
Abstract
Yeast is capable of performing posttranslational modifications, such as N- or O-glycosylation. It has been demonstrated that N-glycans play critical biological roles in therapeutic glycoproteins by modulating pharmacokinetics and pharmacodynamics. However, N-glycan sites on recombinant glycoproteins produced in yeast can be underglycosylated, and hence, not completely occupied. Genomic homology analysis indicates that the Pichia pastoris oligosaccharyltransferase (OST) complex consists of multiple subunits, including OST1, OST2, OST3, OST4, OST5, OST6, STT3, SWP1, and WBP1. Monoclonal antibodies produced in P. pastoris show that N-glycan site occupancy ranges from 75–85 % and is affected mainly by the OST function, and in part, by process conditions. In this study, we demonstrate that N-glycan site occupancy of antibodies can be improved to greater than 99 %, comparable to that of antibodies produced in mammalian cells (CHO), by overexpressing Leishmania major STT3D (LmSTT3D) under the control of an inducible alcohol oxidase 1 (AOX1) promoter. N-glycan site occupancy of non-antibody glycoproteins such as recombinant human granulocyte macrophage colony-stimulating factor (rhGM-CSF) was also significantly improved, suggesting that LmSTT3D has broad substrate specificity. These results suggest that the glycosylation status of recombinant proteins can be improved by heterologous STT3 expression, which will allow for the customization of therapeutic protein profiles.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fungal hosts such as Saccharomyces cerevisiae or the methylotrophic yeast Pichia pastoris have distinct advantages for therapeutic protein expression. For example, they do not typically secrete high amounts of endogenous proteins, utilize strong inducible promoters for producing heterologous proteins, can be grown in defined chemical media without the use of animal-derived products, and can produce relatively high titers of recombinant proteins (Cereghino and Cregg 2000). However, the glycosylation process in yeast is significantly different from that of mammalian cells (Hamilton and Gerngross 2007). Yeast-derived-high mannose N-glycans result in fast serum clearance, thereby adversely affecting the pharmacokinetic properties of recombinant therapeutic glycoproteins (Goetze et al. 2011). Hence, with the exception of granulocyte macrophage colony stimulating factor (GM-CSF), yeast has not generally been used to produce therapeutic glycoproteins intended for human use. However, genetically engineered yeast can produce therapeutic glycoproteins that have predominantly human-like complex glycoforms instead of yeast type N-glycans (Hamilton et al. 2003; Hamilton et al. 2006; Jacobs et al. 2009; Nett et al. 2012). Thus, the opportunity for using glycoengineered yeast to produce therapeutic glycoproteins is now available.
Many therapeutic proteins require the posttranslational addition of N-glycans to specific asparagine residues (Asn-X-Ser/Thr) of the protein to ensure proper biological properties and subsequent stability in human serum (Sinclair and Elliott 2005). N-glycan site occupancy of glycoproteins is affected by the substrate preference of the oligosaccharyltransferase (OST) complex, such as acceptor sequons and lipid-linked oligosaccharide precursors. In other words, the threonine of the Asn-X-Thr sequon is a better substrate than the serine of the Asn-X-Ser sequon (Kaplan et al. 1987; Mellquist et al. 1998; Ben-Dor et al. 2004), and proline residues in the vicinity of the N-glycosylation sites reduce N-glycan site occupancy (Choi and Jiménez-Flores 1996; Mellquist et al. 1998; Choi et al. 2003; Ben-Dor et al. 2004). The X residues of the Asn-X-Ser sequon also affect the glycosylation efficiency (Kasturi et al. 1995; Mellquist et al. 1998). It has been found that while the genetically engineered yeast can produce glycoproteins that have mammalian- or human-like N-glycans, the occupancy of N-glycan attachment sites on glycoproteins varies widely. Depending on the protein being analyzed, N-glycan site occupancy can be lower than the occupancy of these same sites in glycoproteins produced in mammalian cells (Li et al. 2006; Choi et al. 2008). This has been observed for various recombinant antibodies produced in P. pastoris (Jiang et al. 2011; Sethuraman et al. 2011; Zhang et al. 2011) as well as in other fungal systems (Horwitz et al. 1988; Ward et al. 2004). Furthermore, the variability of N-glycan site occupancy has also been observed in mammalian cells. For example, Gawlitzek et al. (2009) demonstrated that specific N-glycosylation site occupancy can vary for particular glycoproteins produced in CHO cells, and that modifications in growth conditions can be made to control occupancy at these sites. In addition, there have been efforts to improve protein N-glycosylation site occupancy of eukaryotic cells using the dolichol-linked oligosaccharide synthesis pathway (Betenbaugh et al. 2006), engineering the OST complex and consensus motif (Jones et al. 2005), and optimizing cell culture conditions (Gawlitzek et al. 2009). However, there still remains a need for increasing N-glycosylation site occupancy of therapeutic proteins produced in recombinant host cells.
Leishmania major (Lm) has four STT3 paralogues (LmSTT3A, B, C, and D) that show high homology at the protein level (55–80 % identity). It has been reported that L. major STT3A, B, and D complemented deletions of essential subunits (STT3, OST1, OST2, WBP1, or SWP1) in S. cerevisiae, where LmSTT3 paralogues function as a single enzyme independently of the S. cerevisiae OST complex (Nasab et al. 2008; Hese et al. 2009). In this study, we demonstrate that overexpression of L. major STT3D in P. pastoris was able to improve N-glycan site occupancy of not only antibodies (>99 %), but also of a non-antibody glycoprotein (GM-CSF).
Materials and methods
Gene synthesis
The open reading frames (ORF) of L. major STT3 paralogues STT3A, STT3B, STT3C, and STT3D were codon optimized for P. pastoris and synthesized by GeneArt AG (Germany). The vectors encoding above genes were designated pGLY6284, pGLY6285, pGLY6286, and pGLY6287, respectively.
