
Abstract
RNA has long been regarded as the important intermediary in the central dogma of gene expression. Recently, the importance of RNAs in the regulation of gene expression became evident with the identification and characterization of non-protein coding transcripts named non-coding RNAs (ncRNAs). The ncRNAs, small and long, are ubiquitously present in all three domains of life and are being recognized for their important roles in genome defense and development. Some of the ncRNAs have been associated with diseases, and therefore, they offer diagnostic and therapeutic potential. In this mini-review, we have highlighted some recent research on the ncRNAs identified in eukaryotic microbes, with special emphasis on fungi that are pathogenic to humans or plants when possible. It is our contention that further elucidation and understanding of ncRNAs will advance our understanding of the development and pathogenesis of eukaryotic microbes and offer alternatives in the diagnosis and treatment of the diseases caused by these pathogens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sequencing of genomes of eukaryotic organisms large and small brought forth the paradox of the number of protein-coding genes not directly correlating with the genetic complexity of the organism. The untranslated “dark matter” of the genome turned out to have functions that contribute to the biological complexity of the organism. It is now apparent that a vast majority of the human genome is transcribed but not translated (Guil and Esteller 2012), and pervasive transcription at both gene-coding and non-coding regions also occurs in most other genomes. The discovery of non-coding RNAs (ncRNAs) with regulatory functions at the transcriptional, translational, and epigenetic level offers a plausible explanation for the differences in genomic and biological complexity among organisms with similar number of protein-coding genes. The recognition of RNAs as a key component of cellular regulatory machinery (ncRNAs), in addition to a primary messenger (mRNAs), represents a major leap in our understanding of gene regulation.
The regulatory ncRNAs are classified based on their size into small ncRNAs and long ncRNAs (lncRNAs). The lncRNAs are defined as non-protein coding RNA transcripts that are greater than 200 nt in length and are transcribed by RNA polymerase II. The ability of ncRNAs to target sequences in a highly specific manner confers precision in their regulatory activity.
The study of ncRNAs gained momentum when they were found to be linked with cancer, autoimmune disorders, cardiovascular, and other debilitating diseases. The most common ncRNAs associated with cancer are the micro RNAs (miRNAs) (Davalos et al. 2011; Manel 2011). Another class of small ncRNAs, called the PIWI RNAs (piRNAs), has also been associated with the development of testicular cancer and other somatic tumors (Manel 2011). LncRNAs, such as HOTAIR, also play important roles in human neoplasia (Gupta et al. 2010). Besides cancer, ncRNAs are also involved in the development of the brain and synaptic plasticity. The deregulations of lncRNAs and small RNAs have been implicated in neurological diseases like Alzheimer’s and Parkinson’s and psychiatric disorders like schizophrenia and depression (Hunsberger et al. 2009; Manel 2011). Given their association with various diseases, ncRNAs hold great potential as targets for disease detection (diagnosis) and/or treatment.
Similar to what has been observed in higher life forms, ncRNAs play a role in the biology of eukaryotic microbes (fungi and protists), which also exhibit complex transcriptional landscapes. The investigation of ncRNAs in these lower eukaryotes, however, has been lagging behind. Given the amenability of some of these eukaryotic microbes at the molecular, cellular, and organismal levels, and the potential roles of ncRNAs in their development and pathogenicity (see below), studying ncRNAs in these systems could expedite the progress in this research area and be exploited for alternative antimicrobial therapies. In this mini-review, we will highlight some recent research on the small and long ncRNAs identified in eukaryotic microbes, with special emphasis on the few ncRNAs that have been functionally characterized in fungi.
Small ncRNAs and their role in microbial genome defense
Mobile DNA elements or transposons pose a significant threat to the stability of the host genome. Such a threat is met with defense mechanisms like methylation or gene silencing of the offender. The RNAi mechanism is a conserved genome defense strategy that is present across the eukaryotic domain where small RNAs are universally used to guide the slicing machinery to the genes to be silenced. RNAi may silence gene expression at the posttranscriptional or at transcriptional levels (Stephane and Robert 2013). The small RNAs involved in RNAi may be produced either by a dsRNA specific RNAse III ribonuclease called the dicer (dicer-dependent biogenesis) or by other RNA processing enzymes (dicer-independent biogenesis). Dicer-dependent small RNAs include miRNAs and small interfering RNAs (siRNAs) (Ghildiyal and Zamore 2009) and are found in protists as well as in fungi (Grimson et al. 2008; Zhao et al. 2007). The dicer-independent small RNAs like piRNA are abundant in higher metazoans but are absent in fungi (Lee et al. 2010).
RNAi mechanism in fungi
In fungi, RNAi is one of the defense mechanisms that silence invading viruses and transposons. The fungal RNAi pathway was first described in the filamentous model fungus Neurospora crassa in 1992 and was called quelling (Romano and Macino 1992). Quelling specifically silences repetitive transgenes during vegetative growth. It is carried out by dicer enzymes and other conserved components, and it produces siRNAs of about 25 nucleotides from abnormal RNAs. The core components of quelling are the RNA-dependent RNA polymerase QDE-1, argonaute protein QDE-2, helicase QDE-3, and two dicer enzymes DCL-1 and DCL-2. Repetitive DNA is recognized by QDE-3, which then recruits QDE-1. QDE-1 initially acts as a DNA-dependent RNA polymerase to produce single-stranded RNA and then acts as a RNA-dependent RNA polymerase to generate a double-stranded RNA (Fig. 1). The dicers then chop the resulting dsRNA into siRNAs. The siRNAs attach to the inactive RNA Induced Silencing Complex (RISC). The clipping of siRNA by QDE-2 and its interacting exonuclease QIP activates the RISC. Once activated, the RISC guided by the siRNA silences the targeted gene. Besides the introduction of repetitive transgenes, DNA-damage that may lead to arrest in cell growth can also trigger RNAi in N. crassa. The DNA-damage-triggered RNAi involves a set of small RNAs called the qiRNAs (21–23 nt) originated from the highly repetitive rDNA locus. The qiRNAs are generated by QDE-1, 3, and the dicers, and they interact with QDE-2. DNA-damage-triggered RNAi is thus similar to quelling and shares the same components in bringing about gene silencing (Heng-Chi et al. 2009).

