
Abstract
In filamentous fungi, the expression of secretory glycoside hydrolase encoding genes, such as those for amylases, cellulases, and xylanases, is generally repressed in the presence of glucose. CreA and CreB have been observed to be regulating factors for carbon catabolite repression. In this study, we generated single and double deletion creA and/or creB mutants in Aspergillus oryzae. The α-amylase activities of each strain were compared under various culture conditions. For the wild-type strain, mRNA levels of α-amylase were markedly decreased in the later stage of submerged culture under inducing conditions, whereas this reduced expression was not observed for single creA and double creA/creB deletion mutants. In addition, α-amylase activity of the wild-type strain was reduced in submerged culture containing high concentrations of inducing sugars, whereas all constructed mutants showed higher α-amylase activities. In particular, the α-amylase activity of the double deletion mutant in a medium containing 5 % starch was >10-fold higher than that of the wild-type strain under the same culture conditions. In solid-state cultures using wheat bran as a substrate, the α-amylase activities of single creA and double deletion mutants were >2-fold higher than that of the wild-type strain. These results suggested that deleting both creA and creB resulted in dramatic improvements in the production of secretory glycoside hydrolases in filamentous fungi.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The filamentous fungus Aspergillus oryzae has been used for producing traditional Japanese fermented foods, such as sake, soy sauce, and miso (soybean paste), for over a thousand years (Machida et al. 2008). A. oryzae can produce considerable amounts of amylolytic enzymes in the presence of maltooligosaccharides, such as starch and maltose, and the induction of the corresponding amylolytic genes is regulated by a fungal-specific Zn2Cys6 transcription activator, AmyR (Gomi et al. 2000; Petersen et al. 1999). In the presence of glucose, however, the production of amylolytic enzymes is repressed, even in the presence of inducing sugars (Gomi et al. 2000; Tada et al. 1989; Tonomura et al. 1961). Therefore, the production levels of amylolytic enzymes are limited by glucose, resulting from the enzymatic hydrolysis of starch or maltose used as inducers.
The glucose-induced repression of amylolytic genes is mediated by a well-known carbon catabolite repression (CCR) system. It is well established that CCR is regulated by the Cys2His2 type transcription factor CreA in filamentous fungi, particularly in Aspergillus nidulans (Dowzer and Kelly 1989, 1991; Kato et al. 1996). In addition to CreA that functions as a transcription repressor, CreB, CreC, and CreD have also been shown to be involved in CCR by modulating the ubiquitination/deubiquitination of the CreA protein in A. nidulans. creB and creC mutants also show defective CCR, and a creD mutation suppresses the creB and creC mutant phenotypes (Hynes and Kelly 1977; Kelly and Hynes 1977).
It has been shown that CreB and CreC form a complex in vivo and that the CreB protein is a ubiquitin processing protease (Lockington and Kelly 2001, 2002). In contrast, CreD belongs to a family of arrestin-like proteins and is presumably involved in recruiting a ubiquitin ligase to a target protein for ubiquitination (Boase and Kelly 2004).
In Trichoderma reesei, Rut-C30 and PC3-7 were identified as hyper-producing mutant strains of cellulases and hemicellulases, and these strains had mutations in cre1, an ortholog of creA (Ilmén et al. 1996; Porciuncula et al. 2013). In addition to these mutants, a cre1 deletion strain produced considerably high levels of these hydrolytic enzymes (Nakari-Setälä et al. 2009). Moreover, disrupting a creB ortholog (cre2) in T. reesei also resulted in increased cellulase activity under inducing conditions (Denton and Kelly 2011). These findings suggest that carbon catabolite derepression is highly effective for improving glycoside hydrolase production by filamentous fungi.
In this study, we generated single and double deletion creA and creB gene mutants in A. oryzae. The α-amylase production capabilities of these deletion mutants under submerged and solid-state cultures were compared with those of the wild-type strain.