Plasmid construction and generation of strains
The plasmids and strains used in this study were generated as follows. For inducible expression of LmSTT3D, pGLY6301 was constructed by cloning of the DNA fragment encoding LmSTT3D ORF (pGLY6287) flanked by EcoRI (5′) and FseI (3′) into pGFI30t at EcoRI (5′) and FseI (3′). pGFI30t is an expression vector, where the expression of LmSTT3D was under the control of the P. pastoris alcohol oxidase 1 (PpAOX1) promoter. Transformants were selected using the S. cerevisiae ARR3 gene. For constitutive expression of LmSTT3D, pGLY6294 was constructed by cloning of the DNA fragment encoding LmSTT3D ORF (pGLY6287) flanked by NotI (5′) and PacI (3′) into pGLY597 at NotI (5′) and FseI (3′). pGLY597 is an expression vector, where expression of LmSTT3D was under the control of the P. pastoris glyceraldehyde-3-phosphate dehydrogenase (GAP) promoter. Transformants were selected using the Streptomyces noursei nourseothricin acetyltransferase (NAT) gene. pGLY7240 was constructed by cloning of the DNA fragment encoding human GM-CSF flanked by AfeI (5′) and FseI (3′) into pGLY5221 at blunted StuI (5′) and FseI (3′). pGLY5221 is an expression vector, where the expression of human GM-CSF was under the control of the PpAOX1 promoter. Transformants were selected using the Zeocin resistance gene (Streptoalloteichus hindustanus ble).
Integration/expression plasmid pGLY6301 or pGLY6294 were linearized with SpeI or SfiI, respectively, and transformed into P. pastoris strain yGLY13992 producing anti-HER2 (Jiang et al. 2011, Zhang et al. 2011) or yGLY14401 producing anti-RSV (Sethuraman et al. 2011). The genomic integration of pGLY6301 at the URA6 locus was confirmed by colony PCR (cPCR) using the primers, PpURA6out/UP (5′-CTGAGGAGTCAGATATCAGCTCAATCTC CAT-3′) and pUC19/LP (5′-TCCGGCTCGTATGTTGTGTGGAATTGT-3′) or ScARR3/UP (5′-GGCAATAGTCGCGAGAATCCTTAAACCAT-3′) and PpURA6out/LP (5′-CTGGATGTTTGATGGGTTCAGTTTCAGCTGGA-3′). The genomic integration of pGLY6294 at the TRP1 locus was confirmed by cPCR using the primers, PpTRP-5′out/UP (5′-CCTCGTAAAGATCTGCGGTTTGCAAAGT-3′) and PpALG3TT/LP (5′-CCTCCCACTGGAACCGATGATATGGAA-3′) or PpTEFTT/UP (5′-GATGCGAAGTTAAGTGCGCAGAAAGTAATATCA-3′) and PpTRP1-3′out/LP (5′-CGTGTGTACCTTGAAACGTCAATGATACTTTGA-3′). The presence of LmSTT3D was confirmed by the primers, LmSTT3D/iUP (5′-GCGACTGGTTCCAATTGACAAGCTT-3′) and LmSTT3D/iLP (5′-CAACAGTAGAACCA GAAGCCTCGTAAGTACAG-3′). The PCR conditions were one cycle of 95 °C for 2 min, 35 cycles of 95 °C for 20 s, 55 °C for 20 s, and 72 °C for 1 min; followed by one cycle of 72 °C for 10 min. For the N-terminal myc-tagged LmSTT3D construct (pGLY10176), the myc tag (EQKLISEEDL) was fused to the N terminus of LmSTT3D, and it was cloned into pGLY8369 at EcoRI and FseI sites. pGLY10176 (SpeI digest) was transformed into yGLY13992 to generate yGLY24589.
Strain yGLY17351 was generated by transforming an expression cassette encoding L. major STT3D (pGLY6301) into YGLY13992, in which L. major STT3D (LmSTT3D) was targeted to the P. pastoris URA6 locus. Strain yGLY17368 was generated by transforming an expression cassette encoding LmSTT3D (pGLY6294) into yGLY13992. Strain yGLY17319 was generated by transforming an expression cassette encoding LmSTT3D (pGLY6301) into an anti-RSV producing strain (yGLY14401). Strain yGLY17354 was generated by transforming an expression cassette encoding LmSTT3D (pGLY6294) into yGLY14401.
For human GM-CSF expression, strain yGLY15660 was generated by transforming an expression cassette encoding human GM-CSF (pGLY7240) into yGLY12900, in which human GM-CSF was targeted to the P. pastoris TRP2 locus (PpTRP2) and was under the control of a PpAOX1 promoter. yGLY12900 is a glycoengineered strain producing complex N-glycans with terminal sialic acids (Hamilton et al. 2006). Strain yGLY16349 was generated by transforming an expression cassette encoding L. major STT3D (pGLY6301) into human GM-CSF producing strain (yGLY15660), in which L. major STT3D (LmSTT3D) was targeted to the P. pastoris URA6 locus. The expression of LmSTT3D was under the control of a PpAOX1 promoter.
Transformation
Pichia transformation was performed as described previously (Choi et al. 2003). Briefly, 100 μL of overnight grown cells (OD600 = 0.2 to 6.0) were transformed with 10 μL linearized DNA (5–10 μg) after 5 min incubation on ice. Electroporation was in a Bio-Rad GenePulser Xcell following the preset P. pastoris protocol (2 kV, 25 μF, 200 Ω), immediately followed by the addition of 1 mL YPDS recovery media (YPD media plus 1 M sorbitol). The transformed cells were allowed to recover for 4 h to overnight at room temperature (24 °C) before plating the cells on selective media.
