
Abstract
Rhodococcus erythropolis WZ010 was capable of producing optically pure (2S,3S)-2,3-butanediol in alcoholic fermentation. The gene encoding an acetoin(diacetyl) reductase from R. erythropolis WZ010 (ReADR) was cloned, overexpressed in Escherichia coli, and subsequently purified by Ni-affinity chromatography. ReADR in the native form appeared to be a homodimer with a calculated subunit size of 26,864, belonging to the family of the short-chain dehydrogenase/reductases. The enzyme accepted a broad range of substrates including aliphatic and aryl alcohols, aldehydes, and ketones. It exhibited remarkable tolerance to dimethyl sulfoxide (DMSO) and retained 53.6 % of the initial activity after 4 h incubation with 30 % (v/v) DMSO. The enzyme displayed absolute stereospecificity in the reduction of diacetyl to (2S,3S)-2,3-butanediol via (S)-acetoin. The optimal pH and temperature for diacetyl reduction were pH 7.0 and 30 °C, whereas those for (2S,3S)-2,3-butanediol oxidation were pH 9.5 and 25 °C. Under the optimized conditions, the activity of diacetyl reduction was 11.9-fold higher than that of (2S,3S)-2,3-butanediol oxidation. Kinetic parameters of the enzyme showed lower K m values and higher catalytic efficiency for diacetyl and NADH in comparison to those for (2S,3S)-2,3-butanediol and NAD+, suggesting its physiological role in favor of (2S,3S)-2,3-butanediol formation. Interestingly, the enzyme showed higher catalytic efficiency for (S)-1-phenylethanol oxidation than that for acetophenone reduction. ReADR-catalyzed asymmetric reduction of diacetyl was coupled with stereoselective oxidation of 1-phenylethanol, which simultaneously formed both (2S,3S)-2,3-butanediol and (R)-1-phenylethanol in great conversions and enantiomeric excess values.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Acetoin(diacetyl) reductase (ADR; also known as 2,3-butanediol dehydrogenase) is one of the key enzymes in the microbial production of 2,3-butanediol, a platform chemical with extensive industrial applications in the production of plastics, printing inks, perfumes, fumigants, spandex, moistening and softening agents, plasticizers, and pharmaceutical carrier (Syu 2001; Celińska and Grajek 2009; Ji et al. 2011). ADRs can reduce diacetyl to acetoin and then to 2,3-butanediol that has three types of stereoisomers: meso-2,3-butanediol, (2R,3R)-2,3-butanediol, and (2S,3S)-2,3-butanediol. Optically pure 2,3-butanediol with two vicinal stereogenic centers can be used as an excellent building block in asymmetric synthesis of chiral compounds. However, native microorganisms usually produce a mixture of two 2,3-butanediol stereoisomers (Yan et al. 2009). Particularly, no native microorganism has been reported to produce optically pure (2S,3S)-2,3-butanediol yet (Ji et al. 2011). Therefore, it is desirable to discover the native producer or its ADR with the capability to form enantiomerically pure (2S,3S)-2,3-butanediol. Due to difficulties in the direct fermentative production of (2S,3S)-2,3-butanediol, researchers have developed different methods for (2S,3S)-2,3-butanediol production. The ADR-overexpressed Escherichia coli cells in conjunction with asymmetric reduction of diacetyl has been approved to be a promising alternative for the production of (2S,3S)-2,3-butanediol (Xiao et al. 2010; Zeng and Sabra 2011; Li et al. 2012).
In terms of stereospecificity, carbonyl reductases such as ADRs are also a group of important biocatalysts for the synthesis of chiral alcohols by either asymmetric reduction or stereoselective oxidation. Rhodococcus erythropolis strains have been reported to contain a large set of oxidoreductases that asymmetrically reduce prochiral ketones to chiral alcohols (de Carvalho and da Fonseca 2005). Two NADP+-dependent short-chain dehydrogenases from R. erythropolis MAK154 and R. erythropolis BCRC 10909 were characterized as promising enzymes for the preparation of double chiral amino alcohols (Kataoka et al. 2006; Lin et al. 2010). The gene encoding an NAD+-dependent medium-chain alcohol dehydrogenase was isolated from the genomic DNA of R. erythropolis DSM 43297, which gene product displayed (S)-stereospecificity for the reduction of acetophenone (Abokitse and Hummel 2003). Another medium-chain alcohol dehydrogenase characterized from R. erythropolis ATCC 4277 was (R)-enantioselective in β-keto esters transformation (Zhu et al. 2012). Up to date, neither ADR nor 2,3-butanediol dehydrogenase has been characterized from R. erythropolis yet.
We previously identified and characterized R. erythropolis WZ010 as a versatile biocatalyst with excellent stereoselectivity in the whole-cell-catalyzed reduction of various aryl ketones (Yang et al. 2012). Here, we report that R. erythropolis WZ010 had the capability to produce optically pure (2S,3S)-2,3-butanediol in alcoholic fermentation. The recombinant NAD+-dependent ADR was strictly (S)-enantioselective in ketone reduction and highly stable at elevated concentrations of dimethyl sulfoxide (DMSO). In addition, the enzyme was used for simultaneous catalysis of both asymmetric reduction of diacetyl and stereoselective oxidation of racemic 1-phenylethanol, which led to efficient recycling of NAD+/NADH and the production of both (2S,3S)-2,3-butanediol and (R)-1-phenylethanol.
