb (1995) for Streptomyces that take into account the nucleotides occurring in 13-nucleotide stretches, including the -10 or -35 consensus hexamers (see “Materials and methods”). Results are summarized in Fig. 6. Only the promoters identified above gave positive results in 5′-RACE experiments.

Transcriptional start site of promoters. The position of the transcriptional start site was determined by 5′ RACE. The putative -10 and -35 hexanucleotides are in bold type and underlined. Scores resulting from the comparison to the matrices reported by Bourn and Babb (1995) for Streptomyces are indicated between brackets. The TSP is indicated by a bent arrow and bold type letter. Nucleotides showing homology with the 16S RNA, which could form a ribosome-binding site, are in bold and labelled RBS. The start codon is boxed. Protected nucleotide sequences (PteF operators) are indicated with shaded boxes
The pteH transcription start point (TSP) is located at a thymine 212 bp upstream from the GTG start codon. Analysis of the region upstream of the TSP revealed that the -10 box with the highest score to the consensus Streptomyces was GAACCT, centred at eight nucleotides from the start site. A search using combined class C-class A matrices (Bourn and Babb 1995) revealed a -35 box ACTACG separated by 19 nucleotides, with a score of 2.02.
The TSP of pteG is located at a guanine 295 bp upstream from the GTG codon. Analysis of the region upstream of the TSP revealed the presence of a -10 box CAGCGC (score 1.62), centred at 12 nucleotides from the start site, and a -35 box CGCACG separated by 15 nucleotides.
A single RACE product of approximately 300 bp was observed for pteF. Interestingly, the TSP transcription start point corresponds to a guanine located 30 bp downstream from the currently annotated ATG start codon, i.e. internal to the pteF coding sequence annotated in the databases, thus proving that such annotation is incorrect (Fig. 6). The sequence GGCCTT (score 1.35), centred at position -10, constitutes the -10 consensus, and a -35 box AGGGCC (score 1.63) was identified at 18-nt distance. Given that pteF was wrongly annotated, we have reassigned pteF start codon to the next ATG methionine-encoding codon located 120 nt downstream. Noteworthy, this new start codon coincides with the one present in the pimM coding sequence. Thus, TSP is located 90 bp upstream the new ATG codon.
In the case of pteR, the TSP was located at a guanine situated 69 bp upstream from the ATG codon. The -10 and -35 boxes (CAGGAT and TTGGAA, respectively) were centred at positions -10 and -33 from the TSP and are separated by 17 nt (Fig. 6).
For pteD, the TSP was identified at a guanine situated 51 bp upstream from the ATG codon. The Patser analysis of the upstream sequence revealed AAACCT and CGCACG as the -10 and -35 boxes (scores 2.68 and 2.28, respectively). Both boxes are separated by 17 nt, with the -10 hexamer centred at 10 nt from the TSP.
pteA3 TSP corresponds to a cytosine located at 196 bp from the ATG start codon, and the analysis of the sequence directly upstream revealed the presence of the -10 box as CAACAT and the -35 box as ACGGTG, with 19 nt separating them (scores 3.05 and 1.26, respectively). Noteworthy, this TSP is located 161 nt upstream from pteA2 stop codon.
In the case of pteA2, a single RACE product of approximately 450 bp was observed, thus supporting the conclusion that this gene has its own promoter. The TSP of pteA2 is located at a guanine 255 bp upstream from the GTG start codon (inside the pteA1 coding sequence). Analysis of the region upstream of the TSP revealed the presence of a -10 box AAGAAT (score 2.18), centred at 10 nucleotides from the start site, and a -35 box TTGCCG (score 3.37) separated by 18 nucleotides (Fig. 6). Noteworthy, one of the protected regions in the coding strand lays 11 nt upstream from the TSP site, covering the -35 hexamer box of the promoter (Fig. 7).

a–d DNase I footprints of the GST-PimMDBD protein bound to the promoter region of pteA2. In each panel, the upper electropherogram (blue line) is the control reaction. The protected nucleotide sequence is boxed; hyper-sensitive sites (arrows) are also indicated. Sequencing reactions are not included except in panel (a). Coordinates are from the translation start point
The pteA1 TSP is located at a guanine 88 bp upstream from the ATG codon, the -10 box CACACT is centred at 10 nt from the TSP and the -35 box CTGCCG is at an 18-nt distance (Fig. 6). Strikingly, and contrary to what occurs in pteA2p and in S. natalensis, in this case, the protected regions did not cover the -35 hexamer of the promoter and were located at 61 and 109 nt from the TSP.