Fermentation
Seed cultures were prepared by adding P. pastoris cells from YSD plates to each well of a Whatman 24 well Uniplate (10 mL, natural polypropylene) containing 3.5 mL of 4 % BMGY medium buffered to pH 6.0 with potassium phosphate buffer. The seed cultures were grown for approximately 65–72 h in a temperature controlled shaker at 24 °C and 650 rpm; 1.0 mL of the 24 well plate grown seed culture and 4.0 mL of 4 % BMGY medium were then used to inoculate each well of a Micro24 plate (Type: REG2). Thirty microliters of Antifoam 204 (1:25 dilution, Sigma-Aldrich, St. Louis, MO) was added to each well. The Micro24 was operated in Microaerobic 1 mode and the fermentations were controlled with 1 vvm constant air flow, pH at 6.5, temperature at 24 °C, and agitation at 800 rpm. The induction phase was initiated upon observance of a DO spike after the growth phase by adding bolus shots of methanol feed solution (100 % [w/w] methanol, 5 mg/L biotin, and 12.5 mL/L PTM2 salts). SixFors were carried out in 0.5 L vessels (Sixfors multi-fermentation system, ATR Biotech, Laurel, MD) as described earlier (Barnard et al. 2010). Three liters of autoclavable glass and 40 L stainless steel SIP bioreactors (Applikon, Foster City, CA) were performed using the protocols described previously (Potgieter et al. 2009). In the present study, martone was replaced with soytone.
N-glycan structure analysis
2-aminobenzamide (2-AB) labeling was used to quantify N-glycan structures as described previously (Choi et al. 2008).
Quadrupole time-of-flight mass spectrometer (Q-TOF) analysis
The high performance liquid chromatography (HPLC) system used in this study consisted of an Agilent 1200 equipped with autoinjector, a column-heating compartment, and a UV detector detecting at 210 and 280 nm. All LC-MS experiments performed with this system were running at 1 mL/min. The flow rate was not split for MS detection. Mass spectrometric analysis was carried out in positive ion mode on Accurate-Mass Q-TOF LC/MS 6520 (Agilent technologies, Santa Clara, CA). The temperature of dual ESI source was set at 350 °C. The nitrogen gas flow rates were set at 13 L/h for the cone and 350 L/h and nebulizer was set at 45 psig with 4,500 volts applied to the capillary. Reference mass of 922.009 was prepared from HP-0921 according to API-TOF reference mass solution kit for mass calibration and the protein mass measurements. The data for ion spectrum range from 300–3,000 m/z were acquired and processed using Agilent Masshunter.
Sample preparation was carried out as follows. Intact antibody or glycoprotein samples (50 μg) were prepared 50 μL 25 mM NH4HCO3, pH 7.8. Reduced antibody or glycoprotein was prepared by adding 1 M DTT to a final concentration of 10 mM to an aliquot of either intact antibody and incubated for 30 min at 37 °C.
Three micrograms of intact sample was loaded into a Poroshell 300SB-C3 column (2.1 × 75 mm, 5 μm; Agilent Technologies) maintained at 40 °C. The protein was first rinsed on the cartridge for 3 min with 90 solvent A (0.1 % HCOOH), 5 % solvent B (90 % Acetonitrile in 0.1 % HCOOH). Elution was then performed using a gradient of 5-80 % of B over 20 min followed by a 7-min regeneration at 80 % B and by a final equilibration period of 10 min at 5 % B.
N-glycan site occupancy analysis
The site occupancy of N-glycan on anti-HER2 or anti-RSV antibodies was determined using capillary electrophoresis (CE). The antibodies were recovered from the cell culture medium and purified by protein A column chromatography. The protein A purified sample (100–200 μg) was concentrated to about 100 μL and then buffer exchanged with 100 mM Tris–HCl pH 9.0 containing 1 % SDS. Then, the sample along with 2 μL of 10 kDa internal standard provided by Beckman was reduced by addition of 5 μL β-mercaptoethanol and boiled for 5 min. About 20 μL of reduced sample was then resolved over a bare-fused silica capillary (about 70 mm, 50 μm I.D.) according to the method recommended by Beckman Coulter.
Microchip CE-SDS (Caliper Life Science, Hopkinton, MA) was also used to determine N-glycan site occupancy. Antibody sample (5 μL) at approximately 1–2 mg/mL was added to 7 μL of sample buffer provided with HT Protein Express LabChip® Kit supplemented with 50 mM 2-mercaptoethanol (Sigma-Aldrich). The sample mixture was then incubated at 75 °C for 15 min. Prior to microchip analysis, deionized HPLC grade water (35 μL) was added to the sample mixture and added onto the instrument for size separation. The N-glycosylation occupancy was determined by percentage of the corrected peak areas corresponding to the glycosylated heavy chain (GHC). The ratio of heavy and light chains (H/L) was calculated from total corrected peak area of GHC and non-glycosylated heavy (NGHC) against that of light chain. The impurity was reported as the total corrected peak area of protein bands that do not belong to GHC, NGHC, or light chain.
Detection of rhGM-CSF
rhGM-CSF was separated by 4–20 % gradient SDS-PAGE and then electroblotted onto nitrocellulose membrane (Schleicher & Schuell Inc., Keene, NH). The membrane was probed with goat anti-human GM-CSF antibody (0.1–0.2 μg/mL; R&D Systems, Inc., Minneapolis, MN) and followed by rabbit anti-goat IgG antibody conjugated with horseradish peroxidase (1:3,000; Sigma-Aldrich). The results were visualized using an ImmunoPure Metal Enhanced DAB Substrate Kit (Thermo Fisher Scientific Inc., Rockford, IL).
Immunofluorescence staining
The immunofluorescence on fixed cells was preformed according to (Swayne et al. 2009). To visualize ER structures, a polyclonal antibody raised against a yeast ER protein, Kar2p, was used (gift of Dr. Elizabeth Miller, Columbia University). To detect Myc-tagged proteins, a monoclonal antibody (9E10) to the Myc epitope was used.