Materials and methods
Chemicals, enzymes, and organisms
All chemicals were purchased from J&K Chemical Ltd. (Shanghai, China) or Shanghai Jingchun Reagent Co., Ltd. (Shanghai, China), unless stated otherwise. Restriction enzymes were purchased from TaKaRa Bio Inc. (Dalian, China). TransStrat™ Taq DNA Polymerase for polymerase chain reaction (PCR) amplification and pEASY-E1 expression vector were purchased from TransGen Biotech Co., Ltd. (Beijing, China). R. erythropolis WZ010 used as the donor of adr gene had been deposited in the China Center for Type Culture Collection (CCTCC 2011336). The strains E. coli DH5α and BL21(DE3) were used for the purposes of cloning and expression, respectively.
Growth conditions
The Luria–Bertani (LB) medium was routinely used for culturing recombinant E. coli and preparing seed inocula of R. erythropolis WZ010. The alcoholic fermentation of R. erythropolis WZ010 was carried out with a simple control strategy of O2 limitation in a previously described fermentation medium containing 110 mM glucose (Jung et al. 2012). During the first 12 h of incubation, R. erythropolis WZ010 was cultivated on a rotary shaker at 200 rpm for sufficient O2 supply. After then, the cells were cultured under static condition for strong O2 limitation. All bacteria were cultured at 30 °C, 200 rpm for 24 h, unless stated otherwise.
Construction of the expression plasmid pEASY-E1-adr
The gene of R. erythropolis WZ010 encoding the putative ADR, named as adr, was amplified from the genomic DNA using forward and reverse primers adrF (5′-ATGAGCATCACGGGCA-3′) and adrR (5′-TCAGCGGTAGACGAGACC-3′). The adr gene was PCR-amplified with the conditions as follows: 94 °C for 4 min for initial denaturation; 30 cycles of 94 °C for 30 s, 52 °C for 30 s, and 72 °C for 50 s; and 72 °C for 10 min for the final extension. The PCR products were purified using the Genomic DNA Fragment Rapid Purification Kit (BioDev-Tech, Beijing, China). According to the instruction of the pEASY-E1 Expression Kit, the targeted DNA fragment was ligated with the expression vector and then the ligated product was transformed into E. coli DH5α competent cells. The plasmid DNA was isolated from positive transformers screened through colony PCRs and further verified by DNA sequencing (Sangon, Shanghai, China). The plasmid harboring the adr gene was designated as pEASY-E1-adr.
Expression and purification of ReADR
The plasmid pEASY-E1-adr was transformed into the strain E. coli BL21(DE3). The recombinant cells were grown at 37 °C in the LB medium until the OD600 reached up to 0.5, when the gene expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside. The cells were subsequently cultured at 28 °C for 16 h and then harvested by centrifugation. The precipitate was resuspended in 50 mM Tris–HCl buffer (pH 8.0) and disrupted through ultrasonication for 8 min. The cell lysate was then centrifuged to remove the cell debris. The obtained supernatant from the crude extract was applied to a Ni-NTA chelating affinity column (Bio-Rad Laboratories, Hercules, CA, USA) equilibrated with the binding buffer (5 mM imidazole and 300 mM NaCl dissolved in 50 mM Tris–HCl, pH 8.0). The bound enzyme was eluted by applying a stepwise gradient of imidazole concentrations from 10 to 250 mM. Fractions containing ReADR were eluted with 100 mM imidazole, desalted with 50 mM Tris–HCl (pH 8.0) by ultrafiltration, and then stored at −20 °C for further characterization. The purity of the purified ReADR was verified using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) as described previously (Laemmli 1970). The molecular mass of the enzyme in the native form was determined using a high-performance liquid chromatography equipped with the size-exclusion column WAT011535 (Waters Corporation, Milford, MA, USA), in which the flow phase was 50 mM phosphate buffer containing 150 mM NaCl (pH 6.8) and the molecular mass standards consist of thyroglobulin (670,000), γ-globulin (158,000), ovalbumin (44,000), myoglobin (17,000), and vitamin B12 (1,350).
Enzyme activity assay of the purified ReADR
The catalytic activity of ReADR was measured by monitoring the substrate-dependent absorbance change of NAD(H) at 340 nm (ε 340 = 6.3 mM−1 cm−1). Unless otherwise specified, the enzyme assay was carried out in duplicate using the assay mixture (2.5 ml) for alcohol oxidation containing 22 mM 2,3-butanediol and 0.64 mM NAD+ at 25 °C in the 50 mM N-cyclohexyl-2-hydroxyl-3-aminopropanesulfonic acid (CAPSO) buffer (pH 9.5). The assay mixture (2.5 ml) for the reduction of ketone/aldehyde contained 22 mM diacetyl and 0.4 mM NADH at 30 °C in the 50 mM piperazine-N,N′-bis(ethanesulfonic acid) buffer (PIPES, pH 7.0). The addition of the purified enzyme (40 μg) initiated the enzyme assay. One unit of the activity is defined as 1 μmol NADH formation or oxidation per minute. The protein concentrations of all samples were determined using the Bradford method, and bovine serum albumin served as the standard protein.