Quelling in N. crassa: The repetitive DNA is identified and targeted by helicase QDE-3, which recruits the RNA-dependent RNA polymerase QDE-1. The aberrant RNA produced by QDE-1 is chopped up into 25 nt siRNAs by the dicers DCL1/DCL2. The siRNAs serve as guide for the RISC complex to target specific sequences for silencing. QDE2 and QIP activate the RISC complex
Prevalence and role of RNAi in fungi
siRNAs connected with the RNAi pathway have been identified in both yeasts and filamentous fungi (Chang et al. 2012). In Schizosaccharomyces pombe, the RNAi machinery is responsible for maintaining centromeres in the heterochromatin stage, which prevents the expression of centromeric repeats that resemble transposons (Volpe et al. 2002). In Cryphonectria parasitica and Aspergillus nidulans, RNAi protects against foreign viral RNA (Dang et al. 2011). Surprisingly, siRNAs and the RNAi pathway are absent in the model yeast Saccharomyces cerevisiae. The loss of the RNAi pathway confers S. cerevisiae tolerance of the killer virus (Drinnenberg et al. 2009), which produces a toxin and consequently offers a competitive advantage to its host over the non-toxin containing relatives (Drinnenberg et al. 2011). The introduction of dicer Dcr1 and argonaute Ago1 to S. cerevisiae restores the RNAi pathway and expectedly causes the loss of the killer virus. Thus, there appears to be a trade-off between the ability to silence active transposons by RNAi and the selective advantage conferred by the killer virus in the absence of RNAi in S. cerevisiae.
Another fungus where the RNAi machinery is absent is Ustilago maydis (Kämper et al. 2006; Laurie et al. 2008), a biotrophic maize pathogen that requires the host plant for the completion of its life cycle. This fungus protects its genome from heterologous introductions by prematurely terminating the transcription of introduced genes (Zarnack et al. 2006). By contrast, Sporisorium reilianum, another biotrophic pathogen related to U. maydis, does carry the core components of RNAi-like dicer and argonaute proteins. Interestingly, the deletion of the dicer gene does not affect the virulence of S. reilianum (Schirawski et al. 2010).
The role of the RNAi pathway has only been investigated in a few human fungal pathogens. In Cryptococcus neoformans, the RNAi machinery consists of two dicer proteins Dcr1 and Dcr2, argonaute Ago1, and the RNA-dependent RNA polymerase Rdp1. Disruption of the RNAi pathway in Cryptococcus does not deleteriously affect its virulence or reproduction, but it does increase the expression and mobilization of transposons during sexual reproduction (Janbon et al. 2010; Wang et al. 2010). This sex-induced silencing (SIS) of transposons is thus a mechanism for genome protection during sexual development. While SIS ensures the genome stability during meiosis, a quelling-like phenomenon called asexual co-suppression ensures the same during mitosis. In addition to the RNAi components used in SIS, an ssDNA-binding complex is involved in asexual co-suppression. The need for the ssDNA-binding complex and the targeting of multiple transgene copies make the co-suppression in Cryptococcus similar to the quelling process in Neurospora. In Cryptococcus, transposon transcripts commonly contain suboptimal introns that stall the spliceosomes. Spliceosome stalling due to inefficient splicing of mRNAs makes these transcript targets of the RNAi pathway (Dumesic et al. 2013). This study elucidates how the RNAi machinery differentiates between the “self” host and the “non-self” transposon sequences and thereby contributes towards the defense of its host genome. Two miRNAs produced from 70 nt pre-miRNA precursors, miR1 and miR2, are also capable of inducing gene silencing through the RNAi pathway in C. neoformans (Jiang et al. 2012). Thus, Cryptococcus uses small RNAs derived from different origins in silencing foreign DNA while using the same core components of the RNAi machinery, elucidating the versatility of this important defense mechanism.
Small ncRNAs in microbial development and stress response
In addition to the siRNAs that mediate the RNAi pathway, other small ncRNAs have also been identified in lower eukaryotes. Differential expression of several kinds of small RNAs was observed in different tissues at various growth stages in Magnaporthe oryzae, suggesting a potential role of small ncRNAs in fungal development (Nunes et al. 2011). Small ncRNAs may also be associated with resistance to biotic and abiotic stresses. For instance, differential expression of a set of miRNAs in resistant or sensitive soybean plants was observed in response to infection by the rust fungus Phakopsora pachyrhizi or to abiotic stresses (Kulcheski et al. 2011). Thus, miRNAs/siRNAs from the host could be used as biomarkers to identify resistant crop strains, while infection-associated ncRNAs expressed from pathogens could be used in disease diagnosis. Such applications are feasible given that miRNAs and other small RNAs linked to diseases in humans have successfully been used in clinical diagnostic assays (Gibb et al. 2011; Lee and Calin 2011). Furthermore, the functional characterization of disease-associated miRNAs and other ncRNAs may open up new avenues for disease control. The knowledge of small ncRNAs is being explored as novel-targeted treatment using antisense transcripts of miRNA, competitive inhibitors of miRNAs, or the restoration of miRNAs with tumor suppressor functions in humans (Manel 2011).