Dnase I protection studies reveal control over pteA2 promoter
The promoter region of pteA2 shown above to be retarded in EMSA was studied by DNase I protection analysis. GST-PimMDBD protein (35 μM) was tested using a 5′-end fluorescein-labelled DNA fragment. Analyses were carried out in triplicate.
Results showed two major protected areas in each strand separated by some 173 nucleotides (Fig. 7), in agreement with the appearance of two retardation bands in EMSA experiments. The region closest to pteA2 coding strand is 31-nucleotide long (positions -92 to -62 with respect to the pteA2 translational GTG start site). The length of the protection of the reverse strand was 38 bp (positions -84 to -47), both regions being slightly displaced. The second protected area extended for 47 bp of the coding strand (positions -312 to -266 with respect to pteA2 translational start site). The length of the protection of the reverse strand of the pteA2 promoter was 43 bp (positions -320 to -278; Fig. 7), and both regions are displaced 8–12 nucleotides. Interestingly, DNase I hypersensitive positions flanked the protected sequence, indicating altered DNA topology after incubation with GST-PimMDBD. Some hypersensitive positions were also found inside the target sequence (Fig. 7), thus suggesting that PimM bends DNA, making those positions accessible to DNase I digestion. As expected, the protected regions contained sequences TTCGGGATTTCCTGAC and CTAGTGGATTCCCTAC that fitted well to the consensus (R i of 13.23 and 11.08, respectively; Table 1).
Discussion
Two distinct regulators of filipin biosynthesis are encoded by genes located in the pte cluster, pteR and pteF. pteR belongs to a particular class of regulators that combine an N-terminal SARP domain with a C-terminal half homologous to guanylate cyclases and LAL regulators (Antón et al. 2004). Regulators of this class not only include other polyene regulators such as the pimaricin regulator PimR (Santos-Aberturas et al. 2012) but also regulators of peptidyl nucleoside antibiotic biosynthesis such as nikkomycins (SanG) and polyoxins (PolR) (Liu et al. 2005; Li et al. 2009). In turn, PteF is a PAS/LuxR regulator. PAS/LuxR regulators combine an N-terminal PAS sensor domain with a C-terminal HTH motif of the LuxR type for DNA binding and are characteristic of polyene gene clusters (Santos-Aberturas et al. 2011b). The PAS domain detects a physical or chemical stimulus and regulates, in response, the activity of the effector domain (Möglich et al. 2009). Unlike most other sensors, proteins containing PAS domains are located in the cytosol, and therefore they detect internal signals, but they can also sense environmental factors that cross the cell membrane. The severe reduction of filipin production upon inactivation of pteF indicates that it behaves as an activator of filipin biosynthesis. Albeit contrary to what occurs in S. natalensis where inactivation of pteF counterpart pimM blocks completely polyene biosynthesis, in S. avermitilis, such inactivation did not halt it. This is reflected in the level of transcription of pte polyketide synthase genes which are still transcribed in the mutant although at a reduced rate. Notably, heterologous complementation of the mutant with the counterpart from the pimaricin gene cluster resulted in restoration of filipin production to parental levels, thus corroborating the functional conservation of these pathway-specific regulators (Santos-Aberturas et al. 2012).
Given the high degree of conservation between PteF and PimM regulators, we searched for the presence of sequences similar to the recently described PimM canonical operator (Santos-Aberturas et al. 2011b) throughout the filipin cluster from S. avermitilis, finding four such operators, two in the promoter region of pteA1 (in the intergenic region between pteA1 and its divergent gene SAV420) and another two in the upstream region of pteA2 (both inside pteA1 coding sequence). The presence of operators inside pteA1 coding sequence was unexpected and suggested the location of pteA2 promoter at that position. Interestingly, identification of the transcriptional start points by 5′-RACE corroborated the location of pteA2 promoter at some 180 nt upstream from pteA1 stop codon. Similarly, the TSP of pteA3 is also located inside the upstream pteA2 gene.