Microscopy and image processing
Images were acquired with an Axioscope 2 Plus microscope (Carl Zeiss, Oberkochen, Germany) by using a Plan-Apochromat 100 × 1.4 numerical aperture objective lens, and a cooled charge-coupled device camera, Orca ER (Hamamatsu, Bridgewater, NJ). Camera control and image collection were performed using OpenLab software (Improvision, Coventry, United Kingdom). z-sections of cells were obtained at 0.4 μm intervals through the entire cell. To remove out-of-focus light, images have been deconvolved using the software program Volocity (Improvision). For visualization, deconvolved images have been projected and rendered with Volocity software.
Results
P. pastoris OST complex
The oligosaccharyltransferase (OST) complex in yeast and higher eukaryotes consists of 8 or 9 subunits and catalyzes the transfer of Dol-PP-GlcNAc2-Man9-Glc3 to an asparagine residue (Asn-X-Ser/Thr) on nascent polypeptide chains. It has been reported that S. cerevisiae has 9 OST subunits (Ost1p, Ost2p, Ost3p, Ost4p, Ost5p, Ost6p, Stt3p, Swp1p, and Wbp1p), where the OST complex is localized at two different SEC61 translocons via Ost3p or Ost6p (Yan and Lennarz 2005). In P. pastoris, we identified all subunits of the OST complex by protein homology search (Table 1), and they all contain predicted transmembrane helices. The highly conserved catalytic subunit, P. pastoris STT3 protein (PpStt3p), shares high homology (65 % identity) with S. cerevisiae STT3 protein (ScStt3p), and contains 13 predicted transmembrane domains (Fig. 1a). It has been reported that Stt3p forms a subcomplex with Ost3p and Ost4p (Karaoglu et al. 1997) and is directly involved in recognition of the target peptide (Nilsson et al. 2003). In addition, the highly conserved domain (WWDYG) is located in the C-terminus of PpStt3p as shown in the protein sequence alignment (Fig. 2). Recently, additional catalytic motifs (DXXK and EXD) on the Stt3p were proposed to form the catalytic center (Liu and Mushegian 2003; Igura et al. 2008; Maita et al. 2010); they were also identified in the PpStt3p (Fig. 2). Yan and Lennarz (2002) have reported that the conserved domain (WWDYG) plays a key role in the recognition of N-glycosylation sites and the glycosylation process.

Predicted transmembrane topology of P. pastoris Stt3p and L. major Stt3Dp. The transmembrane domains were predicted using the TMHMM program. a P. pastoris STT3p: 13 transmenbrane helices. b L. major Stt3Dp: 11 transmenbrane helices. Red rectangle indicates a transmembrane helix

Sequence alignment of Stt3p from S. cerivisiae, P. pastoris, and L. major. The protein alignment was carried out with a multiple sequence alignment program, ClustalW (Chenna et al. 2003). Shaded letters represent identical amino acid residues, and the asterisk indicates the highly conserved motifs, WWDYG, DXXK, and EXD
Subcellular localization of L. major STT3 isoforms
The catalytic subunit (STT3) is an essential protein, and plays a key role in the N-glycosylation of proteins. PpStt3p and LmStt3Dp share 27.8 % sequence identity, and their transmembrane protein topology was predicted using the TMHMM program (Krogh et al. 2001). PpStt3p (Fig. 1a) and LmStt3Dp (Fig. 1b) contain 13 and 11 predicted transmembrane domains, respectively. Both STT3 proteins have the N terminus in the cytosol and the C terminus in the ER lumen.
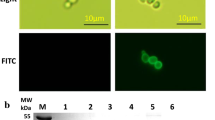
In order to confirm localization of LmSTT3 isoforms (LmSTT3A, B, C, and D) in P. pastoris, the N-terminal myc tag was fused to the coding regions of LmSTT3 isoforms, and their expression was induced in the presence of methanol using the P. pastoris AOX1 promoter. After inducing recombinant protein expression for 1 day, the cells were fixed with paraformaldehyde, LmSTT3 isoforms were stained with anti-myc antibody (red), and the ER was visualized with anti-Kar2p antibody (green). As shown in Fig. 3, LmStt3Dp is mostly localized in nuclear endoplasmic reticulum (nER) rather than cortical ER (cER). This confirms that the recombinant LmStt3Dp is correctly localized in the ER in P. pastoris (see supplementary Fig. S1 for three orthogonal projections). We also confirmed that other L. major STT3 isoforms (LmSTT3A, B, and C) were correctly localized in the ER (data not shown).

Subcellular localization of LmStt3Dp in P. pastoris. a Picha ER was labeled with anti-Kar2p antibody (green). b The N-terminal myc tagged-LmStt3Dp was stained with anti-myc antibody (red). c The nucleus was labeled with DAPI (blue). d Overlay. nER nuclear ER, cER cortical ER, N nucleus
Effect of LmSTT3 isoforms on the N-glycan site occupancy of recombinant antibody
There have been extensive studies aimed toward improving N-glycan site occupancy of therapeutic glycoproteins produced in yeast because N-glycan site occupancy can vary with the N-glycosylation sites not being fully occupied in many cases (Horwitz et al. 1988; Ward et al. 2004; Li et al. 2006; Choi et al. 2008). The N-glycan site occupancy of antibodies produced in P. pastoris is typically in the range of 75 %-85 % depending on process conditions, whereas N-glycan site occupancy of mammalian-produced antibodies is greater than 99 %. In order to test LmSTT3 isoforms on N-glycan site occupancy, the native LmSTT3 isoforms were individually transformed into an anti-HER2 expressing strain (YGLY13992). The expression of the LmSTT3 isoforms was co-induced with a test antibody (anti-HER2) using methanol as a carbon source. Initial analyses were performed using a miniaturized bioreactor, Micro24. Secreted recombinant antibodies were purified using a Protein A column and then subjected for N-glycan site occupancy analysis. The occupancy of N-glycosylation sites on anti-HER2 antibody was determined using capillary electrophoresis (CE) or Microchip CE-SDS (Caliper). As shown in Table 2, LmSTT3C demonstrated that N-glycan site occupancy was slightly lower than that of LmSTT3A and B, but it was still in the range of 75–85 % that was observed in wild-type P. pastoris. Although LmSTT3 isoforms (A, B, and C) showed little or no improvement on N-glycan site occupancy of our test antibody (76.5 ± 1.3 %-80.8 ± 4.1 %), significant improvement was observed with LmSTT3D (>99 %). Statistical analysis (t test) showed no significant differences (p > 0.05) in N-glycan site occupancy of the antibody from LmSTT3 isoforms (A, B, and C) and the control (no LmSTT3 isoforms) except for LmSTT3D.