Long-coding RNAs in eukaryotic genomes
Despite the universal presence of lncRNAs and the recent advancement in their identification by high throughput sequencing and bioinformatics, our knowledge of their function, mode of action, and interacting players are extremely limited. Among the lncRNAs initially discovered were those involved in genomic imprinting and X chromosome inactivation or dosage compensation (Philip and Edith 2001). Genomic imprinting is an epigenetic mechanism that controls gene expression, and almost all known imprinted gene clusters contain lncRNAs in mammals (Koerner et al. 2009). For X chromosome inactivation or dosage compensation in humans, seven regulatory RNAs are involved, including the 17-knt Xist and its antisense transcript Tsix. Although genomic imprinting has been implicated in the mating type inheritance in S. pombe and in other fungi like Aspergillus spp., no lncRNA has yet been associated with this phenomenon. On the other hand, one relatively well-characterized lncRNA in fungi is the telomeric repeat-containing RNA (TERRA). The telomere regulation is highly conserved in eukaryotes. In humans, TERRA forms a parallel G quadruplex structure in vivo and localizes to the telomere. This quadruplex in the presence of sodium ions is said to confer strong resistance to RNase for the repeats in the telomere (Xu et al. 2010). The formation of a hybrid between TERRA and DNA in S. cerevisiae inhibits telomerase activity, which results in the shortening of the telomeres (Luke et al. 2008). Conversely, the degradation of TERRA by 5′ to 3′ exonuclease Rat1p is required for the telomerase activity and the maintenance of telomere length. A recent tiling transcriptional study in the protist Plasmodium falciparum also identified 60 potential ncRNAs, of which 22 were found to be telomere-associated lncRNAs (Broadbent et al. 2011).
In Saccharomyces, transcription from bidirectional promoters produces RNA transcripts like the Cryptic Unstable Transcripts (CUTs) and Stable Un-annotated Transcripts (SUTs) that can be categorized as lncRNA (Zhenyu et al. 2009). Majority of these RNAs are produced from the 5′ Nucleosome Free Regions (NFRs) associated with promoters of genes although antisense transcripts produced from 3′ NFRs are also common. These RNAs exert regulatory functions either by the act of their transcription or by the activity of their transcripts. Another class of unstable transcripts with regulatory activity is the Xrn-1 sensitive Unstable Transcripts (XUTs) (van Dijk et al. 2011). While CUTs are degraded by the 3′-5′ nuclear RNA decay pathway, the XUTs are sensitive to 5′-3′ Xrn1 exonuclease. The XUT antisense transcripts repress gene expression by methylation assisted by histone methyl transferase Set1. In the following sections, we will discuss a few examples to highlight the importance of lncRNAs in eukaryotic microbiology.
Origin and classification of lncRNAs
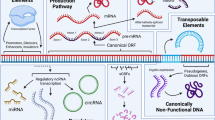
The origin and subcellular localization of lncRNAs vary greatly. LncRNAs often arise from protein-coding regions (e.g., Xist) (Duret et al. 2006; Elisaphenko et al. 2008), or occasionally from non exonic regions through chromosome rearrangement or insertions of transposable elements (Ponting et al. 2009). LncRNAs have been observed to localize to the nucleus, the cytoplasm or both, and their localization is highly dependent on their mode of gene regulation. Based on their physical proximity to protein-coding genes, lncRNAs can be classified as cis, intronic, bidirectional, or overlapping (Fig. 2) (Tim et al. 2009; Wilusz et al. 2008). LncRNAs can also be categorized based on their mode of action as cis- or trans-acting. The trans-acting lncRNAs often act as guides or scaffolds to chromatin remodeling complexes (Guil and Esteller 2012). Most of the regulatory lncRNAs function in cis, but they may differ in their mode of action.

Classification of lncRNAs based on their proximity position relative to the target protein-coding gene. The lncRNAs are marked in gray, and the protein-coding genes in black. The arrows indicate the direction of transcription
LncRNAs in the regulation of microbial growth and development
Undeniably, the largest subset of cis-acting lncRNAs is transcribed in antisense either partially or totally overlapping transcripts originating from the opposite strand. With 50–70 % of sense transcripts transcribed in eukaryotes having antisense partners, and most of these transcripts not coding for proteins, the possibility of them being regulatory RNAs is high (Guil and Esteller 2012). The antisense transcript may regulate gene expression by transcriptional interference, chromatin remodeling, or by forming double strands (Donaldson and Saville 2012). Transcription of antisense may physically dislodge the RNA polymerase II complex from sense strand transcription or create topological constraint on the DNA, preventing the transcription of the sense strand. The Saccharomyces lncRNA RME2 physically interferes with the transcriptional elongation of the IME4 gene. Ime4 is required to activate IME1, the gene encoding the master initiator of meiosis (Gelfand et al. 2011). The expression of these meiosis genes is specific to the a1–α2 diploid cell type resulting from the a–α cell fusion events. Another example of lncRNAs exerting transcriptional interference in yeast is ZRR1, which regulates expression of a zinc-dependent alcohol dehydrogenase ADH1 (Amanda et al. 2006). The expression of this intergenic transcript displaces the transcriptional activator Rap1 from the promoter of ADH1 and consequently prevents the transcription of ADH1.