Materials and methods
Strains and media
An A. oryzae ∆ligD::loxP pyrG-deficient strain (∆ligD::loxP, niaD −, sC −, pyrG −), which was derived from a ∆ligD::loxP strain (Mizutani et al. 2012) and provided by Dr. Mizutani at the National Research Institute of Brewing (Higashi-hiroshima, Japan), was used as the recipient strain for gene deletion. The ∆ligD::loxP strain was constructed from A. oryzae NS4 (niaD −, sC −) (Yamada et al. 1997; National Research Institute of Brewing Stock Culture) derived from the wild-type strain, A. oryzae RIB40 (National Research Institute of Brewing Stock Culture). Escherichia coli DH5α (endA1, hsdR17, supE44, recA1, gyrA96, thi-1, relA1, deoR, Φ80lacZΔM15, Δ(lacZYA − argF)U169) was used for the construction and propagation of plasmid DNAs. Saccharomyces cerevisiae BY4741 (MATa his3Δ leu2Δ met15Δ ura3Δ) was used for in vivo plasmid construction for gene deletion. The minimal medium (MM) for A. oryzae culture was Czapek–Dox (CD) medium, which contained 0.5 % (NH4)2SO4; 0.05 % KCl; 0.2 % KH2PO4; 0.05 % MgSO4; trace amounts of FeSO4, ZnSO4, CuSO4, MnSO4, Na2B4O7, and (NH4)6Mo7O24; and 1 % sugar, and supplemented with 0.0003 % (0.02 mM) methionine. For culture of the ∆ligD::loxP pyrG − strain, 0.2 % uracil was added to the medium. To assess α-amylase production in liquid medium, YP complete medium containing 0.5 % yeast extract and 1 % Bacto-peptone supplemented with a 1 or 5 % carbon source was used. For solid-state culture, 5 g of wheat bran (small particles were removed through a 10 standard mesh sieve; opening 1.7 mm) was moistened with 2 ml of distilled water after sterilization.
Construction of plasmid DNAs
The plasmids for creA and creB deletions were constructed as follows: To generate a plasmid for creA deletion, the 5′- and 3′-flanking regions of creA were amplified using PCR with primer sets P1 + P2 and P3 + P4 using the genomic DNA of A. oryzae RIB40 as the template. An A. nidulans sC cassette encoding ATP sulfurylase was prepared from pUSC (Yamada et al. 1997) digested with BamHI and PstI. A yeast vector, pYES2 (Life Technologies, Carlsbad, CA), was digested with EcoRI and BamHI. These four DNA fragments were assembled in S. cerevisiae using an endogenous homologous recombination system (Colot et al. 2006; Oldenburg et al. 1997), which resulted in ΔcreA/pYES2.
To generate a plasmid for creB deletion using sC as a selectable marker, the 3′-flanking region of creB was amplified by PCR with primers (designated creBdownsenPstI and creBdownanti) using the genomic DNA of A. oryzae RIB40 as the template. This amplified fragment was digested with PstI and inserted into PstI-digested pUSC, which resulted in creBdown/pUSC. The 5′-flanking region of creB was also amplified by PCR using the primers creBupsen and creBupantiBamHI and digested with KpnI and BamHI. This fragment was subsequently cloned into KpnI/BamHI-digested creBdown/pUSC, which generated ∆creB/pUSC.
To generate a plasmid for creB deletion using the orotidine-5′-decarboxylase gene, pyrG, as a selectable marker, a pyrG fragment was amplified by PCR with A. nidulans genomic DNA using the primers AnpyrGsen and AnpyrGantiBglII. The pyrG fragment was digested with BglII and ligated to BamHI-digested ∆creB/pUSC, which resulted in a ∆creB/pyrG plasmid.
To construct plasmid DNA for complementation of the pyrG-deficient strain, a pyrG fragment was amplified by PCR with A. nidulans genomic DNA using the primers AnpyrGsen and AnpyrGantiPstI. The fragment obtained was digested with PstI and BglII and inserted into PstI/BamHI-digested pUC119, which gave pUC/pyrG. A niaD fragment encoding nitrate reductase was obtained from a pNGA142 vector (Tamalampudi et al. 2007) by digestion with NruI and inserted into SmaI-digested pUC/pyrG, which generated pUC/pyrG/niaD. The nucleotide sequences of all primers used for plasmid construction are shown in Supplemental Table S1.
Fungal transformation
Transformation of A. oryzae was performed according to the method of Gomi et al. (1987). To obtain the DNA fragments for deleting creA and creB, ∆creA/pYES2 and ∆creB/pyrG plasmids were digested with NotI/KpnI and EcoRI/SphI, respectively. The pUC/pyrG/niaD plasmid was introduced into the recipient pyrG-deficient strain and ∆creA mutant for complementation of pyrG deficiency.
Southern blot analysis
A. oryzae genomic DNA preparation and Southern blot analysis were performed as previously described (Tanaka et al. 2012).
α-Amylase activity
α-Amylase activity was determined using the iodine–starch method (Sato et al. 2011). In a submerged culture of A. oryzae, secreted α-amylase is absorbed onto the fungal cell wall (Sato et al. 2011). Therefore, the mycelia harvested by filtration through Miracloth (EMD Chemicals Inc., San Diego, CA) were incubated in 0.1 M phosphate buffer (pH 7.0) for 60 min to release α-amylase that had been adsorbed on the fungal cell wall. The α-amylase activities in the extraction buffer and culture supernatant were summed for total α-amylase activity. After incubation in phosphate buffer, the mycelia were dried in a vacuum freeze drier (VD-550R; TAITEC, Saitama, Japan), after which the dry mycelia weight was determined. The α-amylase activity in submerged cultures was expressed as the activity per dry mycelia weight of the respective strains. To determine α-amylase activity in solid-state culture, wheat bran, on which A. oryzae had grown, was suspended in 50 ml of 10-mM acetate buffer (pH 5.0) and incubated for 3 h at room temperature to extract secreted proteins. Subsequently, wheat bran was removed by filtration through Miracloth, and α-amylase activity in the filtered sample was determined.
