
Abstract
Yeast host–vector systems have been very successful in expressing recombinant proteins. However, because there are some proteins that cannot be expressed with existing systems, there is a need for new yeast expression systems. Here we describe a new host–vector system based on the basidiomycetous yeast Cryptococcus sp. strain S-2 (S-2). Two advantages of S-2 are that it naturally produces some very useful enzymes, so it would be a good system for expressing multiple copies of some of its genes, and that, it is a nonhazardous species. The orotate phosphoribosyltransferase (OPRTase, EC 2.4.2.10) gene (URA5) was selected as a selectable marker for transformation in the new host–vector system. URA5 was isolated and introduced into a uracil auxotroph of S-2 by electroporation. To demonstrate the S-2 system, we selected one of its unique enzymes, a plastic-degrading cutinase-like enzyme (CLE). We were able to insert multiple copies of the CLE gene (CLE1) into the chromosomes in a high fraction of the targeted cells. Under optimal conditions, one transformant exhibited 3.5 times higher CLE activity than the wild type. Expression vectors, including an inducible promoter (the promoter for the xylanase or α-amylase gene), were constructed for recombinant protein production, and green fluorescent protein was expressed under the control of these promoters. The xylanase promoter was more tightly controlled. Furthermore, putting CLE1 under the control of the xylanase promoter, which is induced by xylose, increased CLE activity of the culture medium to approximately 15 times greater than that of the wild type.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Yeast host–vector systems are widely used to produce recombinant proteins. Some of the best known systems are based on Schizosaccharomyces pombe (Beach and Nurse 1981), Pichia pastoris (Cregg et al. 1985), Kluyveromyces lactis (Das and Hollenberg 1982), Saccharomyces cerevisiae (Hinnen et al. 1978), and Candida maltosa (Takagi et al. 1986). Such systems can be used for the production of recombinant heterologous enzymes or to enhance the ability of yeast to produce homologous enzymes. For the production of recombinant proteins, most yeast host–vector systems, like the ones mentioned above, use ascomycetous yeasts (Böer et al. 2007; Buckholz and Gleeson 1991; Domînguez et al. 1998). However, 1,312 yeast species, including 464 basidiomycetous yeast species, have been identified so far (Kurtzman et al. 2011), so there may be many other yeast expression systems that are better than the present ones. Basidiomycetous yeasts tend to have higher GC contents than ascomycetous yeasts. Thus, they also tend to have different codon usage than ascomycetous yeasts and might be better suited for expressing some recombinant proteins. Two species of basidiomycetous yeasts, Pseudozyma flocculosa and Pseudozyma antarctica, have recently been used to construct host–vector systems for heterologous gene expression using either heterologous promoters (heat shock protein 70, glyceraldehyde-3-phosphate dehydrogenase promoters from Ustilago maydis and α-glucosidase promoter from Pseudozyma tsukubaensis) or a homologous promoter (actin promoter) (Avis et al. 2005, 2008; Cheng et al. 2008; Neveu et al. 2007).
Although many recombinant protein production systems have been developed, none are universally applicable. Furthermore, synthesis of several enzymes by existing protein expression systems is difficult. Since the efficiency of protein production can vary considerably depending on the host or production system, different approaches are used for optimization. Modification of the host strain is one such strategy (Gasser et al. 2007; Valkonen et al. 2003). Another strategy is to construct a new host–vector system in a new microorganism for recombinant protein production. Mass production of enzymes has many uses in industry and applied microbiology. Therefore, the development of new systems for recombinant protein production should accelerate the application of enzymatic technology.
The basidiomycetous yeast Cryptococcus sp. strain S-2 (S-2) was isolated from the air in Japan and stored in our laboratory (Iefuji et al. 1994). S-2 produces certain unique extracellular enzymes, for example, acid cellulase (Thongekkaew et al. 2008), acid xylanase (Iefuji et al. 1996a), raw-starch-degrading α-amylase (Iefuji et al. 1996b), and plastic-degrading cutinase-like enzyme (CLE) (Kawai et al. 2011; Kodama et al. 2009; Masaki et al. 2005). Each of these enzymes was expressed and secreted into culture media under their respective optimum conditions. CLE has attracted much interest because of its potential use in industry and environmental preservation, e.g., in biodiesel production (Kamini and Iefuji 2001), as an additive in laundry detergent (Thirunavukarasu et al. 2008), and in plastic degradation (Kawai et al. 2011; Masaki et al. 2005). Because of the usefulness of CLE, we wished to genetically modify S-2 to produce more CLE in this study.