Catalytic properties of ReADR
The effect of temperature on the enzyme activity was examined at temperatures from 15 to 50 °C. The effect of pH on the enzyme activity was determined over a range of pH 6.1 to 11. The buffers (50 mM) used were PIPES (pH 6.1 to 7.5), Tris–HCl (pH 7.5 to 9.0), CAPSO (pH 9.0 to 10.0), and 3-(cyclohexylamino)-1-propanesulfonic acid (pH 10.0 to 11.0). Substrate specificity was determined using primary and secondary alcohols, diols, polyols, and aromatic alcohols or aldehydes and ketones under standard assay conditions. The effect of cations, ethylenediaminetetraacetic acid (EDTA), and organic solvents on enzyme activity was examined by adding each compound at a final concentration of 1 mM (metal ions and EDTA) or 10 % (v/v) (organic solvents). The residual enzyme activity was determined by measuring the reduction of diacetyl in the 50 mM PIPES (pH 7.0), and the control enzyme activity was assayed in the absence of any test compound. The stability at elevated concentrations of DMSO was carried out by incubating the enzyme in the 50 mM Tris–HCl buffer (pH 8.0) with DMSO ranging from 10 to 40 % (v/v) at 4 °C for 4 h and then the residual activity for diacetyl reduction was determined.
Enzyme kinetic parameters were determined using different substrates and coenzymes (NAD+ or NADH). Various substrate concentrations were used for determining the corresponding activities at the appropriate temperature (30 °C for ketone reduction and 25 °C for alcohol oxidation) when concentrations of corresponding coenzymes were kept constant. Substrates and coenzymes used were NAD+ (0, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, and 0.8 mM), (2S,3S)-2,3-butanediol (0, 0.45, 2.3, 5.8, 11.6, 23.2, 34.8, 46.4, 58, and 116 mM), (S)-1-phenylethanol (0, 1.16, 2.3, 4.0, 6.9, 11.6, 23.2, 46.4, and 92.8 mM), NADH (0, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, and 0.6 mM), diacetyl (0, 0.45, 2.3, 5.8, 11.6, 23.2, 34.8, 46.4, and 92.8 mM), and acetophenone (0, 0.58, 2.0, 4.6, 8.4, 11.6, 23.2, 34.8, 46.4, and 92.8 mM). Apparent values of K m and V max were calculated by fitting the data into the Michaelis–Menten equation using SigmaPlot (Systat Software Inc., San Jose, CA, USA). All the reactions followed Michaelis–Menten-type kinetics.
Analyses of end products in alcoholic fermentation
The possible end products, such as acetoin and 2,3-butanediol, were quantitated using a gas chromatography (GC) method as follows: instrument, Shimadzu GC-2014; column, Agilent HP-5, 30 m × 0.32 mm × 0.25 μm; carrier gas, nitrogen at a flow rate of 20 ml/min; split ratio, 1:50; oven temperature program, isotherm at 50 °C for 3 min, a 10 °C/min ramp to 200 °C, and isotherm at 200 °C. Prior to the analyses, the culture medium was centrifuged at 10,000×g for 10 min and the supernatant was filtered through a 0.45-μm membrane to remove the residual cells. After filtration, the supernatant was mixed with methanol at a ratio of 1:1 and 1 μl treated sample was applied for GC analyses. The stereoselectivity of acetoin and 2,3-butanediol was determined using a chiral GC method described in the succeeding section.
Asymmetric reduction of diacetyl and acetoin
The reduction mixture (2 ml) containing 10 mM diacetyl or racemic acetoin, 50 mM 2-butanol as cosubstrate, 1 mM NADH, and 300 μg purified enzyme in the 50 mM PIPES buffer (pH 7.0). The reaction was carried out at 30 °C for 36 h under static condition, unless otherwise specified. The reactants were determined with a Shimadzu GC-2014 gas chromatograph equipped with a chiral GC column (Varian CP7502, 25 m × 0.25 mm × 0.39 mm). The GC analyses were performed under the following conditions: carrier gas, nitrogen at a flow rate of 20 ml/min; split ratio, 1:50; oven temperature program, isotherm at 80 °C for 5 min, 30 °C/min ramp to 120 °C, 15 °C/min ramp to 180 °C, and then isotherm at 180 °C for 5 min; detector, flame ionization detector (250 °C). The 2-ml reaction mixture was extracted with 1 ml 1-butanol under strong vibration. The reaction mixture after extraction was dehydrated with anhydrous sodium sulfate and then 1 μl dehydrated sample was directly applied onto the injector (250 °C) for GC analyses. The peak areas were quantitated using specific external standards. Retention times of the reactants were listed as follows: 3.25 min for diacetyl, 6.30 min for (R)-acetoin, 6.60 min for (S)-acetoin, 9.16 min for (2S,3S)-2,3-butanediol, 9.25 min for (2R,3R)-2,3-butanediol, and 9.42 min for meso-2,3-butanediol.