Two examples of non-coding antisense transcripts of important protein-coding genes that affect major developmental processes are seen in Neurospora and in the plant pathogen Cochliobolus heterostrophus. The conidial pigmentation in C. heterostrophus requires the activity of MAP Kinases Chk1 and Mps1 and the transcription factor Cmr1. In addition to the sense transcript, the CMR1 gene also encodes a shorter antisense transcript (Eliahu et al. 2007). The expression of this antisense lncRNA was almost undetectable when the components of MAPK pathway were deleted in the fungus. The expression of the cluster of melanin biosynthesis genes is dependent on the expression of CMR1. It is speculated, based on the higher expression of the antisense lncRNA compared to the sense strand in the presence of functional MAPK pathway, that this antisense lncRNA works together with the MAPK pathway to regulate the transition of the melanin gene cluster region between euchromatin and heterochromatin. In Neurospora, the antisense transcripts of circadian clock core regulator gene FRQ are required for rhythmic conidiation of the fungus and their expression is responsive to environmental stimuli-like light (Cas et al. 2003). Functional characterization of these transcripts may give more insight into the regulatory intricacies of circadian rhythm.
The control of the variegated expression of the FLO11 gene in S. cerevisiae by two lncRNAs, ICR1 and PWR1, offers a complex regulatory model of cis-acting lncRNAs’ interference with gene transcription by preventing the recruitment of general transcription factors (Bumgarner et al. 2009). Flo11 is a key protein that is required for haploid adhesion and for diploid pseudohyphal growth (Fig. 3). Transcription factor Sfl1 binds to the promoter of FLO11 and represses its expression while another transcription factor Flo8 activates it. The lncRNA ICR1 (3.2 knt) is transcribed in same direction as FLO11 from the upstream intergenic region. PWR1 (1.2 knt) is antisense to ICR1. The transcription of ICR1 is hypothesized to prevent the recruitment of general transcription factors and chromatin remodelers and consequently to prevent the transcription of FLO11. The expression of PWR1 in antisense promotes FLO11 expression by interfering with ICR1. Chromatin remodeler Rpd3L represses ICR1, and thereby, it acts as an indirect activator of FLO11 (Bumgarner et al. 2009). The competition between the transcription factors Flo8 and Sfl1 determines which of the two lncRNAs is expressed. The toggling effect of these two lncRNAs on the promoter of FLO11 is not observed when these RNAs are overexpressed in trans. This model of Flo11 regulation based on genetic and gene expression studies is further supported by another study using FISH microscopy and computational modeling analysis (Bumgarner et al. 2012).

The regulation of transcription of the FLO11 gene in S. cerevisiae by two lncRNAs ICR1 and PWR1: The transcription of PWR1 inhibits the transcription of ICR1 and thereby leaves the promoter of FLO11 open for the recruitment of transcription activators. Transcription activator Flo8 and repressor Sfl1 compete for the expression of the lncRNAs. In the absence of PWR1 expression, ICR1 is expressed, leading to dislodging of the transcription machinery from the promoter of FLO11 and thereby the repression of the FLO11 expression
The act of transcription of lncRNAs could cause chromatin remodeling and subsequently the opening up of promoters of nearby protein-coding genes. Glucose starvation in S. pombe induces the expression of FBP1 gene that encodes a fructose-1,6-bis phosphatase (Hirota et al. 2008). Under glucose rich conditions, the global transcriptional repressor Tup1 prevents the expression of FBP1. The transcription of the upstream lncRNA sequences under the glucose depleted condition is accelerated by the transcription activator Atf1. This activity removes the repressive effect of Tup1. The disruption of chromatin structure at the promoter region of FBP1 due to the transcription activity in the upstream sequences makes the promoter available for the binding by another transcription factor Rst1. This leads to the subsequent production of a stable long transcript of FBP1 (Hirota et al. 2008). Similar transcription of a lncRNA upstream of gene of interest, leading to chromatin adjustment and activation, is also seen in the case of PHO5 in S. cerevisiae (Uhler et al. 2007). Antisense transcript-mediated chromatin remodeling may occur in cis or in trans. The cryptic antisense transcripts of the retrovirus-like element Ty1, Ty1AS, inhibit its retro-transposition in S. cerevisiae posttranscriptionally (Matsuda and Garfinkel 2009). The loss of the antisense transcript leads to increased copy number of Ty1 and increased transposition. Thus, the antisense transcripts inhibit Ty1 transposition in trans.
In S. pombe, lncRNAs (meiRNA-S and L) working with an RNA-binding protein Mei2p are essential for reductive division in meiosis I (Ding et al. 2012; Watanabe and Yamamoto 1994). Mei2p is required for both pre-meiotic DNA synthesis and meiosis I, while the lncRNAs are only involved in the latter process. During meiotic prophase, Mei2p together with its bound RNA transcripts accumulate at the SME2 locus that encodes the lncRNAs, forming a dot in the elongated horsetail-shaped nucleus (Shimada et al. 2003). The ncRNA transcripts encoded by the SME2 locus were recently found to improve the homologous pairing of chromosomes during meiosis (Ding et al. 2012). The involvement of lncRNA in homologous pairing of chromosome is another new function discovered in the ever expanding role of ncRNAs.
Virulence-associated lncRNAs in eukaryotic microbial pathogens
lncRNAs, especially those transcribed in antisense, have been considered as drug targets to mitigate human diseases such as colon cancer (Yamamoto et al. 2002) and lymphoma (Hammond 2006; Wahlestedt 2006). It is our conviction that some of the transcripts produced by disease-causing infectious agents could also be potential targets for new drugs. For instance, viral ncRNAs can suppress host RNAi defense, maintain viral latency, and prevent the host apoptosis (Scaria and Pasha 2013). Bacteria also produce virulence-associated ncRNAs (Gong et al. 2011; Weissenmayer et al. 2011). However, studies on the roles of ncRNAs in eukaryotic pathogens are still lagging behind, hindering our effort in developing alternative management strategies in combating these diseases.