Quantitative RT-PCR analysis
To prepare total RNA, mycelia grown in liquid cultures were harvested and dried in a vacuum freeze drier. Total RNA was extracted from a freeze-dried mycelia suspension and disrupted in ISOGEN (Nippon Gene, Tokyo, Japan), according to the manufacturer's instructions. After treating total RNA with DNase I (TaKaRa Bio, Otsu, Japan), first-strand cDNAs were synthesized using M-MLV reverse transcriptase (Life Technologies, Carlsbad, CA) with an oligo(dT) primer, after which synthesized cDNA was treated with RNase H (Life Technologies, Carlsbad, CA). PCR reactions and subsequent analyses were performed using SYBR® Green PCR Master Mix and a StepOnePlus™ Real-Time PCR system (Life Technologies, Carlsbad, CA). The histone H4 gene was used as reference, and relative mRNA expression was calculated by the − ∆∆C T method (Livak and Schmittgen 2001). The nucleotide sequences of primers used for quantitative RT-PCR analysis are shown in Supplemental Table S1.
Results
Construction of single and double creA and creB deletion mutants
To investigate the effect of carbon catabolite derepression on amylolytic enzyme production, we generated gene deletion mutants for creA and creB orthologs in A. oryzae. The amino acid identities of A. oryzae CreA (AO090026000464) and CreB (AO090009000320) to those of A. nidulans were 80.6 and 70.5 %, respectively. DNA fragments for deleting creA and creB using the A. nidulans sC and pyrG genes as selectable markers were individually constructed, and the resulting DNA fragments were introduced into a recipient strain (Fig. 1a). These gene deletions were confirmed by PCR (data not shown) and Southern blot analysis (Fig. 1b). The creA deletion cassette was introduced into the ∆creB mutant, and a double deletion mutant (∆creA∆creB) was successfully obtained. For complementation of pyrG deficiency, pUC/pyrG/niaD plasmid DNA was introduced into the recipient and ∆creA mutant strains. For this study, the pyrG-complemented recipient was defined as the wild-type strain.

Construction of single and double deletion strains. a Strategies for creA and creB deletion by homologous recombination using A. nidulans sC and pyrG genes as selectable markers. ∆ligD::loxP pyrG − was used as the host strain for single deletion strains. To generate a double deletion strain, a DNA fragment for creA deletion was introduced into a creB deletion mutant. b Southern blot analysis of single and double deletion strains. The genomic DNAs of wild type (lanes 1 and 4), ∆creA (lane 2), ∆creA∆creB (lane 3), and ∆creB (lane 5) were digested with HpaI or NcoI, electrophoresed on a 0.8 % agarose gel, and transferred onto a nylon membrane. The labeled DNA fragments of the creA or creB upstream region were used as a probe
Because it is known that deleting creA causes severe morphological defects in filamentous fungi, we examined the growth characteristics of these gene deletion mutants. On MM agar plates, the ∆creA and ∆creA∆creB mutants showed distinctive growth defects compared with the wild-type strain (Fig. 2a). In contrast, the ∆creB mutant showed no apparent growth abnormalities (Fig. 2a). These phenotypes were identical to those of the creA and creB disruptants of other filamentous fungal species, such as A. nidulans, Neurospora crassa, and T. reesei (Denton and Kelly 2011; Hynes and Kelly 1977; Nakari-Setälä et al. 2009; Ziv et al. 2008). However, in submerged cultures with YP plus 1 % glucose (YPD) medium, the wild-type and ∆creB mutant strains generally grew in compact hyphal pellets, whereas the ∆creA and ∆creA∆creB mutants were more likely to grow in pellets of smaller size or with a pulpy-like morphology.

Growth of single and double deletion strains. a Growth on minimal agar plate. Approximately 1 × 104 conidiospores of each strain were grown on a minimal agar plate containing 1 % glucose as the sole carbon source at 30 °C for 3 days. WT wild type. b Growth in submerged culture. Approximately 2 × 106 conidiospores of each strain were grown in YP + 1 % glucose (YPD) medium at 30 °C, after which mycelia dry weights were determined. Error bars indicate the standard errors of three independent experiments
Mycelia weights of the ∆creA and ∆creA∆creB mutants in 24-h culture were relatively lower than those of the wild-type and ∆creB mutant strains, which was consistent with the growth defects observed on agar medium. However, after an additional 48-h culture, the ∆creA and ∆creA∆creB mutants showed rather better growth, which resulted in an increase in their dry mycelia weights compared with that of the wild-type and ∆creB mutant strains. This was presumably due to the differences in the growth morphologies of these mutants (Fig. 2b).