The improvement of N-glycan site occupancy of the antibody was further confirmed using quadrupole time-of-flight mass spectrometer (Q-TOF) analysis. Figure 4 shows a comparison of N-glycan site occupancy of the anti-HER2 antibody produced in YGLY17351 to that of a commercially available anti-HER2 antibody produced in CHO cells (Herceptin). Anti-HER2 antibody produced in YGLY17351 showed N-glycan site occupancy similar to that of an anti-HER2 antibody produced in CHO cells (>99 %), whereas the antibody from the control strain (no LmSTT3D) showed that 85 % of the antibody contained two N-glycans (fully occupied 2N-glycosylation sites), and the rest (15 %) has a single N-glycan, where 1N-glycosylation site was occupied. Unglycosylated antibodies were not detected.

Q-TOF analysis of anti-HER produced in P. pastoris and Chinese hamster ovary (CHO). The N-glycan site occupancy of anti-HER2 was confirmed using Q-TOF. a Commercial Herceptin (CHO produced anti-HER2). b Anti-HER2 produced in P. pastoris in the absence of LmSTT3D. c Anti-HER2 produced in P. pastoris in the presence of LmSTT3D under the control of the AOX1 promoter. 1N-linked: anti-HER2 occupied with 1N-glycosylation site; 2N-linked: anti-HER2 occupied with 2N-glycosylation sites. Multiple peaks within 1N-linked or 2N-linked indicate different N-glycoforms
Interestingly, the N-terminal myc-tagged LmStt3Dp is fully functional, but the C-terminal myc tag seems to inhibit its activity according to the N-glycan site occupancy data (Table 2). That is, the N-glycan site occupancy of an anti-HER2 was greater than 99 % in the strain carrying the N-terminal myc-tagged LmStt3Dp, whereas no N-glycan site occupancy improvement was observed with the C-terminal myc-tagged LmStt3Dp compared to the control strain without LmStt3Dp overexpression. This suggests that the C-terminal region of LmStt3Dp is functionally involved, although the C-terminal tag did not abolish its activity as shown in a study by Nasab et al. (2008)), where the C-terminal HA-tagged LmStt3Dp complemented a deletion of yeast STT3. However, we did not observe any gain-of-function with the C-terminal myc-tagged LmStt3Dp.
Inducible AOX1 versus constitutive GAP promoters on N-glycan site occupancy
Two different promoters, P. pastoris inducible AOX1 and constitutive GAP promoters, were evaluated for N-glycan site occupancy of two different antibodies (anti-HER2 and anti-RSV). As shown in Table 3, even though N-glycan site occupancy of both antibodies was improved with expression of LmStt3Dp under the GAP promoter (94 %), higher N-glycan site occupancy (99 %) was obtained under the AOX1 promoter. Interestingly, this high occupancy was observed when LmStt3Dp and antibodies were co-induced. The GAP promoter is known to be less active in the presence of methanol (Waterham et al. 1997). This suggests that the LmStt3Dp expression may not be sufficient under the GAP promoter when test antibodies were induced with methanol as a sole carbon source.
N-glycan profiles
Since the quality of N-glycans is critical in glycoengineered P. pastoris, the N-glycan composition of the anti-HER2 (YGLY17351) and anti-RSV (YGLY17319) antibodies produced in strains that overexpress LmStt3Dp, compared to strains that do not overexpress LmSTT3D (YGLY13992 and YGLY14401), was evaluated. As shown in Table 4, the quality of N-glycans derived from antibodies produced in LmSTT3D overexpressing strains was comparable to that of strains that did not overexpress LmSTT3D. Overall, overexpression of LmStt3Dp did not appear to significantly affect the N-glycan composition of the antibodies. The complex N-glycans of antibodies produced in the glycoengineered P. pastoris strains, which have been genetically engineered to make galactose-terminated N-glycans, ranges from 80.8 % to 82.8 % with the LmStt3Dp overexpression, and from 79.8 % to 81.6 % without the LmStt3Dp overepression.
Scalability of N-glycan site occupancy in various bioreactors
In addition to the OST complex and protein substrate in yeast, N-glycan site occupancy of glycoproteins is affected by bioprocess conditions and the scale or size of the bioreactors used for cultivation. Consistent and robust N-glycan site occupancy is desirable for the production of therapeutic glycoproteins in order to maintain desired product profiles. As such, the scalability of N-glycan site occupancy would be critical for maintaining both protein quality and biological activity. Thus, we tested the anti-HER2 expressing strain carrying LmSTT3D (YGLY17351) in different bioreactor scales ranging from 5 mL through 40 L. Table 5 demonstrates the scalability of N-glycan site occupancy on a recombinant immunoglobulin G1 (IgG1) monoclonal antibody produced in P. pastoris overexpressing LmSTT3D. As mentioned earlier, it has been observed that N-glycan site occupancy of glycoproteins produced in glycoengineered P. pastoris varies with the process conditions. However, the LmSTT3D overexpressing strains showed very consistent N-glycan site occupancy of antibody (>99 %) regardless of the scale of bioreactors and process conditions that were tested. Thus, the expression of LmSTT3D enables the production of recombinant antibodies in P. pastoris, in which the N-glycan site occupancy of the recombinant antibodies is comparable to that observed in mammalian expression systems, such as CHO cells. The anti-HER2 produced in the glycoengineered strain was extensively evaluated in vitro and in vivo (preclinical study), and its biological activity was comparable to Herceptin (Jiang et al 2011; Zhang et al 2011).