Asymmetric reduction of diacetyl and stereoselective oxidation of 1-phenylethanol simultaneously catalyzed by ReADR
The reaction mixture (2 ml) containing 60 mM diacetyl, 300 mM racemic 1-phenylethanol, 1 mM NADH, and 1.8 mg purified enzyme in the 50 mM PIPES buffer (pH 7.0). The reaction was carried out at 30 °C for 60 h under static condition. To investigate the effect of different ketone/alcohol ratios on conversion and stereoselectivity, the concentration of diacetyl was fixed at 60 mM but that of racemic 1-phenylethanol varied over a range of 120 to 480 mM. The chiral GC method used for stereoselectivity determination was the same as that described in the previous section. In addition, the retention times for acetophenone and 1-phenylethanol enantiomers were listed as follows: 10.35 min for acetophenone, 11.68 min for (R)-1-phenylethanol, and 11.81 min for (S)-1-phenylethanol.
Nucleotide sequence accession number
The nucleotide sequence of ReADR has been submitted to the GenBank database under accession number KC508606.
Results
Identification, cloning, and expression of ReADR
During alcoholic fermentation, R. erythropolis WZ010 produced optically pure (S)-acetoin and (2S,3S)-2,3-butanediol in small amounts (<0.5 mM) as end products, suggesting the presence of the entire metabolic pathway of (2S,3S)-2,3-butanediol formation and corresponding stereospecific ADR (Yan et al. 2009; Ji et al. 2011; Li et al. 2012). The mining of the genome sequence of R. erythropolis PR4 revealed two genes involving the interconversion between acetoin/diacetyl and 2,3-butanediol: a putative medium-chain (2R,3R)-2,3-butanediol dehydrogenase (GenBank accession number BAH34736) and an ADR (GenBank accession number BAH31367) (Sekine et al. 2006), the latter of which was studied further in detail. The adr gene encoding the putative ADR was PCR-amplified from the genomic DNA of R. erythropolis WZ010, of which a 780-bp PCR product encoded 259 amino acid residues with a deduced molecular mass of 26,864. The gene adr was subsequently overexpressed in E. coli cells using the pEASY-E1 expression vector. The recombinant ReADR with an N-terminal His-tag was purified to electrophoretic homogeneity by nickel affinity chromatography. The specific activity of the purified enzyme was 9.16 U/mg using diacetyl as substrate, indicating a 4.0-fold purification from the crude extract. The purified enzyme migrated as a single band with a size of 29,000 ± 1,000 on SDS-PAGE (Fig. 1) and its native molecular mass was determined using gel filtration to be 70,000 ± 2,000. Thus, the purified ReADR in the native form could be a homodimer.

SDS-PAGE (12.5 %) analysis of the purified ReADR. Lane 1 molecular weight marker, lane 2 4 μg ReADR
The nucleotide sequence of ReADR showed 97 % identity with its homologue in R. erythropolis PR4 (Sekine et al. 2006), with two amino acid residues Arg51 and Val60 in ReADR different from Gln51 and Ile60 in R. erythropolis PR4 ADR. The deduced amino acid sequence of ReADR also showed high overall identities to ADRs from other Rhodococcus species, e.g., ADRs from Rhodococcus opacus B4 (79 % identity; BAH53568), Rhodococcus jostii RHA1 (78 % identity; ABG97060), Rhodococcus sp. JVH1 (78 % identity; EJI95910), Rhodococcus wratislaviensis IFP 2016 (77 % identity; ELB92199), and Rhodococcus imtechensis RKJ300 (77 % identity; EID77487). The enzyme ReADR showed high sequence homology to two NAD+-dependent stereospecific 2,3-butanediol dehydrogenases with known three-dimensional structure, and the sequence alignment revealed that the enzyme harbored the highly conserved amino acid residues for all “classical” short-chain dehydrogenases/reductases (SDRs) (Fig. 2). The coenzyme binding motif T11G12XXXG16XG18 was conserved in the N-terminal region, which is the glycine-rich consensus sequence in all “classical” SDRs (Kavanagh et al. 2008). The conserved catalytic tetrad Asn113-Ser142-Tyr155-Lys159 was also identified with the active site motif Y155XXXK159.

Structure-related sequence alignment between ReADR and homologous proteins. The homologues of ReADR were identified by performing BLASTP searches (Altschul et al. 1997). The alignment of ReADR and its close homologues of SDR families were carried out using the program ClustalW and subsequently visualized using ESPript 2.2 (Thompson et al. 1994; Gouet et al. 1999). PDB codes and DDBJ accession numbers for the sequences are as follows: 1GEG (D86412), meso-2,3-butanediol dehydrogenase from K. pneumoniae IAM 1063 (Ui et al. 1997; Otagiri et al. 2001); 3A28 (AB009078), (2S,3S)-2,3-butanediol dehydrogenase from B. saccharolyticum C-1012 (Ui et al. 1998; Otagiri et al. 2010). Shown above the alignments are elements of the secondary structure of 1GEG. The numbering shown is from 1GEG. Blue triangles critical cofactor binding residues, red stars putative catalytic residues located at the corresponding positions of the conserved catalytic residues N113, S142, Y155, and K159. Strictly conserved residues are highlighted with red boxes
Catalytic properties of ReADR
The enzyme ReADR was strictly NAD+-dependent. The enzyme activity was not detectable when NADP(H) was used as coenzyme. ReADR was active in the range of 10–50 °C. The optimal temperatures of the enzyme was found to be 25 °C for the oxidation of 2,3-butanediol and 30 °C for the reduction of diacetyl (Fig. 3a). The enzyme could also be adapted to a broader pH range from 6.1 to 11.0. The enzyme exhibited the highest activity at pH 9.5 for the oxidation reaction, of which the optimal pH was higher than that for the reduction reaction (pH 7.0) (Fig. 3b). Under the optimized conditions, the highest activity of diacetyl reduction was 11.9 times higher than that of (2S,3S)-2,3-butanediol oxidation, revealing the nature of reductase.