Carbon catabolite derepression by creA and creB deletions
To investigate the effects of deleting the creA and/or creB genes on CCR, each of these strains was grown on MM agar plates containing 1 % starch, with or without 1 % glucose. Clear zones formed by starch degradation were observed. On both types of agar plate, 0.25 % Triton X-100 was added to limit the colony size so that the clear zone could be clearly seen. The wild-type and all mutant strains showed clear zones around the colonies on the agar plates, containing starch as the sole carbon source (Fig. 3). In comparison, the formation of clear zones around wild-type strain colonies was strongly inhibited by adding glucose. In contrast, all of the deletion mutants formed distinct clear zones on starch medium supplemented with glucose (Fig. 3). This clearly indicated that CCR was relieved by deleting creB and creA.

Effects of gene deletions on carbon catabolite repression. Approximately 1 × 104 conidiospores of each strain were grown on minimal agar medium containing 1 % starch supplemented with or without 1 % glucose at 30 °C for 2 days. In both agar media, 0.25 % Triton X-100 was added to more clearly discriminate the clear zones
Comparisons of α-amylase activities in submerged culture
To examine the effects of the creA and/or creB deletions on α-amylase production, all transformants were grown in liquid YP medium supplemented with 1 % sugar, after which secreted α-amylase activity was measured. In YPD medium, the wild-type and ∆creB mutant strains produced small amounts of α-amylase, whereas the ∆creA and ∆creA∆creB mutants showed significantly high α-amylase activities (Fig. 4a). This indicated that deleting the creA gene resulted in inducing α-amylase production concomitant with CCR relief. This also suggested that glucose could act as an inducer for α-amylase production, as reported for A. nidulans by Murakoshi et al. (2012). Another study using carbon-limited chemostat cultures also suggested that α-amylase production by A. oryzae was induced by glucose (Carlsen and Nielsen 2001).

α-Amylase activities in submerged culture. Approximately 2 × 106 conidiospores of each strain were grown either in a YP + 1 % glucose (YPD) medium at 30 °C for 24 and 48 h or in b YP + 1 % starch (YPS) and YPS + 1 % glucose media for 48 h at 30 °C. Harvested mycelia were incubated in 100 mM phosphate buffer for 60 min to release α-amylase bound to the cell wall. The α-amylase activities in the culture broth and phosphate buffer were measured, and total activity was divided by the mycelia dry weight. Error bars indicate the standard errors of three independent experiments
In YP plus 1 % starch (YPS) medium after 48 h of culture, the α-amylase activities of the single ∆creA or ∆creB and double ∆creA∆creB mutants were approximately 1.5-, 2-, and 3-fold higher, respectively, compared with that of the wild-type strain. In addition, in YPS medium supplemented with the repressing sugar glucose, the single ∆creA and double ∆creA∆creB mutants retained high α-amylase activity similar to that in YPS medium without glucose, whereas the wild-type strain showed significantly reduced α-amylase activity (Fig. 4b). This indicated that the creA deletion resulted in carbon catabolite derepression of α-amylase production in submerged culture, as described above using the plate assay (Fig. 3).
In the presence of glucose, the ΔcreB mutant showed decreased α-amylase activity. However, the degree of reduced α-amylase activity by the ΔcreB mutant was not comparable with that of the wild-type strain, which suggested that the creB deletion partially resulted in carbon catabolite derepression. Together with the results for α-amylase production in YPD and YPS media, the creA and creB double deletion was the most effective for improving α-amylase production in submerged culture.
Comparisons of α-amylase activities in submerged cultures containing high concentrations of inducing sugars
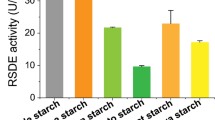
We subsequently examined α-amylase production in liquid medium containing 5 % inducing sugars maltose and starch to determine whether higher production levels could be achieved with these conditions. As shown in Fig. 5, all deletion mutants had significantly higher α-amylase activity than the wild-type strain. In YP plus 5 % maltose (YP5M) medium in the 48-h culture, both the single ∆creA and ∆creB mutants had approximately 4-fold higher α-amylase activity compared with the wild-type strain. Under the same conditions, the double ∆creA∆creB mutant had an approximately 7-fold higher activity than the wild-type strain (Fig. 5a). Further, culture for 72 h in YP5M resulted in reduced α-amylase activity in all strains, although the activity of the deletion mutants remained at a higher level compared with the wild-type strain, for which the activity in the medium was markedly reduced (Fig. 5a).