The existence of promoters inside upstream gene coding sequences is not unprecedented. Recently, Robles-Reglero et al. (2013) described the internal promoters for several hlm genes for holomycin production in Streptomyces clavuligerus. An internal promoter essential for sporulation has also been described in the spoIIIAF gene of Bacillus subtilis (Guillot and Moran 2007), and several internal promoters have been described in Escherichia coli, including the P2 promoter in the trp operon, which is located inside trpD (Horowitz and Platt 1982), among others.
Electrophoretic mobility shift assays were used to prove the direct binding of PimM to those regions. Two shift bands were observed upon incubation of PimM with the pteA1 promoter, thus confirming that there were two operators in such region. Similarly, two retardation bands were observed for the pteA2 promoter, thus suggesting that PteF controls the transcription of both genes directly. EMSAs with a different DNA probe covering pteA1p had been previously carried out with identical results (Santos-Aberturas et al. 2011b), whereas the unexpected location of pteA2 promoter well inside the coding region of pteA1 explains why previous attempts to perform EMSA with PimM and the intergenic region between pteA1 and pteA2 were unsuccessful (Santos-Aberturas et al. 2011b).
The lack of binding of PimM to the promoter regions of pteA3, which is thought to drive the expression of the multicistronic operon pteA3A4A5BC, pteD, which drives the expression of the bicistron pteDE, pteR or pteG suggests that the differential expression of these genes upon gene disruption of pteF is mediated by the action of another hierarchical regulator/s which would be activated by PteF. To our knowledge, there are no precedents for such conjecture, although it is conceivable given that the cross-talk between regulators has been described in the literature in several occasions (Huang et al. 2005; Santos-Beneit et al. 2009). Further experimental analyses will be required to test these hypotheses.
Footprinting analysis of pteA2p revealed two protected sequences in each strand. The protected regions in the sense strand were accompanied by protections in the complementary strand, both protected sequences being slightly displaced. This is in agreement with the binding of one monomer of GST-PimMDBD to each strand of the operator. Protected sequences were flanked by DNase I hypersensitive positions, suggesting an altered DNA topology after incubation with the protein. Some hypersensitive positions were also found inside the target sequences, thus suggesting that PimM bends DNA, making those positions accessible to DNase I digestion, as it does with its natural operators in S. natalensis (Santos-Aberturas et al. 2011a). Identification of the transcriptional start point in the pteA2 promoter revealed that one of the regulator binding sites overlaps with the putative -35 region, as occurs in S. natalensis; however, identification of the TSP in pteA1p unveiled that the two protected regions were far away from the -35 hexamer of the promoter. Although this interaction still corresponds to a class II activation mechanism, where PteF would contact domain 4 of the RNA polymerase σ subunit resulting in recruitment of RNA polymerase to the promoter, the significance of this variability is unclear (Browning and Busby 2004).
Transcription start point analysis of the pteF gene identified a TSP internal to the pteF coding sequence annotated in the databases, thus suggesting that the currently annotated sequence of pteF is incorrect. The coding sequence deposited in the databases starts with an ATG start codon 120 nt away from the new ATG proposed here. Hence, this new start codon would yield a protein 40 amino acid residues shorter. This circumstance was already pinpointed by Antón et al. (2007) and is corroborated here. Besides, this TSP is preceded by conserved -10 and -35 hexamer sequences (Bourn and Babb 1995). Bearing in mind that several of the pteF homologues deposited in the databases are likely to have been annotated by comparison with pteF, based on our results, we encourage the revision of their starting codons.
Interestingly, besides being an activator of polyene production by recruiting RNA polymerase to given cluster promoters such as pteA1p or pteA2p, our results indicate that PteF must activate or repress other regulators, hence controlling indirectly the expression of other genes of the pte cluster. This pleiotropic behaviour is also reflected in the differential spore formation pace observed between the mutant and the parental strain. The possibility that the severe reduction on filipin production is the consequence of the delay in sporulation is unlikely given that the mutant and the wild-type strain show identical growth profiles and that the final spore titres after 14 days of growth are identical. Rather, it is conceivable that this regulator could bind operators in other sites of the chromosome, hence affecting other processes besides polyene biosynthesis, and those could be involved in sporulation. It could also be argued that filipin is required for a normal sporulation profile, but the existence of filipin non-producing mutants not affected in sporulation precludes such possibility (unpublished).