Improvement of N-glycan site occupancy of non-antibody glycoprotein
In this study, it has been demonstrated that LmSTT3D can improve N-glycan site occupancy of antibodies greater than 99 %. In order to confirm that the improvement of N-glycan site occupancy is reproducible on a non-antibody glycoprotein, recombinant human granulocyte macrophage colony-stimulating factor (rhGM-CSF) was evaluated for N-glycan site occupancy by overexpressing LmStt3Dp in the presence of intact Pichia OST complex. rhGM-CSF contains 2N-glycosylation sites (Asn27-Leu-Ser, Ans37-Glu-Thr), which are not fully glycosylated in wild-type P. pastoris. As demonstrated on Q-TOF and Western blot analysis (Fig. 5), the predominant rhGM-CSF produced in wild-type P. pastoris contains a single N-glycan, while the rest is unglycosylated or has two N-glycans (Fig.5b, d; lane 1). However, the LmStt3Dp overexpressing strain was able to produce rhGM-CSF, most of which contained two N-glycans (Fig. 5c and d, lane 2). Commercial rhGM-CSF (Leukine) produced in S. cerevisiae contains predominantly unglycosylated protein (Fig. 5a). Again, the enhanced N-glycan site occupancy of rhGM-CSF supports that LmStt3Dp has broad substrate specificity.

Q-TOF analysis of rhGM-CSF produced in glycoengineered P. pastoris with or without the LmSTT3D overexpression. rhGM-CSF was purified using CaptoMMC column, and then non-reduced rhGM-CSF was analyzed using Q-TOF. a Commercial rhGM-CSF (Leukine). b rhGM-CSF produced in P. pastoris in the absence of the LmStt3Dp expression. c rhGM-CSF produced in P. pastoris overexpressing the LmSTT3D under the control of the AOX1 promoter. Molecular mass of rhGM-CSF is as follows; unglycosylated: 14430–14432; 1N-linked: 16636; 2N-linked: 18842. Other minor peaks are degraded rhGM-CSF. d Western blot analysis of rhGM-CSF produced in P. pastoris. rhGM-CSF is labeled with anti-human GM-CSF antibody. Lane 1 rhGM-CSF produced without LmSTT3D overexpression. Lane 2 rhGM-CSF produced with LmSTT3D overexpression. 0N unglycosylated, 1N 1N-linked glycan, 2N 2N-linked glycans
Discussion
There has been great interest in modulating the N-glycan site occupancy in yeast, triggering numerous efforts to improve N-glycan site occupancy comparable to one in mammalian cells (Jones et al. 2005; Betenbaugh et al. 2006; Schulz et al. 2009). Here, we show that LmSTT3D is active and its overexpression in Pichia with a functional native OST complex further increases N-glycan site occupancy of antibodies greater than 99 % and of a non-antibody glycoprotein as well.
The correct subcellular targeting of the heterologous membrane proteins has been challenged in P. pastoris to acquire optimal enzymatic activity, which has led to extensive studies on the optimization of the targeting domains to the ER and Golgi (choi et al. 2003; Hamilton et al. 2003; Hamilton et al. 2006). However, the subcellular localization study confirmed that the native LmStt3Dp were correctly localized in the ER membrane. In addition, further improvement of N-glycan site occupancy supports that LmStt3Dp is enzymatically active and functional. This also indicates that localization of LmStt3Dp is associated with the SEC61 translocon, where protein is translocated into the lumen of the ER and N-glycosylation occurs co-translationally by the OST complex. The OST complex is localized at the SEC61 translocons via Ost3p or Ost6p in yeast (Yan and Lennarz 2005). However, it is not clear how LmStt3Dp interacts with the SEC61 translocons in P. pastoris because the majority of LmStt3Dp was not associated with the OST complex in S. cerevisiae and functions as a single protein (Nasab et al. 2008; Hese et al. 2009). In the light of the function of the OST and SEC61 translocon, it can be speculated that LmStt3Dp contains the domain that mediates the interaction with SEC61 translocon.
In our study of heterologous STT3 overexpression, in which individual STT3 proteins were expressed under the control of the AOX1 promoter in glycoengineered P. pastoris producing a recombinant IgG1, the STT3 subunits from human, mouse, yeast, Caenorhabditis elegans, and protozoa (Trypanosoma cruzi, Trypanosoma brucei, Toxoplasma gondii, and three L. major paralogues (A, B, and C)) did not show any further improvement on N-glycan site occupancy of an anti-HER2 in the presence of an endogenous P. pastoris Stt3p. Only LmSTT3D demonstrated a gain-of-function. This finding is possibly explained by protein substrate preference studies of individual LmSTT3 paralogues in S. cerevisiae (Nasab et al. 2008), in which LmSTT3D demonstrated broad peptide substrate specificity and better enzyme activity among other LmSTT3 paralogues, although more detailed analysis will define the gain-of-function. This also supports that LmSTT3D is a good substitute for yeast STT3.
Production scales and process conditions often affect N-glycan site occupancy of proteins of interest as observed in P. pastoris (75–85 %). Our scale-up study indicates that the function of LmStt3Dp is very consistent and reproducible and is not affected by different process conditions and bioreactor scales as demonstrated in the production scales (5 mL–40 L). This genetic approach offers a significant benefit in maintaining protein quality over various process conditions. In addition, N-glycan profiles are consistent between strains with or without LmStt3Dp overexpression, suggesting that LmStt3Dp did not negatively influence N-glycan processing. This is highly advantageous in developing industrial scales of protein production.
This successful demonstration of fully occupied N-glycan site occupancy of antibodies, by the LmStt3Dp overexpression, will further mature Pichia expression platform suitable for the production of therapeutic antibodies or other glycoproteins that require consistent product quality. In particular, our study strongly indicates that all therapeutic antibodies produced in the LmSTT3D-engineered P. pastoris would have high N-glycan site occupancy (>99 %), equivalent to that of CHO cells, because the Fc regions of IgGs bear a highly conserved and identical N-glycosylation site (Asn297-Ser-Thr) located on a CH2 domain.