Effect of temperature (a) and pH (b) on the activities of ReADR. The relative activity of 100 % represents 9.16 U/mg for diacetyl reduction at 30 °C and pH 7.0. Solid symbols reduction, empty symbols oxidation
The substrate specificity of the recombinant enzyme was determined using a set of alcohols, aldehydes, and ketones (Table 1). In the oxidation reaction, the enzyme was able to transform a broad range of primary and secondary alcohols including aromatic alcohols. The enzyme could oxidize 2,3-butanediol but not acetoin. Among the tested alcohols, the enzyme exhibited the highest activity for 2-pentanol as substrate. In the reduction reaction, the enzyme exhibited the ability to reduce diacetyl, acetoin, and various aromatic ketones and aldehydes. Differing from the oxidation reaction, the enzyme showed the highest activity on diacetyl but not acetophenone in the reduction reaction.
Enzymes resistant to environmental factors such as metal ions or organic solvents are of great interest for the purpose of practical applications. The addition of EDTA or the cations (1 mM) such as Na+, K+, Mn2+, Mg2+, and Ca2+ had no significant effect on the activity of ReADR, whereas 2 mM cations including Na+, K+, and Mn2+ significantly increased the activity by 201.6 to 265.6 %. On the other hand, the cations Zn2+ and Fe2+ decreased the activity to 5.4 and 8.4 % of the control enzyme activity, respectively, and Al3+ thoroughly inhibited the enzyme activity. When 10 % (v/v) organic solvent such as methanol, ethanol, acetone, or acetonitrile was added into the assay mixture, the residual activity remained at 35.8–57.2 % of the control enzyme activity. Interestingly, when DMSO at a final concentration of 30 % (v/v) was added into the assay mixture, the activity was increased up to 120 % of the control enzyme activity. To further investigate its stability at elevated concentrations of DMSO, the enzyme was incubated with 30 % (v/v) DMSO at 4 °C for 4 h and the residual activity was 53.6 % of the initial activity.
Stereoselectivity and kinetic parameters of ReADR
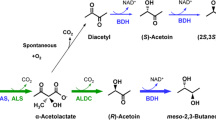
In the oxidation of alcohols, ReADR demonstrated activities on meso-2,3-butanediol, (2S,3S)-2,3-butanediol, and (S)-1-phenylethanol, but not (2R,3R)-2,3-butanediol and (R)-1-phenylethanol, suggesting that the enzyme stereoselectively functioned on the (S)-hydroxyl group of 2,3-butanediol and 1-phenylethanol as substrate (Table 1). To further approve its stereoselectivity, the corresponding ketone reduction and its products were further investigated. In the reduction reaction, the ReADR-catalyzed reduction of diacetyl led to optically pure (S)-acetoin and (2S,3S)-2,3-butanediol with 34 and 60 % yields, respectively. Moreover, the reduction of racemic (R/S)-acetoin formed (2S,3S)-2,3-butanediol and meso-2,3-butanediol, which were presumably originated from (S)-acetoin and (R)-acetoin, respectively, indicating that the reduction of diacetyl and acetoin was also (S)-stereoselective.
The apparent K m value for the coenzyme NADH was 3.6 times lower than that for the coenzyme NAD+ (Table 2). The specificity constant K cat/K m for NADH as an electron donor in the diacetyl reduction (8,519 s−1 M−1) was over 30 times higher than that of the electron acceptor NAD+ in the oxidation of corresponding alcohol (269 s−1 M−1). These catalytic properties suggest that the enzyme was more likely to be involved in the oxidation of NADH rather than the reduction of NAD+ in vivo. In the interconversion between diacetyl and (2S,3S)-2,3-butanediol, the enzyme had higher catalytic efficiency for diacetyl as compared to (2S,3S)-2,3-butanediol. Since the diacetyl reduction requires the coupling of the NADH oxidation, the kinetic parameters of the enzyme supported that its physiological role was very likely to be responsible for the formation of (2S,3S)-2,3-butanediol rather than its oxidation. Different from the interconversion between (2S,3S)-2,3-butanediol and diacetyl, the enzyme showed lower K m and higher catalytic efficiency for (S)-1-phenylethanol in comparison to those for acetophenone.