It is interesting to note that polyene production is very low in S. avermitilis. Filipin III production in the wild-type strain ranged from 45 to 150 ng/ml at early stationary phase in complex media such as YEME without sucrose (Fig. 3). These values are extremely low when we compare them with the 800–1,000 μg pimaricin/ml of a standard fermentation of S. natalensis in the same growth medium (Recio et al. 2006). This represents some four orders of magnitude less than other polyene producers. Several factors may be the reason for such low production, the first one maybe the reduced expression of pte genes (unpublished), although other reasons cannot be excluded such as the high variability of fermentation yields. This variability maybe due to the described genetic instability of S. avermitilis (Novák et al. 1993; Chen et al. 2010), which is particularly relevant for the filipin gene cluster given its situation near one of the ends of the chromosome. Additionally, the relaxed control of PteF over its cognate promoters compared to that of PimM may also contribute to the low yields observed. Future studies will hopefully provide an answer to this question.
References
Antón N, Mendes MV, Martín JF, Aparicio JF (2004) Identification of PimR as a positive regulator of pimaricin biosynthesis in Streptomyces natalensis. J Bacteriol 186:2567–2575. doi:10.1128/JB.186.9.2567-2575.2004
Antón N, Santos-Aberturas J, Mendes MV, Guerra SM, Martín JF, Aparicio JF (2007) PimM, a PAS domain positive regulator of pimaricin biosynthesis in Streptomyces natalensis. Microbiology 53:3174–3183. doi:10.1099/mic.0.2007/009126-0
Aparicio JF, Mendes MV, Antón N, Recio E, Martín JF (2004) Polyene macrolide antibiotic biosynthesis. Curr Med Chem 11:1645–1656. doi:10.2174/0929867043365044
Bergy ME, Eble TE (1968) The filipin complex. Biochemistry 7:653–659. doi:10.1021/bi00842a021
Bi C, Rogan PK (2006) BIPAD: a web server for modeling bipartite sequence elements. BMC Bioinforma 7:76. doi:10.1186/1471-2105-7-76
Bibb MJ (2005) Regulation of secondary metabolism in Streptomycetes. Curr Opin Microbiol 8:208–215. doi:10.1016/j.mib.2005.02.016
Bourn WR, Babb B (1995) Computer assisted identification and classification of streptomycete promoters. Nucleic Acids Res 23:3696–3703. doi:10.1093/nar/23.18.3696
Browning DF, Busby SJ (2004) The regulation of bacterial transcription initiation. Nat Rev Microbiol 2:57–65. doi:10.1038/nrmicro787
Chen W, He F, Zhang X, Chen Z, Wen Y, Li J (2010) Chromosomal instability in Streptomyces avermitilis: major deletion in the central region and stable circularized chromosome. BMC Microbiol 10:198. doi:10.1186/1471-2180-10-198
Enríquez LL, Mendes MV, Antón N, Tunca S, Guerra SM, Martín JF, Aparicio JF (2006) An efficient gene transfer system for the pimaricin producer Streptomyces natalensis. FEMS Microbiol Lett 257:312–318. doi:10.1111/j.1574-6968.2006.00189.x
Gimpl G, Gehrig-Burger K (2011) Probes for studying cholesterol binding and cell biology. Steroids 76:216–231. doi:10.1016/j.steroids.2010.11.001
Guillot C, Moran CP (2007) Essential internal promoter in the spoIIIA locus of Bacillus subtilis. J Bacteriol 189:7181–7189. doi:10.1128/JB.00915-07
Gust B, Challis GL, Fowler K, Kieser T, Chater KF (2003) PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A 100:1541–1546. doi:10.1073/pnas.0337542100
Hertz GZ, Stormo GD (1999) Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15:563–577. doi:10.1093/bioinformatics/15.7.563
Higgens CE, Hamill RL, Sands TH, Hoehn MM, Davis NE (1974) The occurrence of deacetoxycephalosporin C in fungi and streptomycetes. J Antibiot 27:298–300. doi:10.7164/antibiotics.27.298