N-glycan site occupancy of therapeutic glycoproteins plays a critical role in the pharmacokinetics (PK) and pharmacodynamics (PD) of biological molecules when N-glycans are biologically important. Thus, it is desirable to maintain the highest N-glycan site occupancy of glycoproteins for maximal efficacy. In addition, N-glycan site occupancy frequently influences glycoprotein folding if the N-glycosylation sites are normally occupied and involved in protein folding. Therefore, the expression of a single oligosaccharyltransferase, LmSTT3D, is an important application to increase the N-glycan site occupancy of glycoproteins. Furthermore, as demonstrated by the scalability study, this genetic basis for improving N-glycan site occupancy is very robust, thus providing a benefit to glycoprotein production at industrial scales.
References
Barnard GC, Kull AR, Sharkey NS, Shaikh SS, Rittenhour AM, Burnina I, Jiang Y, Li F, Lynaugh H, Mitchell T, Nett JH, Nylen A, Potgieter TI, Prinz B, Rios SE, Zha D, Sethuraman N, Stadheim TA, Bobrowicz P (2010) High-throughput screening and selection of yeast cell lines expressing monoclonal antibodies. J Ind Microbiol Biotechnol 37:961–971
Ben-Dor S, Esterman N, Rubin E, Sharon N (2004) Biases and complex patterns in the residues flanking protein N-glycosylation sites. Glycobiology 14:95–101
Betenbaugh MJ, Viswanathan K, Krag SS, Jones JG (2006) Improving protein N-glycosylation of eukaryotic cells using dolichol-linked oligosaccharide synthesis pathway, other N-glycosylation-increasing methods, and engineered hosts expressing products with increased N-glycosylation. International Published Application No. WO 2006107990
Cereghino LJ, Cregg JM (2000) Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 24:45–66
Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497–3500
Choi BK, Jiménez-Flores R (1996) The study of putative glycosylation sites in bovine β-casein introduced by PCR-based site-directed mutagenesis. J Agric Food Chem 44:358–364
Choi BK, Bobrowicz P, Davidson RC, Hamilton SR, Kung DH, Li H, Miele RG, Nett JH, Wildt S, Gerngross TU (2003) Use of combinatorial genetic libraries to humanize N-linked glycosylation in the yeast Pichia pastoris. Proc Natl Acad Sci USA 100:5022–5027
Choi BK, Actor JK, Rios S, d’Anjou M, Stadheim TA, Warburton S, Giaccone E, Cukan M, Li H, Kull A, Sharkey N, Gollnick P, Kocieba M, Artym J, Zimecki M, Kruzel ML, Wildt S (2008) Recombinant human lactoferrin expressed in glycoengineered Pichia pastoris: effect of terminal N-acetylneuraminic acid on in vitro secondary humoral immune response. Glycoconj J 25:581–593
Gawlitzek M, Estacio M, Fürch T, Kiss R (2009) Identification of cell culture conditions to control N-glycosylation site-occupancy of recombinant glycoproteins expressed in CHO cells. Biotechnol Bioeng 103:1164–1175
Goetze AM, Liu YD, Zhang Z, Shah B, Lee E, Bondarenko PV, Flynn GC (2011) High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 21:949–959
Hamilton SR, Gerngross TU (2007) Glycosylation engineering in yeast: the advent of fully humanized yeast. Curr Opin Biotechnol 18:387–392
Hamilton SR, Bobrowicz P, Bobrowicz B, Davidson RC, Li H, Mitchell T, Nett JH, Rausch S, Stadheim TA, Wischnewski H, Wildt S, Gerngross TU (2003) Production of complex human glycoproteins in yeast. Science 301:1244–1246
Hamilton SR, Davidson RC, Sethuraman N, Nett JH, Jiang Y, Rios S, Bobrowicz P, Stadheim TA, Li H, Choi BK, Hopkins D, Wischnewski H, Roser J, Mitchell T, Strawbridge RR, Hoopes J, Wildt S, Gerngross TU (2006) Humanization of yeast to produce complex terminally sialylated glycoproteins. Science 313:1441–1443
Hese K, Otto C, Routier FH, Lehle L (2009) LmSTT3 parologs complemented Sc OST subunits. Glycobiology 19:160–171
Horwitz AH, Chang P, Better M, Hellstrom KE, Robinson RR (1988) Secretion of functional antibody and Fab fragment from yeast cells. Proc Natl Acad Sci USA 85:8678–8682
Igura M, Maita N, Kamishikiryo J, Yamada M, Obita T, Maenaka K, Kohda D (2008) Structure-guided identification of a new catalytic motif of oligosaccharyltransferase. EMBO J 27:234–243
Jacobs PP, Geysens S, Vervecken W, Contreras R, Callewaert N (2009) Engineering complex-type N-glycosylation in Pichia pastoris using GlycoSwitch technology. Nat Protoc 4:58–70
Jiang Y, Li F, Zha D, Potgieter TI, Mitchell T, Moore R, Cukan M, Houston-Cummings NR, Nylen A, Drummond JE, McKelvey TW, d’Anjou M, Stadheim TA, Sethuraman N, Li H (2011) Purification process development of a recombinant monoclonal antibody expressed in glycoengineered Pichia pastoris. Protein Expr Purif 76:7–14
Jones J, Krag SS, Betenbaugh MJ (2005) Controlling N-linked glycan site occupancy. Biochim Biophys Acta 1726:121–137
Kaplan HA, Welply JK, Lennarz WJ (1987) Oligosaccharyltransferase: the central enzyme in the pathway of glycoprotein assembly. Biochim Biophys Acta 906:161–173
Karaoglu D, Kelleher DJ, Gilmore R (1997) The highly conserved Stt3 protein is a subunit of the yeast oligosaccharyltransferase and forms a suncomplex with Ost3p and Ost4p. J Biol Chem 272:32513–32520