Asymmetric reduction of diacetyl and stereoselective oxidation of 1-phenylethanol simultaneously catalyzed by ReADR
Both asymmetric reduction of diacetyl and stereoselective oxidation of racemic 1-phenylethanol were coupled together to achieve not only two chiral alcohols from the single-enzyme-catalyzed system but also efficient coenzyme recycling (Fig. 4). The concentration of diacetyl was fixed as 60 mM and that of racemic 1-phenylethanol varied. When the ratio of ketone/alcohol was 1:5, diacetyl was completely reduced to optically pure (2S,3S)-2,3-butanediol meanwhile the enzyme oxidized (S)-enantiomer of racemic 1-phenylethanol (300 mM) to acetophenone in 49.3 % conversion and afforded the unoxidized (R)-1-phenylethanol in 97 % enantiomeric excess (e.e.) value (Table 3). In the coupled reaction, the concentration of coenzyme used was only 1 mM and its total turnover number (TTN) was calculated to be 147.9 based on the oxidation of 1-phenylethanol, indicating that the recycling of NAD+/NADH was highly efficient. When the concentration of racemic 1-phenylethanol was reduced to 120 mM, the conversion of diacetyl reduction was decreased to 81 % and the product was a mixture of (S)-acetoin and (2S,3S)-2,3-butanediol. The TTN value of the coenzyme was lowered to be 60, suggesting that the incomplete conversion of diacetyl and the emergence of (S)-acetoin might be attributed to the relatively lower level of the coenzyme regeneration. On the other hand, the elevated concentration of racemic 1-phenylethanol (480 mM) lowered its conversion (24.7 %) and the optical purity of (R)-1-phenylethanol (32.9 % e.e.), although the yield and optical purity of (2S,3S)-2,3-butanediol remained excellent.

Scheme of asymmetric reduction of diacetyl and stereoselective oxidation of racemic 1-phenylethanol simultaneously catalyzed by ReADR
Discussion
The enzyme ReADR shares common features with other homologues in the SDR family. Short-chain ADRs from bacteria have highly conserved residues and similar overall three-dimensional structures. They usually are NAD+-dependent, of which coenzyme discrimination is determined by an acidic residue located about 20 residues downstream of the coenzyme-binding motif (Lesk 1995; Kavanagh et al. 2008). Using the meso-2,3-butanediol dehydrogenase from Klebsiella pneumoniae IAM 1063 (PDB code 1GEG) as template, the secondary and tertiary structures of ReADR were predicted using the SWISS-MODEL server (Arnold et al. 2006) and visualized using the software program PyMOL (Delano 2002). The three-dimensional modeling of the structure indicated that the enzyme folding is highly similar to that found in other SDRs (Otagiri et al. 2001, 2010). Seven parallel β-sheets (β1–β7) flanked by seven parallel α-helices (α1–α6, α9) were the core of the domain constituting the putative dinucleotide and substrate binding motifs, while two α-helices (α7 and α8) formed a small lobe on top of the core structure. Similar to other ADRs (Giovannini et al. 1996; González et al. 2000; Nicholson 2008; Ying and Ma 2011), ReADR could not oxidize acetoin to diacetyl and the reduction of diacetyl to acetoin is irreversible. In terms of stereospecificity, the SDR-type ADRs in the alcohol oxidation are commonly stereospecific for meso-2,3-butanediol and/or (2S,3S)-2,3-butanediol, while (2R,3R)-2,3-butanediol dehydrogenases are often clustered in the family of medium-chain zinc-containing alcohol dehydrogenases (González et al. 2000; Takeda et al. 2011; Ying and Ma 2011).
ReADR had distinct catalytic characteristics. The amino acid sequence of ReADR shared 57 and 52 % identities to those of meso-2,3-butanediol dehydrogenase from K. pneumoniae IAM 1063 and (2S,3S)-2,3-butanediol dehydrogenase from Brevibacterium saccharolyticum C-1012, respectively. The triple mutant of meso-2,3-butanediol dehydrogenase from K. pneumoniae IAM 1063 (Q140I, N146F, and W190H) was prepared, and the stereospecificity of the triple mutant was shifted from the meso-type to the (2S,3S)-type (Otagiri et al. 2010). ReADR harbored the same conserved residues I143, F149, and W193 as (2S,3S)-2,3-butanediol dehydrogenase from B. saccharolyticum C-1012.3D. However, the enzyme ReADH displayed the oxidizing activities using either meso-2,3-butanediol or (2S,3S)-2,3-butanediol as substrate. This indicates that substrate recognition was not simply determined by the three residues I143, F149, and W193 (numbering in ReADR), and it seems necessary to further investigate the subtle differences in the environment around the catalytic cleft. Furthermore, the enzyme activity of ReADR was severely inhibited by the cations Zn2+ and Fe2+, implying that the residues involved in the binding of Zn2+ and Fe2+ might also be critical for the activity (Ying et al. 2009; Ying and Ma 2011). In addition, the organic solvent tolerance of ReADR proved to be special. DMSO at a final concentration of 30 % (v/v) slightly increased the enzyme activity and the enzyme incubated in a 30 % (v/v) DMSO solution exhibited remarkably high stability. Owing to the DMSO tolerance of ReADR, the use of nonaqueous solvent such as DMSO would provide the advantages to solubilize more hydrophobic substrates but also make it possible to carry out ReADR-catalyzed redox reactions in nonconventional microaqueous media (Lavandera et al. 2008; Jakoblinnert et al. 2011).