Horowitz H, Platt T (1982) Identification of trp-p2, an internal promoter in the tryptophan operon of Escherichia coli. J Mol Biol 156:257–267. doi:10.1016/0022-2836(82)90327-8
Huang J, Shi J, Molle V, Sohlberg B, Weaver D, Bibb MJ, Karoonuthaisiri N, Lih CJ, Kao CM, Buttner MJ, Cohen SN (2005) Cross-regulation among disparate antibiotic biosynthetic pathways of Streptomyces coelicolor. Mol Microbiol 58:1276–1287. doi:10.1111/j.1365-2958.2009.06624.x
Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S (2003) Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol 21:526–531. doi:10.1038/nbt820
Ikeda H, Shin-ya K, Omura S (2014) Genome mining of the Streptomyces avermitilis genome and development of genome-minimized hosts for heterologous expression of biosynthetic gene clusters. J Ind Microbiol Biotechnol 41:233–250. doi:10.1007/s10295-013-1327-x
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. John Innes Foundation, Norwich
Kim JD, Han JW, Hwang IC, Lee D, Kim BS (2012) Identification and biocontrol efficacy of Streptomyces miharaensis producing filipin III against Fusarium wilt. J Basic Microbiol 52:150–159. doi:10.1002/jobm.201100134
Kruth HS, Comlyg ME, Butlerll JD, Vanierl MT, Finks JK, Wenger DA, Patel S, Pentchev PG (1986) Type C Niemann-Pick disease: abnormal metabolism of low density lipoprotein in homozygous and heterozygous fibroblasts. J Biol Chem 261:16769–16774
Li R, Xie Z, Tian Y, Yang H, Chen W, You D, Liu G, Deng Z, Tan H (2009) polR, a pathway-specific transcriptional regulatory gene, positively controls polyoxin biosynthesis in Streptomyces cacaoi subsp. asoensis. Microbiology 155:1819–1831. doi:10.1099/mic.0.028639-0
Liu G, Tian Y, Yang H, Tan H (2005) A pathway-specific transcriptional regulatory gene for nikkomycin biosynthesis in Streptomyces ansochromogenes that also influences coloy development. Mol Microbiol 55:1855–1866. doi:10.1111/j.1365-2958.2005.04512.x
Liu G, Chater KF, Chandra G, Niu G, Tan H (2013) Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol Mol Biol Rev 77:112–143. doi:10.1128/MMBR.00054-12
Martín JF, Liras P (2010) Engineering of regulatory cascades and networks controlling antibiotic biosynthesis in Streptomyces. Curr Opin Microbiol 13:263–273. doi:10.1016/j.mib.2010.02.008
Martín JF, Gutiérrez S, Aparicio JF (2000) Secondary metabolites. In: Lederberg J (ed) Encyclopedia of microbiology, 2nd edn. Academic, San Diego, pp 213–236, Vol. 4
Möglich A, Ayers RA, Moffat K (2009) Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 17:1282–1294. doi:10.1016/j.str.2009.08.011
Novák J, Kopecký J, Kofronová O, Vanĕk Z (1993) Instability of the production of avermectins, sporulation, and pigmentation in Streptomyces avermitilis. Can J Microbiol 39:265–267. doi:10.1139/m93-038
Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, Hattori M (2001) Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc Natl Acad Sci U S A 98:12215–12220. doi:10.1073/pnas.211433198
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi:10.1093/nar/29.9.e45
Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30:e36. doi:10.1093/nar/30.9.e36
Rasmussen R (2000) Quantification on the LightCycler. In: Meuer S, Wittwer C, Nakagawara K (eds) Rapid cycle real-time PCR, methods and applications. Springer, Heidelberg, pp 21–34
Recio E, Aparicio JF, Rumbero A, Martín JF (2006) Glycerol, ethylene glycol and propanediol elicit pimaricin biosynthesis in the PI-factor defective strain Streptomyces natalensis npi287 and increase polyene production in several wild type actinomycetes. Microbiology 152:3147–3156. doi:10.1099/mic.0.28953-0