Kasturi L, Eshleman JR, Wunner WH, Shakin-Eshleman SH (1995) The hydroxy amino acid in an Asn-X-Ser/Thr sequon can influence N-linked core glycosylation efficiency and the level of expression of a cell surface glycoprotein. J Biol Chem 270:14756–14761
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580
Li H, Sethuraman N, Stadheim TA, Zha D, Prinz B, Ballew N, Bobrowicz P, Choi BK, Cook WJ, Cukan M, Houston-Cummings NR, Davidson R, Gong B, Hamilton SR, Hoopes JP, Jiang Y, Kim N, Mansfield R, Nett JH, Rios S, Strawbridge R, Wildt S, Gerngross TU (2006) Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat Biotechnol 24:210–215
Liu J, Mushegian A (2003) Three monophyletic superfamilies account for the majority of the known glycosyltransferases. Protein Sci 12:1418–1431
Maita N, Nyirenda J, Igura M, Kamishkiryo J, Kohda D (2010) Comparative structural biology of eubacterial and archaeal oligosaccharyltransferases. J Biol Chem 285:4941–4950
Mellquist JL, Kasturi L, Spitalnik SL, Shakin-Eshleman SH (1998) The amino acid following an Asn-X-Ser/Thr Sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 37:6833–6837
Nasab FP, Schulz BL, Gamarro F, Parodi AJ, Aebi M (2008) All in one: leishmania major STT3 proteins substitute for the whole oligosaccharyltransferase complex in Saccharomyces cerevisiae. Mol Biol Cell 19:3758–3768
Nett JH, Gomathinayagam S, Hamilton SR, Gong B, Davidson RC, Du M, Hopkins D, Mitchell T, Mallem MR, Nylen A, Shaikh SS, Sharkey N, Barnard GC, Copeland V, Liu L, Evers R, Li Y, Gray PM, Lingham RB, Visco D, Forrest G, DeMartino J, Linden T, Potgieter TI, Wildt S, Stadheim TA, d’Anjou M, Li H, Sethuraman N (2012) Optimization of erythropoietin production with controlled glycosylation-PEGylated erythropoietin produced in glycoengineered Pichia pastoris. J Biotechnol 157:198–206
Nilsson I, Kelleher DJ, Miao Y, Shao Y, Kreibich G, Gilmore R, von Heijne G, Johnson AE (2003) Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J Cell Biol 161:715–725
Potgieter TI, Cukan M, Drummond JE, Houston-Cummings NR, Jiang Y, Li F, Lynaugh H, Mallem M, McKelvey TW, Mitchell T, Nylen A, Rittenhour A, Stadheim TA, Zha D, d’Anjou M (2009) Production of monoclonal antibodies by glycoengineered Pichia pastoris. J Biotechnol 139:318–325
Schulz BL, Stirnimann CU, Grimshaw JPA, Brozzo MS, Fritsch F, Mohorko E, Capitani G, Glockshuber R, Grütter MG, Aebi M (2009) Oxidoreductase activity of oligosaccharyltransferase subunits Ost3p and Ost6p defines site-specific glycosylation efficiency. Proc Natl Acad Sci USA 106:11061–11066
Sethuraman N, Choi BK, Prinz B, Meehl M, Stadheim TA (2011) Methods for increasing N-glycosylation site occupancy on therapeutic glycoproteins produced in Pichia pastoris. Patent application number US2011/025879
Sinclair AM, Elliott S (2005) Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J Pharm Sci 94:1626–1635
Swayne TC, Boldogh IR, Pon LA (2009) Imaging of the cytoskeleton and mitochondria in fixed budding yeast cells. Method Mol Biol 586:171–184
Ward M, Lin C, Victoria DC, Fox BP, Fox JA, Wong DL, Meerman HJ, Pucci JP, Fong RB, Heng MH, Tsurushita N, Gieswein C, Park M, Wang H (2004) Characterization of humanized antibodies secreted by Aspergillus niger. Appl Environ Microbiol 70:2567–2576
Waterham HR, Digan ME, Koutz PJ, Lair SV, Cregg JM (1997) Isolation of the Pichia pastoris glyceraldehyde-3-phosphate dehydrogenase gene and regulation and use of its promoter. Gene 186:37–44
Yan A, Lennarz WJ (2002) Studies on the function of oligosaccharyl transferase subunits. Stt3p is directly involved in the glycosylation process. J Biol Chem 277:47692–47700
Yan A, Lennarz WJ (2005) Two oligosaccharyl transferase complexes exist in yeast and associate with two different translocons. Glycobiology 15:1407–1415
Zhang N, Liu L, Dumitru CD, Houston-Cummings NR, Cukan M, Jiang Y, Li Y, Li F, Mitchell T, Mallem MR, Ou Y, Patel RN, Vo K, Wang H, Burnina I, Choi BK, Huber H, Stadheim TA, Zha D (2011) Glycoengineered Pichia produced anti-HER2 is comparable to trastuzumab in preclinical study. Mabs 3:289–298
Acknowledgments
The authors would like to thank Nathan Sharkey and Seemab Shaikh for DasGip and Micro24 runs, Nga Rewa Houston-Cummings and Dongxing Zha for the strain YGLY13992, Bianka Prinz for the strain YGLY14401, Daniel Hopkins and Stephan Hamilton for the strain YGLY12900, Alissa Rittenhour for analytical support, and Erin Giaccone, Sujatha Gomathinayagam, and Sandra Rios for protein purification.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 67 kb)
Rights and permissions
About this article
Cite this article
Choi, BK., Warburton, S., Lin, H. et al. Improvement of N-glycan site occupancy of therapeutic glycoproteins produced in Pichia pastoris . Appl Microbiol Biotechnol 95, 671–682 (2012). https://doi.org/10.1007/s00253-012-4067-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4067-3