α-Amylase activities in liquid YP + 5 % sugar media. Approximately 2 × 106 conidiospores of each strain were grown in a YP5M or b YP5S medium for 48 and 72 h at 30 °C. Total α-amylase activities were determined as described in Fig. 4
In the presence of a high concentration of maltose, the wild-type and deletion mutant strains continued to grow during the entire culture period, although their α-amylase production was repressed by CCR induced by glucose resulting from maltose degradation. Thus, the α-amylase activity per dry mycelia weight of the wild-type strain was significantly reduced compared with activities of the deletion mutants. In comparison, in YP plus 5 % starch (YP5S) medium, α-amylase production by all of the strains was considerably higher than in YP5M medium for the same culture period (Fig. 5b). In particular, the single ∆creA, ∆creB, and double ∆creA∆creB mutants had approximately 2-, 3-, and 4-fold higher α-amylase activity, respectively, than the wild-type strain. Furthermore, the wild-type strain showed a remarkable decrease in activity in a 72-h culture, similar to that in YP5M medium, whereas α-amylase production by all of the deletion mutants in the 72-h culture was almost equal to that in the 48-h culture (Fig. 5b). These results clearly indicated that carbon catabolite derepression was considerably effective for improving α-amylase production in submerged cultures with high concentrations of inducing sugars.
To investigate whether the increased α-amylase production by the ∆creA∆creB mutant was due to an increase in transcript levels, α-amylase mRNA expression in each strain grown in YP5M medium was compared by quantitative RT-PCR (qRT-PCR) analysis (Fig. 6). The recipient strain used in this study was originated from RIB40 and contained three α-amylase genes (amyA, amyB, and amyC) with almost the same nucleotide sequence in the genome (Tada et al. 1989). Thus, the primers used in qRT-PCR were designed to amplify DNA fragments corresponding to mRNAs transcribed from all three genes. The α-amylase mRNA levels in the ∆creA and ∆creA∆creB mutants were notably higher than that in the wild-type strain during the entire culture period. The single ∆creB mutant also showed an approximately 2-fold higher transcription level than the wild-type strain, although its transcription level was considerably lower compared with those in the ∆creA and ∆creA∆creB mutants. These results suggested that under culture conditions with high concentrations of inducing sugars, the creA deletion was considerably more effective for enhancing the transcription level of the α-amylase gene and resulted in high-level production of secreted α-amylase compared with the wild-type strain. The creA and creB double deletion further increased the α-amylase mRNA level and resulted in an additional increase in α-amylase production. However, although it had a higher transcription level, the single creA deletion did not result in an increased yield of α-amylase compared with the single ∆creB mutation (Figs. 5a and 6).

Quantitative RT-PCR analysis for α-amylase mRNA. Approximately 2 × 106 conidiospores of each strain were grown in YP5M medium. Total RNA was prepared from harvested mycelia, and the resulting cDNA was subjected to qRT-PCR analysis using specific primers for α-amylase genes. The relative expression level of α-amylase genes was normalized to that of histone H4 gene. Error bars indicate the standard errors of three independent experiments
Comparisons of α-amylase activities in solid-state cultures
It is well known that A. oryzae produces larger amounts of proteins when in solid-state culture compared with submerged culture (Machida et al. 2008). Thus, we assessed α-amylase production by deletion mutants in solid-state culture using wheat bran as the substrate. In solid-state culture, there were no apparent differences in growth between the wild-type and deletion mutant strains (data not shown). After the 48-h culture of wheat bran, secreted proteins were extracted with acetate buffer, and α-amylase activity was determined. The α-amylase activity of the ∆creB mutant was not high and was comparable with that of the wild-type strain. However, the ∆creA and ∆creA∆creB mutants had approximately 2–2.5-fold higher activity than the wild-type strain (Fig. 7). This suggested that α-amylase production in solid-state culture improved after the creA deletion, although the creB deletion had no apparent additive effects.

α-Amylase activities in solid-state culture. Approximately 1 × 106 conidiospores of each strain were grown in solid-state culture with wheat bran as a substrate at 30 °C for 48 h. Secreted proteins were extracted with acetate buffer, after which the α-amylase activity in the acetate buffer was determined. Error bars indicate the standard errors of three independent experiments
Discussion
In this study, we generated single and double deletion mutants for the genes involved in CCR, creA and creB, in A. oryzae. The α-amylase activity of these mutant strains was compared under various culture conditions to evaluate the effect of these gene deletions on α-amylase production.
Deleting creA resulted in reduced growth on MM agar medium, consistent with observations of other filamentous fungi in which their orthologous genes had been deleted. The reduced growth of the ∆creA mutants was assumed to render them unsuitable for industrial use in enzyme production. However, in submerged cultures using YPD medium, the growth of mutants with a creA deletion ultimately exceeded that of the wild-type and ∆creB mutant strains in the 48-h culture, even though their growth in the early culture period of 24 h was relatively worse compared with that of the wild-type strain. This was also observed for submerged cultures with high concentrations of inducing sugars using YP5M and YP5S media (data not shown).