The protozoan malarial parasite Plasmodium falciparum lacks many conventional regulatory components like sequence-specific transcription factors. Its genome does accommodate ncRNAs with potential regulatory functions, and virulence-associated ncRNAs have been reported in this parasite (Kyes et al. 2003; Li et al. 2008). A recent tiling transcriptional study identified 60 potential ncRNAs (Broadbent et al. 2011). Some of the lncRNAs (lncTARE-3 and TARE-6) are expressed specifically in the asexual blood-stage of the parasite (Sierra-Miranda et al. 2012). They are expressed in the early ring stage, suppressed during the intermediate trophozoite stage, and reappeared in the schizont stage. Such phase-specific expression implicates these RNAs in phase transitions. TARE-6 has the potential to form a stem loop structure, indicating its ability to act as a scaffold. Taken together with its histone-binding activity, TARE-6 may epigenetically regulate gene expression. Further functional studies of these RNAs are needed to assess their importance in the differentiation and pathogenesis of this parasite.
Infection-associated lncRNAs have also been identified in oomycetes such as Phytophthora infestans. This eukaryotic microbe infects potato and causes late blight that was responsible for the well-known Irish famine. Pinc1-1 and Pinc1-5, two experimentally proved ncRNAs belonging to the gene family Pinc1, are upregulated during this oomycete infection and growth in planta. The Pinc1 ncRNA members have a predicted secondary structure that precludes the RNA being processed into miRNAs (Avrova et al. 2007). Thus, they most likely function as regulatory lncRNAs. These ncRNAs might contribute to the evasion of host detection and the biotrophic survival of this organism during infection. Understanding their mode of action could help develop strategies to facilitate host detection and resistance to Phytophthora.
LncRNAs are also involved in fungal dimorphism, a trait that is important for the virulence of many fungal pathogens. For instance, U. maydis infects and produces corn smut disease only when this fungus has undergone mating and changed its morphology from yeast to dikaryotic hyphae. An intergenic lncRNA transcribed in antisense from its two neighboring genes um02150 and um02151 increases in expression during filamentation and during meiotic spore production and in planta (Morrison et al. 2012). Consistently, the loss of this lncRNA significantly reduces the virulence of the fungus. Surprisingly, this lncRNA was not present in the closely related fungus Sporisorium reilianum, which also causes head smut in maize with different manifestations. Thus, lncRNAs, similar to many fast evolving proteins, are likely involved in species-specific or even strain-specific virulence functions. Recently, another lncRNA antisense to gene um02151 was found to be essential for Ustilago virulence (Donaldson and Saville 2013). The expression of this antisense transcript and the RNA hybrid it formed with its sense strand is high in resting spores and teliospores of U. maydis. In comparison, the expression of the antisense transcript is low in metabolically active haploid yeast cells. When the NAT is expressed, the sense transcription and the RNA hybrid production increase, without increasing the um02151 protein level. Thus, this antisense transcript must be preventing the translation of um02151 in dormant spores. Recently, our laboratory has identified an lncRNA through random mutagenesis screens and found that this lncRNA is critical for the morphologic switch between the yeast and the hyphal form in Cryptococcus (unpublished results). This lncRNA, termed RZE1, functions upstream of the irreplaceable regulator of morphogenesis and a key mediator of virulence in Cryptococcus, Znf2 (Lin et al. 2010; Wang et al. 2012; Zhai et al. 2013). As the expression of RZE1 is upregulated under condition relevant to host physiology, RZE1 might mediate the ability of Cryptococcus to cause disease, making it a potentially important regulator of morphogenesis and virulence.
Conclusions
ncRNAs form a vital part of the transcriptomes of eukaryotic microbes. With the availability of high throughput sequencing and tiling array technology, the task of identification of a variety of ncRNAs is becoming easier. One major challenge is to functionally characterize the regulatory ncRNAs. The non-conservative nature of ncRNA sequences and the vast possibility in their mode of action make it difficult to predict their functions based on bioinformatics compared to protein-coding genes. The development-specific and condition-specific expression of many ncRNAs, as described earlier, also renders it a rather daunting task to catalog all the potential functional ncRNAs encoded in the genome. Despite these challenges, there is enormous potential for the application of the knowledge of ncRNAs and the challenges could very well be effectively addressed with the growth of this relatively new field.