In the organic syntheses, stereospecific oxidoreductases or related cells can be employed for ketone/alcohol conversions in both the oxidative and reductive directions. As a whole-cell biocatalyst, R. erythropolis WZ010 stereoselectively oxidized 120 mM 1-phenylethanol, affording 49.4 % (R)-1-phenylethanol yield and 99.9 % e.e. value after 48 h, while the bioreduction of 80 mM acetophenone after the same reaction time only gave 64.5 % (S)-1-phenylethanol yield (Yang et al. 2012). In comparison with the whole-cell-catalyzed synthesis of chiral aryl alcohols, the ReADR-based biocatalysis could deal with higher concentrations of 1-phenylethanol as substrate and afford greater yield and optical purity of (2S,3S)-2,3-butanediol as product. When 1 mM NADH was used, the coupled redox process led to the production of 58.8 mM (2S,3S)-2,3-butanediol from 60 mM diacetyl and stereoselective oxidation of 147.3 mM (S)-enantiomer from 300 mM racemic 1-phenylethanol. The coupled reaction provides an efficient approach to simultaneously obtain (2S,3S)-2,3-butanediol and (R)-1-phenylethanol, both of which are relatively easy to be separated. The reaction setup requires stereospecific carbonyl reductase and a suitable substrate pair of ketone and alcohol. The model reaction also rendered a means for in situ recycling of NAD+/NADH in enzymatic redox reactions useful in organic syntheses. In the stoichiometry of the coupled reaction, 1 mol diacetyl can be completely reduced to (2S,3S)-2,3-butanediol by ReADR together with 2 mol NADH, which is expected to originate from the stereoselective oxidation of 4 mol racemic 1-phenylethanol. Both (2S,3S)-2,3-butanediol and (R)-1-phenylethanol were formed in great yields and e.e. values when the ketone/alcohol ratio was set as 1:5, a value very close to the stoichiometric value 1:4, indicating that the reaction equivalent in favor of the synthesis of (2S,3S)-2,3-butanediol and (R)-1-phenylethanol was not necessary to be driven by a high concentration of cosubstrate. In comparison to conventional coenzyme recycling in which the concentration of the cosubstrate (e.g., isopropanol) is usually ten times higher than that of the substrate (Lavandera et al. 2008), the single-enzyme-catalyzed redox process for two chiral alcohols would be more economical in practical applications.
References
Abokitse K, Hummel W (2003) Cloning, sequence analysis, and heterologous expression of the gene encoding a (S)-specific alcohol dehydrogenase from Rhodococcus erythropolis DSM 43297. Appl Microbiol Biotechnol 62:380–386
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL Workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201
Celińska E, Grajek W (2009) Biotechnological production of 2,3-butanediol—current state and prospects. Biotechnol Adv 27:715–725
de Carvalho CCCR, da Fonseca MMR (2005) The remarkable Rhodococcus erythropolis. Appl Microbiol Biotechnol 67:715–726
Delano WL (2002) The PyMOL molecular graphics system. Delano Scientific, Palo Alto
Giovannini PP, Medici A, Bergamini CM, Rippa M (1996) Properties of diacetyl(acetoin) reductases from Bacillus stearothermophilus. Bioorg Med Chem 4:1197–1201
González E, Fernández MR, Larroy C, Solà L, Pericàs MA, Parés X, Biosca JA (2000) Characterization of a (2R,3R)-2,3-butanediol dehydrogenase as the Saccharomyces cerevisiae YAL060W gene product. J Biol Chem 275:35876–35885
Gouet P, Courcelle E, Stuart DI, Metoz F (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308
Jakoblinnert A, Mladenov R, Paul A, Sibilla F, Schwaneberg U, Ansorge-Schumacher MBA, de María PD (2011) Asymmetric reduction of ketones with recombinant E. coli whole cells in neat substrates. Chem Commun 47:12230–12232
Ji X, Huang H, Ouyang P (2011) Microbial 2,3-butanediol production: a state-of-the-art review. Biotechnol Adv 29:351–364
Jung M, Ng CY, Song H, Lee J, Oh M (2012) Deletion of lactate dehydrogenase in Enterobacter aerogenes to enhance 2,3-butanediol production. Appl Microbiol Biotechnol 95:461–469
Kataoka M, Nakamura Y, Urano N, Ishige T, Shi G, Kita S, Sakamoto K, Shimizu S (2006) A novel NADP+-dependent l-1-amino-2-propanol dehydrogenase from Rhodococcus erythropolis MAK154: a promising enzyme for the production of double chiral aminoalcohols. Lett Appl Microbiol 43:430–435
Kavanagh KL, Jörnvall H, Persson B, Oppermann U (2008) The SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol Life Sci 65:3895–3906
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lavandera I, Kern A, Schaffenberger M, Gross J, Glieder A, de Wildeman S, Kroutil W (2008) An exceptionally DMSO-tolerant alcohol dehydrogenase for the stereoselective reduction of ketones. ChemSusChem 1:431–436