For T. reesei, a cre1 disruption mutant grew either poorly or failed to grow in liquid glucose or maltose medium, respectively (Denton and Kelly 2011). The media used for T. reesei were based on MM, whereas we used YP as the liquid medium in this study. Thus, it is likely that the ∆creA mutants can grow normally in media rich in nitrogen and vitamins. In addition, there were no apparent differences in growth among the wild-type strain and deletion mutants under solid-state culture conditions. These results indicated that the creA deletion would not be disadvantageous for industrial applications.
The plate assay for α-amylase activity showed that a creA or creB deletion resulted in relief from CCR induced by glucose. Similarly, the α-amylase activities of the deletion mutants in YPS medium supplemented with glucose were considerably higher than that of the wild-type, which also indicated that a creA or creB deletion resulted in carbon catabolite derepression. In the presence of glucose, the double deletion mutant showed the highest α-amylase production and suggested that deleting both creA and creB was most effective for enhancing α-amylase production, possibly by completely circumventing glucose repression.
The α-amylase gene expression level by the creA deletion mutants (∆creA and ∆creA∆creB) in YP5M liquid medium was highly increased compared with that of the wild-type strain. In particular, a significant reduction in the α-amylase mRNA level in the 72-h culture was observed for the wild-type but not for the ∆creA mutants. These results also indicated that expression of the α-amylase gene was repressed by glucose derived from maltose and that the creA deletion resulted in carbon catabolite derepression. In contrast to the creA deletion, the single creB deletion mutant showed only a slightly increased α-amylase gene expression. This suggested that the creB deletion caused partial CCR relief at the transcriptional level.
As reported for A. nidulans, CCR is directly regulated by CreA, the stability of which is modulated by its ubiquitination and deubiquitination. Under CCR conditions, ubiquitinated CreA is deubiquitinated by a CreB ubiquitin processing protease, which allows it to escape degradation by the proteasome; thus, it can exert its repressor function. In contrast, a defect in CreB results in the ubiquitination and destabilization of CreA, and proteolytic degradation of CreA results in derepression of CCR (Lockington and Kelly 2002). Because CreA is directly involved in CCR, the absence of CreA could result in nearly complete CCR relief.
In contrast, even in the absence of CreB, if a small amount of deubiquitinated CreA may be present, it could repress the expression of genes that are subject to CreA-mediated CCR. This may be a reason why creB deletion did not result in an increased transcript level of the α-amylase gene. However, despite similar expression levels of the α-amylase gene for the single ∆creA and the double ∆creA∆creB mutants, the double mutant had considerably higher α-amylase activity than the single ∆creA mutant in the submerged cultures examined in this study. Thus, creB deletion appeared to have an additive effect on carbon catabolite derepression.
In addition, the ∆creB mutant had a higher level of α-amylase production than the ∆creA mutant in YP5M medium, although the α-amylase mRNA level in the single ∆creB mutant was not increased to a level comparable to that in the ∆creA mutant. This was also observed when using cultures with YPS and YP5S media. In both cultures, the α-amylase mRNA levels in the ∆creB mutant were notably lower than those in the ∆creA mutant in YP5M medium (data not shown). These observations raise the intriguing possibility that creB deletion has an effect on α-amylase production other than CCR relief through the destabilization of ubiquitinated CreA.
Because CreB has been inferred to be a deubiquitinating enzyme, it is likely that there are several target proteins for CreB other than CreA. An as yet unidentified target protein(s) may be involved in inhibiting α-amylase production, presumably at the posttranscriptional level. Thus, creB deletion may cause an increased level of α-amylase. In A. nidulans, CreB has been proposed to be involved in the expression of genes required for utilizing proline and quinate as carbon or nitrogen sources by regulating either transcription activators or enzymes and permeases (Lockington and Kelly 2002). However, the possible pleiotropic functions of CreB are uncertain. Thus, further analysis of CreB function will be necessary. In this context, because it was reported that CreB functions as a deubiquitinating enzyme with another component CreC (Lockington and Kelly 2002), we are now constructing a creC deletion mutant to examine whether a similar phenomenon is observed in the creC mutant. Furthermore, we are also planning the microarray analyses of the mutant strains obtained in this study to find the putative target gene(s) or protein(s) of CreA and CreB.