References
Amanda JB, Mat G, David JE, Dennis RW (2006) Repression of ADH1 and ADH3 during zinc deficiency by Zap1-induced intergenic RNA transcripts. EMBO J 25(24):5726–5734. doi:10.1038/sj.emboj.7601453
Avrova AO, Whisson SC, Pritchard L, Venter E, De Luca S, Hein I, Birch PR (2007) A novel non-protein-coding infection-specific gene family is clustered throughout the genome of Phytophthora infestans. Microbiology 153(Pt 3):747–59. doi:10.1099/mic.0.2006/002220-0
Broadbent KM, Park D, Wolf AR, Van Tyne D, Sims JS, Ribacke U, Volkman S, Duraisingh M, Wirth D, Sabeti PC, Rinn JL (2011) A global transcriptional analysis of Plasmodium falciparum malaria reveals a novel family of telomere-associated lncRNAs. Genome Biol 12(6):R56. doi:10.1186/gb-2011-12-6-r56
Bumgarner Stacie L, Neuert G, Voight Benjamin F, Symbor-Nagrabska A, Grisafi P, van Oudenaarden A, Fink Gerald R (2012) Single-cell analysis reveals that noncoding RNAs contribute to clonal heterogeneity by modulating transcription factor recruitment. Mol Cell 45(4):470–482. doi:10.1016/j.molcel.2011.11.029
Bumgarner SL, Dowell RD, Grisafi P, Gifford DK, Fink GR (2009) Toggle involving cis-interfering noncoding RNAs controls variegated gene expression in yeast. Proc Natl Acad Sci U S A 106(43):18321–18326. doi:10.1073/pnas.0909641106
Cas K, Jennifer JL, Jay CD, Susan KC (2003) Role for antisense RNA in regulating circadian clock function in Neurospora crassa. Nature 421(6926):948–952. doi:10.1038/nature01427
Chang S-S, Zhang Z, Liu Y (2012) RNA interference pathways in fungi: mechanisms and functions. Annu Rev Microbiol 66(1):305–323. doi:10.1146/annurev-micro-092611-150138
Dang YK, Yang QY, Xue ZH, Liu Y (2011) RNA interference in fungi: pathways, functions, and applications. Eukaryot Cell 10(9):1148–1155. doi:10.1128/Ec.05109-11
Davalos A, Goedeke L, Smibert P, Ramirez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, Esplugues E, Fisher EA, Penalva LO, Moore KJ, Suarez Y, Lai EC, Fernandez-Hernando C (2011) miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci U S A 108(22):9232–7. doi:10.1073/pnas.1102281108
Ding D-Q, Okamasa K, Yamane M, Tsutsumi C, Haraguchi T, Yamamoto M, Hiraoka Y (2012) Meiosis-specific noncoding RNA mediates robust pairing of homologous chromosomes in meiosis. Science 336(6082):732–736. doi:10.1126/science.1219518
Donaldson ME, Saville BJ (2012) Natural antisense transcripts in fungi. Mol Microbiol 85(3):405–417. doi:10.1111/j.1365-2958.2012.08125.x
Donaldson ME, Saville BJ (2013) Ustilago maydis natural antisense transcript expression alters mRNA stability and pathogenesis. Mol Microbiol 89(1):29–51. doi:10.1111/mmi.12254
Drinnenberg IA, Weinberg DE, Xie KT, Mower JP, Wolfe KH (2009) RNAi in budding yeast. Science 326:544. doi:10.1126/science.1176945
Drinnenberg IA, Fink GR, Bartel DP (2011) Compatibility with killer explains the rise of RNAi-deficient fungi. Science 333(6049):1592. doi:10.1126/science.1209575
Dumesic Phillip A, Natarajan P, Chen C, Drinnenberg Ines A, Schiller Benjamin J, Thompson J, Moresco James J, Yates Iii John R, Bartel David P, Madhani Hiten D (2013) Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell 152(5):957–968. doi:10.1016/j.cell.2013.01.046
Duret L, Chureau C, Samain S, Weissenbach J, Avner P (2006) The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 312(5780):1653–5. doi:10.1126/science.1126316
Eliahu N, Igbaria A, Rose MS, Horwitz BA, Lev S (2007) Melanin biosynthesis in the maize pathogen Cochliobolus heterostrophus depends on two mitogen-activated protein kinases, Chk1 and Mps1, and the transcription factor Cmr1. Eukaryot Cell 6(3):421–429. doi:10.1128/Ec.00264-06
Elisaphenko EA, Kolesnikov NN, Shevchenko AI, Rogozin IB, Nesterova TB, Brockdorff N, Zakian SM (2008) A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PLoS One 3(6):e2521. doi:10.1371/journal.pone.0002521
Gelfand B, Mead J, Bruning A, Apostolopoulos N, Tadigotla V, Nagaraj V, Sengupta AM, Vershon AK (2011) Regulated antisense transcription controls expression of cell-type-specific genes in yeast. Mol Cell Biol 31(8):1701–1709. doi:10.1128/mcb.01071-10
Ghildiyal M, Zamore PD (2009) Small silencing RNAs: an expanding universe. Nat Rev Genet 10(2):94–108. doi:10.1038/nrg2504
Gibb EA, Enfield KS, Tsui IF, Chari R, Lam S, Alvarez CE, Lam WL (2011) Deciphering squamous cell carcinoma using multidimensional genomic approaches. J Skin Cancer 2011:541405. doi:10.1155/2011/541405
Gong H, Vu GP, Bai Y, Chan E, Wu R, Yang E, Liu F, Lu S (2011) A Salmonella small non-coding RNA facilitates bacterial invasion and intracellular replication by modulating the expression of virulence factors. PLoS Pathog 7(9):e1002120. doi:10.1371/journal.ppat.1002120
Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP (2008) Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 455(7217):1193–7. doi:10.1038/nature07415
Guil S, Esteller M (2012) Cis-acting noncoding RNAs: friends and foes. Nat Struct Mol Biol 19(11):1068–75. doi:10.1038/nsmb.2428
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464(7291):1071–6. doi:10.1038/nature08975