Lesk AM (1995) NAD-binding domains of dehydrogenases. Curr Opin Struct Biol 5:775–783
Li L, Wang Y, Zhang L, Ma C, Wang A, Tao F, Xu P (2012) Biocatalytic production of (2S,3S)-2,3-butanediol from diacetyl using whole cells of engineered Escherichia coli. Bioresour Technol 115:111–116
Lin W, Chen C, Chen H, Hsu W (2010) Enantioselective synthesis of (S)-phenylephrine by whole cells of recombinant Escherichia coli expressing the amino alcohol dehydrogenase gene from Rhodococcus erythropolis BCRC 10909. Process Biochem 45:1529–1536
Nicholson WL (2008) The Bacillus subtilis ydjL (bdhA) gene encodes acetoin reductase/2,3-butanediol dehydrogenase. Appl Environ Microbiol 74:6832–6838
Otagiri M, Kurisu G, Ui S, Takusagawa Y, Ohkuma M, Kudo T, Kusunoki M (2001) Crystal structure of meso-2,3-butanediol dehydrogenase in a complex with NAD+ and inhibitor mercaptoethanol at 1.7 Å resolution for understanding of chiral substrate recognition mechanisms. J Biochem 129:205–208
Otagiri M, Ui S, Takusagawa Y, Ohtsuki T, Kurisu G, Kusunoki M (2010) Structural basis for chiral substrate recognition by two 2,3-butanediol dehydrogenase. FEBS Lett 584:219–223
Sekine M, Tanikawa S, Omata S, Saito M, Fujisawa T, Tsukatani N, Tajima T, Sekigawa T, Kosugi H, Matsuo Y, Nishiko R, Imamura K, Ito M, Narita H, Tago S, Fujita N, Harayama S (2006) Sequence analysis of three plasmids harboured in Rhodococcus erythropolis strain PR4. Environ Microbiol 8:334–346
Syu MJ (2001) Biological production of 2,3-butanediol. Appl Microbiol Biotechnol 55:10–18
Takeda M, Muranushi T, Inagaki S, Nakao T, Motomatsu S, Suzuki I, Koizumi J (2011) Identification and characterization of a mycobacterial (2R,3R)-2,3-butanediol dehydrogenase. Biosci Biotechnol Biochem 75:2384–2389
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Ui S, Okajima Y, Minura A, Kanai H, Kobayashi T, Kudo T (1997) Sequence analysis of the gene for and characterization of d-acetoin forming meso-2,3-butanediol dehydrogenase of Klebsiella pneumoniae expressed in Escherichia coli. J Ferment Bioeng 83:32–37
Ui S, Okajima Y, Mimura A, Dohmae N, Takio K, Ohkuma M, Kudo T (1998) Cloning, expression and nucleotide sequence of the l-2,3-butanediol dehydrogenase gene from Brevibacterium saccharolyticum C-1012. J Ferment Bioeng 86:290–295
Xiao Z, Lv C, Gao C, Qin J, Ma C, Liu Z, Liu P, Li L, Xu P (2010) A novel whole-cell biocatalyst with NAD+ regeneration for production of chiral chemicals. PLoS One 5:e8860
Yan Y, Lee C, Liao JC (2009) Enantioselective synthesis of pure (R,R)-2,3-butanediol in Escherichia coli with stereospecific secondary alcohol dehydrogenases. Org Biomol Chem 7:3914–3917
Yang C, Ying X, Yu M, Zhang Y, Xiong B, Song Q, Wang Z (2012) Towards the discovery of alcohol dehydrogenases: NAD(P)H fluorescence-based screening and characterization of the newly isolated Rhodococcus erythropolis WZ010 in the preparation of chiral aryl secondary alcohols. J Ind Microbiol Biotechnol 39:1431–1443
Ying X, Ma K (2011) Characterization of a zinc-containing alcohol dehydrogenase with stereoselectivity from the hyperthermophilic archaeon Thermococcus guaymasensis. J Bacteriol 193:3009–3019
Ying X, Grunden AM, Nie L, Adams MWW, Ma K (2009) Molecular characterization of the recombinant iron-containing alcohol dehydrogenase from the hyperthermophilic archaeon, Thermococcus strain ES1. Extremophiles 13:299–311
Zeng A, Sabra W (2011) Microbial production of diols as platform chemicals: recent progresses. Curr Opin Biotechnol 22:749–757
Zhu Q, Jia H, Li Y, Jia L, Ma Y, Wei P (2012) Cloning, expression and characterization of chiral alcohol dehydrogenase from Rhodococcus erythropolis ATCC 4277. Acta Microbiol Sin 52:83–89
Acknowledgments
This work was financially supported by Natural Science Foundation of Zhejiang Province (nos. LY12B06011 and Y4110468), Zhejiang Provincial Top Academic Discipline of Biomedical Engineering (no. SWYX0905), Research Foundation of Education Bureau of Zhejiang Province (no. Y201122330), and National High Technology Research and Development Program of China (no. 2011AA02A210).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, Z., Song, Q., Yu, M. et al. Characterization of a stereospecific acetoin(diacetyl) reductase from Rhodococcus erythropolis WZ010 and its application for the synthesis of (2S,3S)-2,3-butanediol. Appl Microbiol Biotechnol 98, 641–650 (2014). https://doi.org/10.1007/s00253-013-4870-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-4870-5