It is known that CCR in filamentous fungi is generally circumvented by solid-state fermentation (Viniegra-González and Favela-Torres 2006). However, the α-amylase activities of the ∆creA and ∆creA∆creB mutants in solid-state culture using wheat bran were approximately 2–2.5-fold higher than that of the wild-type strain. This suggested that deleting creA was also effective for improving the production of enzyme proteins in solid-state culture, whereas no synergistic effect of creA and creB double deletion was observed. The reason why no effect of creB deletion on the α-amylase production was observed in the single and double deletion mutants remains unclear. However, since the transcription profiles of many genes, such as glaB encoding glucoamylase B, pepA encoding aspartic protease, and melB encoding secretory tyrosinase, are known to be different in between solid-state culture and submerged culture (Hata et al. 1998; Kitano et al. 2002; Obata et al. 2004), there might be a different effect of the creB deletion on the protein production under both culture conditions. Because the yield of A. oryzae-secreted proteins in solid-state culture was considerably higher than that in submerged culture, an increase in protein production during solid-state fermentation by creA deletion would be advantageous for enzyme protein production.
In conclusion, we report that a creA and creB double deletion is highly effective for improving α-amylase production by A. oryzae. The maximum α-amylase activity of this double deletion mutant was achieved at 72 h of culture with YP5S medium, and its α-amylase activity was more than 10-fold higher than that of the wild-type strain. It was recently reported that a creB deletion also resulted in carbon catabolite derepression in A. oryzae, although no experimental results for single creA or double creA and creB deletions were reported (Hunter et al. 2013). Based on our results, we anticipate that a creA and creB double deletion would be extremely effective for improving the production of hydrolytic enzymes in filamentous fungi, such as cellulases and xylanases of T. reesei. In addition to the production of amylolytic enzymes, A. oryzae has attracted attention as a host organism for recombinant protein production. Because the promoters for amylolytic genes have been widely used as expression promoters for heterologous genes, we also anticipate that a creA and creB double deletion strain could be useful as a suitable host strain for recombinant protein production.
References
Boase N, Kelly JM (2004) A role for creD, a carbon catabolite repression gene from Aspergillus nidulans, in ubiquitination. Mol Microbiol 53:929–940
Carlsen M, Nielsen J (2001) Influence of carbon source on α-amylase production by Aspergillus oryzae. Appl Microbiol Biotechnol 57:346–349
Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, Litvinkova L, Weiss RL, Borkovich KA, Dunlap JC (2006) A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci U S A 103:10352–10357
Denton JA, Kelly JM (2011) Disruption of Trichoderma reesei cre2, encoding an ubiquitin C-terminal hydrolase, results in increased cellulose activity. BMC Biotechnol 11:103
Dowzer CE, Kelly JM (1989) Cloning of the creA gene from Aspergillus nidulans: a gene involved in carbon catabolite repression. Curr Genet 15:457–459
Dowzer CE, Kelly JM (1991) Analysis of the creA gene, a regulator of carbon catabolite repression in Aspergillus nidulans. Mol Cell Biol 11:5701–5709
Gomi K, Iimura Y, Hara S (1987) Integrative transformation of Aspergillus oryzae with a plasmid containing the Aspergillus nidulans argB gene. Agric Biol Chem 51:2549–2555
Gomi K, Akeno T, Minetoki T, Ozeki K, Kumagai C, Okazaki N, Iimura Y (2000) Molecular cloning and characterization of a transcriptional activator gene, amyR, involved in the amylolytic gene expression in Aspergillus oryzae. Biosci, Biotechnol, Biochem 64:816–827
Hata Y, Ishida H, Ichikawa E, Kawato A, Suginami K, Imayasu S (1998) Nucleotide sequence of an alternative glucoamylase-encoding gene (glaB) expressed in solid-state culture of Aspergillus oryzae. Gene 207:127–134
Hunter AJ, Morris TA, Jin B, Saint CP, Kelly JM (2013) Deletion of creB in Aspergillus oryzae increases secreted hydrolytic enzyme activity. Appl Environ Microbiol 79:5480–5487
Hynes MJ, Kelly JM (1977) Pleiotropic mutants of Aspergillus nidulans altered in carbon metabolism. Mol Gen Genet 150:193–204
Ilmén M, Thrane C, Penttilä M (1996) The glucose repressor gene cre1 of Trichoderma: isolation and expression of a full-length and a truncated mutant form. Mol Gen Genet 251:451–460
Kato M, Sekine K, Tsukagoshi N (1996) Sequence-specific binding sites in the Taka-amylase A G2 promoter for the CreA repressor mediating carbon catabolite repression. Biosci, Biotechnol, Biochem 60:1776–1779