Hammond SM (2006) MicroRNAs as oncogenes. Curr Opin Genet Dev 16(1):4–9. doi:10.1016/j.gde.2005.12.005
Heng-Chi L, Shwu-Shin C, Swati C, Antti PA, Mekhala M, Dennis HB, Yi L (2009) qiRNA is a new type of small interfering RNA induced by DNA damage. Nature 459(7244):274–277. doi:10.1038/nature08041
Hirota K, Miyoshi T, Kugou K, Hoffman CS, Shibata T, Ohta K (2008) Stepwise chromatin remodelling by a cascade of transcription initiation of non-coding RNAs. Nature 456(7218):130–134. doi:10.1038/nature07348
Hunsberger J, Austin D, Chen G, Manji H (2009) MicroRNAs in mental health: from biological underpinnings to potential therapies. Neuromol Med 11(3):173–182. doi:10.1007/s12017-009-8070-5
Janbon G, Maeng S, Yang DH, Ko YJ, Jung KW, Moyrand F, Floyd A, Heitman J, Bahn YS (2010) Characterizing the role of RNA silencing components in Cryptococcus neoformans. Fungal Genet Biol 47(12):1070–80. doi:10.1016/j.fgb.2010.10.005
Jiang N, Yang Y, Janbon G, Pan J, Zhu X (2012) Identification and functional demonstration of miRNAs in the fungus Cryptococcus neoformans. PLoS One 7(12):e52734. doi:10.1371/journal.pone.0052734
Kämper J, Kahmann R, Bölker M, Ma L-J, Brefort T, Saville BJ, Banuett F, Kronstad JW, Gold SE, Müller O, Perlin MH, Wösten HAB, de Vries R, Ruiz-Herrera J, Reynaga-Peña CG, Snetselaar K, McCann M, Pérez-Martín J, Feldbrügge M, Basse CW, Steinberg G, Ibeas JI, Holloman W, Guzman P, Farman M, Stajich JE, Sentandreu R, González-Prieto JM, Kennell JC, Molina L, Schirawski J, Mendoza-Mendoza A, Greilinger D, Münch K, Rössel N, Scherer M, Vraneš M, Ladendorf O, Vincon V, Fuchs U, Sandrock B, Meng S, Ho ECH, Cahill MJ, Boyce KJ, Klose J, Klosterman SJ, Deelstra HJ, Ortiz-Castellanos L, Li W, Sanchez-Alonso P, Schreier PH, Häuser-Hahn I, Vaupel M, Koopmann E, Friedrich G, Voss H, Schlüter T, Margolis J, Platt D, Swimmer C, Gnirke A, Chen F, Vysotskaia V, Mannhaupt G, Güldener U, Münsterkötter M, Haase D, Oesterheld M, Mewes H-W, Mauceli EW, DeCaprio D, Wade CM, Butler J, Young S, Jaffe DB, Calvo S, Nusbaum C, Galagan J, Birren BW (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444(7115):97–101. doi:10.1038/nature05248
Koerner MV, Pauler FM, Huang R, Barlow DP (2009) The function of non-coding RNAs in genomic imprinting. Development 136(11):1771–83. doi:10.1242/dev.030403
Kulcheski FR, de Oliveira LF, Molina LG, Almerao MP, Rodrigues FA, Marcolino J, Barbosa JF, Stolf-Moreira R, Nepomuceno AL, Marcelino-Guimaraes FC, Abdelnoor RV, Nascimento LC, Carazzolle MF, Pereira GA, Margis R (2011) Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genomics 12:307. doi:10.1186/1471-2164-12-307
Kyes SA, Christodoulou Z, Raza A, Horrocks P, Pinches R, Rowe JA, Newbold CI (2003) A well-conserved Plasmodium falciparum var gene shows an unusual stage-specific transcript pattern. Mol Microbiol 48(5):1339–48. doi:10.1046/j.1365-2958.2003.03505.x
Laurie J, Linning R, Bakkeren G (2008) Hallmarks of RNA silencing are found in the smut fungus Ustilago hordei but not in its close relative Ustilago maydis. Curr Genet 53(1):49–58. doi:10.1007/s00294-007-0165-7
Lee SK, Calin GA (2011) Non-coding RNAs and cancer: new paradigms in oncology. Discov Med 11(58):245–54
Lee HC, Li L, Gu W, Xue Z, Crosthwaite SK (2010) Diverse pathways generate microRNA-like RNAs and dicer-independent small interfering RNAs in fungi. Mol Cell 38:803. doi:10.1016/j.molcel.2010.04.005
Li F, Sonbuchner L, Kyes SA, Epp C, Deitsch KW (2008) Nuclear non-coding RNAs are transcribed from the centromeres of Plasmodium falciparum and are associated with centromeric chromatin. J Biol Chem 283(9):5692–8. doi:10.1074/jbc.M707344200
Lin X, Jackson JC, Feretzaki M, Xue C, Heitman J (2010) Transcription factors Mat2 and Znf2 operate cellular circuits orchestrating opposite- and same-sex mating in Cryptococcus neoformans. PLoS Genet 6(5):e1000953. doi:10.1371/journal.pgen.1000953
Luke B, Panza A, Redon S, Iglesias N, Li Z, Lingner J (2008) The Rat1p 5' to 3' exonuclease degrades telomeric repeat-containing RNA and promotes telomere elongation in Saccharomyces cerevisiae. Mol Cell 32(4):465–77. doi:10.1016/j.molcel.2008.10.019
Manel E (2011) Non-coding RNAs in human disease. Nat Rev Genet 12(12):861–874. doi:10.1038/nrg3074
Matsuda E, Garfinkel DJ (2009) Posttranslational interference of Ty1 retrotransposition by antisense RNAs. Proc Natl Acad Sci U S A 106(37):15657–62. doi:10.1073/pnas.0908305106
Morrison EN, Donaldson ME, Saville BJ (2012) Identification and analysis of genes expressed in the Ustilago maydis dikaryon: uncovering a novel class of pathogenesis genes. Can J Plant Pathol 34(3):417–435. doi:10.1080/07060661.2012.697077
Nunes CC, Gowda M, Sailsbery J, Xue M, Chen F, Brown DE, Oh Y, Mitchell TK, Dean RA (2011) Diverse and tissue-enriched small RNAs in the plant pathogenic fungus. Magnaporthe oryzae. BMC Genomics 12:288. doi:10.1186/1471-2164-12-288
Philip A, Edith H (2001) X-chromosome inactivation: counting, choice and initiation. Nat Rev Genet 2(1):59–67. doi:10.1038/35047580
Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136(4):629–641. doi:10.1016/j.cell.2009.02.006