Robles-Reglero V, Santamarta I, Álvarez-Álvarez R, Martin JF, Liras P (2013) Transcriptional analysis and proteomics of the holomycin gene cluster in overproducer mutants of Streptomyces clavuligerus. J Biotechnol 163:69–76. doi:10.1016/j.jbiotec.2012.09.017
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, pp 365–386
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. N.Y. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Santos-Aberturas J, Vicente CM, Guerra SM, Payero TD, Martín JF, Aparicio JF (2011a) Molecular control of polyene macrolide biosynthesis: direct binding of the regulator PimM to eight promoters of pimaricin genes and identification of binding boxes. J Biol Chem 286:9150–9161. doi:10.1074/jbc.M110.182428
Santos-Aberturas J, Payero TD, Vicente CM, Guerra SM, Cañibano C, Martín JF, Aparicio JF (2011b) Functional conservation of PAS-LuxR transcriptional regulators in polyene macrolide biosynthesis. Metab Eng 13:756–767. doi:10.1016/j.ymben.2011.09.011
Santos-Aberturas J, Vicente CM, Payero TD, Martín-Sánchez L, Cañibano C, Martín JF, Aparicio JF (2012) Hierarchical control on polyene macrolide biosynthesis: PimR modulates pimaricin production via the PAS-LuxR transcriptional activator PimM. PLoS One 7:e38536. doi:10.1371/journal.pone.0038536
Santos-Beneit F, Rodríguez-García A, Sola-Landa A, Martín JF (2009) Cross-talk between two global regulators in Streptomyces: PhoP and AfsR interact in the control of afsS, pstS and phoRP transcription. Mol Microbiol 72:53–68. doi:10.1111/j.1365-2958.2009.06624.x
Schneider TD (1997) Information content of individual genetic sequences. J Theor Biol 189:427–441. doi:10.1006/jtbi.1997.0540
Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:3. doi:10.1186/1471-2199-7-3
van Helden J (2003) Regulatory sequence analysis tools. Nucleic Acids Res 31:3593–3596. doi:10.1093/nar/gkg567
Vicente CM, Santos-Aberturas J, Guerra SM, Payero TD, Martín JF, Aparicio JF (2009) PimT, an amino acid exporter controls polyene production via secretion of the quorum sensing pimaricin-inducer PI-factor in Streptomyces natalensis. Microb Cell Fact 8:33. doi:10.1186/1475-2859-8-33
Whitfield GB, Brock TD, Ammann A, Gottlieb D, Carter HE (1955) Filipin, an antifungal antibiotic: isolation and properties. J Am Chem Soc 77:4799–4801. doi:10.1021/ja01623a032
Xu LH, Fushinobu S, Takamatsu S, Wakagi T, Ikeda H, Shoun H (2010) Regio- and stereospecificity of filipin hydroxylation sites revealed by crystal structures of cytochrome P450 105P1 and 105D6 from Streptomyces avermitilis. J Biol Chem 285:16844–16853. doi:10.1074/jbc.M109.092460
Yoo HG, Kwon SY, Kim S, Karki S, Park ZY, Kwon HJ (2011) Characterization of 2-octenoyl-CoA carboxylase/reductase utilizing pteB from Streptomyces avermitilis. Biosci Biotechnol Biochem 75:1191–1193. doi:10.1271/bbb.110003
Acknowledgments
This work was supported by the Spanish Ministerio de Economía y Competitividad (Grant BIO2010-19911 to JFA), F.P.U. fellowships of the Ministerio de Educación, Cultura y Deporte (AP2005-3644 to JSA, AP2007-02055 to TDP), a contract from the Junta de Castilla y León co-financed by the European Social Fund (to EGB) and a fellowship from the Portuguese Fundação para a Ciência e a Tecnologia (SFRH/BD/64006/2009 to CMV). We thank Prof. Haruo Ikeda from the Kitasato Institute for Life Sciences for providing cosmid CL240_D05.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 193 kb)
Rights and permissions
About this article

Cite this article
Vicente, C.M., Santos-Aberturas, J., Payero, T.D. et al. PAS-LuxR transcriptional control of filipin biosynthesis in S. avermitilis . Appl Microbiol Biotechnol 98, 9311–9324 (2014). https://doi.org/10.1007/s00253-014-5998-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5998-7