Kelly JM, Hynes MJ (1977) Increased and decreased sensitivity to carbon catabolite repression of enzymes of acetate metabolism in Aspergillus nidulans. Mol Gen Genet 156:87–92
Kitano H, Kataoka K, Furukawa K, Hara S (2002) Specific expression and temperature-dependent expression of the acid protease-encoding gene (pepA) in Aspergillus oryzae in solid-state culture (Rice-Koji). J Biosci Bioeng 93:563–567
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆C T method. Methods 25:402–408
Lockington RA, Kelly JM (2001) Carbon catabolite repression in Aspergillus nidulans involves deubiquitination. Mol Microbiol 40:1311–1321
Lockington RA, Kelly JM (2002) The WD40-repeat protein CreC interacts with and stabilizes the deubiquitinating enzyme CreB in vivo in Aspergillus nidulans. Mol Microbiol 43:1173–1182
Machida M, Yamada O, Gomi K (2008) Genomics of Aspergillus oryzae: learning from the history of koji mold and exploration of its future. DNA Res 15:173–183
Mizutani O, Masaki K, Gomi K, Iefuji H (2012) Modified Cre-loxP recombination in Aspergillus oryzae by direct introduction of Cre recombinase for marker gene rescue. Appl Environ Microbiol 78:4126–4133
Murakoshi Y, Makita T, Kato M, Kobayashi T (2012) Comparison and characterization of α-amylase inducers in Aspergillus nidulans based on nuclear localization of AmyR. Appl Microbiol Biotechnol 94:1629–1635
Nakari-Setälä T, Paloheimo M, Kallio J, Vehmaanperä J, Penttilä M, Saloheimo M (2009) Genetic modification of carbon catabolite repression in Trichoderma reesei for improved protein production. Appl Environ Microbiol 75:4853–4860
Obata H, Ishida H, Hata Y, Kawato A, Abe Y, Akao T, Akita O, Ichishima E (2004) Cloning of a novel tyrosinase-encoding gene (melB) from Aspergillus oryzae and its overexpression in solid-state culture (Rice Koji). J Biosci Bioeng 97:400–405
Oldenburg KR, Vo KT, Michaelis S, Paddon C (1997) Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res 25:451–452
Petersen KL, Lehmbeck J, Christensen J (1999) A new transcriptional activator for amylase genes in Aspergillus. Mol Gen Genet 262:668–676
Porciuncula JO, Furukawa T, Mori K, Shida Y, Hirakawa H, Tashiro K, Kuhara S, Nakagawa S, Morikawa Y, Ogasawara W (2013) Single nucleotide polymorphism analysis of a Trichoderma reesei hyper-cellulolytic mutant developed in Japan. Biosci, Biotechnol, Biochem 77:534–543
Sato H, Toyoshima Y, Shintani T, Gomi K (2011) Identification of potential cell wall component that allows Taka-amylase A adsorption in submerged cultures of Aspergillus oryzae. Appl Microbiol Biotechnol 92:961–969
Tada S, Iimura Y, Gomi K, Takahashi K, Hara S, Yoshizawa K (1989) Cloning and nucleotide sequence of the genomic Taka-amylase A gene of Aspergillus oryzae. Agric Biol Chem 53:593–599
Tamalampudi S, Talukder MM, Hama S, Tanino T, Suzuki Y, Kondo A, Fukuda H (2007) Development of recombinant Aspergillus oryzae whole-cell biocatalyst expressing lipase-encoding gene from Candida antarctica. Appl Microbiol Biotechnol 75:387–395
Tanaka M, Tokuoka M, Shintani T, Gomi K (2012) Transcripts of a heterologous gene encoding mite allergen Der f 7 are stabilized by codon optimization in Aspergillus oryzae. Appl Microbiol Biotechnol 96:1275–1282
Tonomura K, Suzuki H, Nakamura N, Kuraya K, Tanabe O (1961) On the inducers of α-amylase formation in Aspergillus oryzae. Agric Biol Chem 25:1–6
Viniegra-González G, Favela-Torres E (2006) Why solid-state fermentation seems to be resistant to catabolite repression? Food Technol Biotechnol 44:397–406
Yamada O, Lee BR, Gomi K (1997) Transformation system for Aspergillus oryzae with double auxotrophic mutations, niaD and sC. Biosci, Biotechnol, Biochem 61:1367–1369
Ziv C, Gorovits R, Yarden O (2008) Carbon source affects PKA-dependent polarity of Neurospora crassa in a CRE-1-dependent and independent manner. Fungal Genet Biol 45:103–116
Acknowledgments
We thank Osamu Mizutani for kindly providing the ∆ligD::loxP pyrG − mutant strain. We thank Youhei Kudo and Tetsuya Hiramoto for ∆creA/pYES2 plasmid construction. This study was supported by the Program for Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN).
Author information
Authors and Affiliations
Corresponding author
Additional information
Sakurako Ichinose and Mizuki Tanaka contributed equally to this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 77 kb)
Rights and permissions
About this article
Cite this article
Ichinose, S., Tanaka, M., Shintani, T. et al. Improved α-amylase production by Aspergillus oryzae after a double deletion of genes involved in carbon catabolite repression. Appl Microbiol Biotechnol 98, 335–343 (2014). https://doi.org/10.1007/s00253-013-5353-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5353-4