Romano N, Macino G (1992) Quelling: transient inactivation of gene expression in Neurospora crassa by transformation with homologous sequences. Mol Microbiol 6:3343. doi:10.1111/j.1365-2958.1992.tb02202.x
Scaria V, Pasha A (2013) Long noncoding RNAs in infection biology. Front Genet 3 doi:10.3389/fgene.2012.00308
Schirawski J, Mannhaupt G, Münch K, Brefort T, Schipper K, Doehlemann G, Di Stasio M, Rössel N, Mendoza-Mendoza A, Pester D, Müller O, Winterberg B, Meyer E, Ghareeb H, Wollenberg T, Münsterkötter M, Wong P, Walter M, Stukenbrock E, Güldener U, Kahmann R (2010) Pathogenicity determinants in smut fungi revealed by genome comparison. Science 330(6010):1546–1548. doi:10.1126/science.1195330
Shimada T, Yamashita A, Yamamoto M (2003) The fission yeast meiotic regulator Mei2p forms a dot structure in the horse-tail nucleus in association with the sme2 locus on chromosome II. Mol Biol Cell 14(6):2461–9. doi:10.1091/mbc.E02-11-0738
Sierra-Miranda M, Delgadillo DM, Mancio-Silva L, Vargas M, Villegas-Sepulveda N, Martinez-Calvillo S, Scherf A, Hernandez-Rivas R (2012) Two long non-coding RNAs generated from subtelomeric regions accumulate in a novel perinuclear compartment in Plasmodium falciparum. Mol Biochem Parasitol 185(1):36–47. doi:10.1016/j.molbiopara.2012.06.005
Stephane EC, Robert AM (2013) RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet 14(2):100–112. doi:10.1038/nrg3355
Tim RM, Marcel ED, John SM (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10(3):155–159. doi:10.1038/nrg2521
Uhler JP, Hertel C, Svejstrup JQ (2007) A role for noncoding transcription in activation of the yeast PHO5 gene. Proc Natl Acad Sci 104(19):8011–8016. doi:10.1073/pnas.0702431104
van Dijk EL, Chen CL, d’Aubenton-Carafa Y, Gourvennec S, Kwapisz M, Roche V, Bertrand C, Silvain M, Legoix-Ne P, Loeillet S, Nicolas A, Thermes C, Morillon A (2011) XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature 475(7354):114–7. doi:10.1038/nature10118
Volpe TA, Kidner C, Hall IM, Teng G, Grewal SIS, Martienssen RA (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297(5588):1833–1837. doi:10.1126/science.1074973
Wahlestedt C (2006) Natural antisense and noncoding RNA transcripts as potential drug targets. Drug Discov Today 11(11–12):503–8. doi:10.1016/j.drudis.2006.04.013
Wang X, Hsueh YP, Li W, Floyd A, Skalsky R, Heitman J (2010) Sex-induced silencing defends the genome of Cryptococcus neoformans via RNAi. Genes Dev 24(22):2566–82. doi:10.1101/gad.1970910
Wang L, Zhai B, Lin X (2012) The link between morphotype transition and virulence in Cryptococcus neoformans. PLoS Pathog 8(6):e1002765. doi:10.1371/journal.ppat.1002765
Watanabe Y, Yamamoto M (1994) S. pombe mei2+ encodes an RNA-binding protein essential for premeiotic DNA synthesis and meiosis I, which cooperates with a novel RNA species meiRNA. Cell 78(3):487–498. doi:10.1016/0092-8674(94)90426-X
Weissenmayer BA, Prendergast JG, Lohan AJ, Loftus BJ (2011) Sequencing illustrates the transcriptional response of Legionella pneumophila during infection and identifies seventy novel small non-coding RNAs. PLoS One 6(3):e17570. doi:10.1371/journal.pone.0017570
Wilusz JE, Freier SM, Spector DL (2008) 3' end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 135(5):919–932. doi:10.1016/j.cell.2008.10.012
Xu Y, Suzuki Y, Ito K, Komiyama M (2010) Telomeric repeat-containing RNA structure in living cells. Proc Natl Acad Sci U S A 107(33):14579–84. doi:10.1073/pnas.1001177107
Yamamoto T, Manome Y, Nakamura M, Tanigawa N (2002) Downregulation of survivin expression by induction of the effector cell protease receptor-1 reduces tumor growth potential and results in an increased sensitivity to anticancer agents in human colon cancer. Eur J Cancer 38(17):2316–24. doi:10.1016/S0959-8049(02)00247-2
Zarnack K, Maurer S, Kaffarnik F, Ladendorf O, Brachmann A, Kämper J, Feldbrügge M (2006) Tetracycline-regulated gene expression in the pathogen Ustilago maydis. Fungal Genet Biol 43(11):727–738. doi:10.1016/j.fgb.2006.05.006
Zhai B, Zhu P, Foyle D, Upadhyay S, Idnurm A, Lin X (2013) Congenic strains of the filamentous form of Cryptococcus neoformans for studies of fungal morphogenesis and virulence. Infect Immun. doi:10.1128/IAI.00259-13
Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D (2007) Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 129(2):303–17. doi:10.1016/j.cell.2007.03.030
Zhenyu X, Wu W, Julien G, Fabiana P, Sandra C-M, Jurgi C, Elisa G, Françoise S, Wolfgang H, Lars MS (2009) Bidirectional promoters generate pervasive transcription in yeast. Nature 457(7232):1033–1037. doi:10.1038/nature07728
Acknowledgments
This work was supported by NIAID/NIH (grant R01AI097599 to XL). The funder had no role in the decision to publish or in preparation of the manuscript. We thank Lin lab members for critical reading and helpful suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chacko, N., Lin, X. Non-coding RNAs in the development and pathogenesis of eukaryotic microbes. Appl Microbiol Biotechnol 97, 7989–7997 (2013). https://doi.org/10.1007/s00253-013-5160-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5160-y