Safe handling of host strain to produce enzymes is also important in industrial use. S-2 appears to be a safe strain to work with because it cannot grow at 37°C (Iefuji et al. 1994) and feeding to mice produced no ill effects (H. Yamaguchi and K. Uchida, personal communication). Currently, 70 species in the genus of Cryptococcus have been described (Fonseca et al. 2011). The ITS1 and ITS2 sequences of S-2 (119 and 172 bp) are 100% and 97%, identical, respectively, to those of Cryptococcus flavus. On the other hand, the two species have different GC content (67% and 55%) (Iefuji et al. 1994). In a phylogenetic analysis of 33 species of Cryptococcus and related genera (Takashima and Nakase 1999), C. flavus was obviously different from Cryptococcus neoformans and Cryptococcus gattii that are known as pathogenetic yeasts. S-2 is thought to be closely related to C. flavus based on their physiological, biochemical, and chemotaxonomic properties (Iefuji et al. 1994) as well as their ITS sequences. In this paper, we report the construction of a new host–vector system that uses the basidiomycetous yeast S-2 and one of its powerful promoters for recombinant protein expression.
Materials and methods
Strains, vectors, and media
The wild-type Cryptococcus sp. strain S-2 (S-2) is deposited in the International Patent Organism Depositary (accession number: FERM ABP-10961). S-2 was cultured at 25 °C in liquid or solid (2.0% agar) yeast malt medium (YM) containing 1.0% glucose, 0.5% peptone, 0.3% yeast extract, and 0.3% malt extract, unless otherwise stated. Uracil-free synthetic complete media, SC(−ura), contains 0.67% yeast nitrogen base without amino acids (Difco Laboratories, Detroit, USA), 2% dextrose, and 0.077% dropout supplements without uracil (Clontech, Mountain View, USA). Escherichia coli K12 strain DH5α (Nippon gene, Tokyo, Japan) was used as the host for cloning and plasmid amplification. pUC19 was used for construction of the yeast transformation system. The vector pGEM-T Easy (Promega, Madison, USA) was used for subcloning and nucleotide sequencing. The green fluorescent protein (GFP) gene was amplified from pEGFP (Clontech, Mountain View, USA).
Isolation of the uracil auxotroph and sequencing of the orotate phosphoribosyltransferase (OPRTase) gene (URA5) of S-2
To prepare host strains for a transformation system for this yeast, uracil auxotrophs were obtained by using 5-fluoro-orotic acid (5-FOA) (Boeke et al. 1984). Spontaneous 5-FOA-resistant mutants were isolated from the medium containing 5-FOA. The isolated mutant U-5 was inoculated in minimum medium containing orotic acid or orotidine. To sequence the orotate phosphoribosyltransferase (OPRTase) gene (URA5) by PCR, four degenerate primers were designed from highly conserved regions of 10 OPRTase genes from the following microbes: C. neoformans (CAD88274); S. cerevisiae (P13298); P. pastoris (AAQ72470); S. pombe (O94331); K. lactis (CAA04694); Yarrowia lipolytica (CAA80294); E. coli (AAG58786); Azotobacter vinelandii (COG0461); Pseudomonas aeruginosa (AAC44427); and Vibrio cholerae (AAF93387). (Protein IDs from DDBJ/EMBL/Swiss-Prot follow microbe names.) Two sense degenerate primers, 5′-CCHTAYTTCTTYAAYKYBGG-3′ (primer-1F) and 5′-TTYGGYCCBGCYTAYAARGG-3′ (primer-2F), and two antisense degenerate primers, 5′-CCDGMRGTSAKVACRTCRTC-3′ (primer-1R) and 5′-AKRBTDCCDCCYTCRCCRTG-3′ (primer-2R), were used to derive a part of URA5 of S-2. PCR was performed using these primers, ExTaq (Takara Bio, Shiga, Japan) and genomic DNA from S-2. An approximately 400-bp fragment amplified by primer-1F and primer-2R was purified using QIAquick Gel extraction kit (Qiagen, Hilden, Germany), and the sequence was analyzed using ABI PRISM 310 (Applied Biosystems, Foster City, USA). The acquired sequence was used for subsequent primer designs.
Total RNA was extracted from yeast that was cultured at 25°C in uracil-free medium by the hot phenol method (Köhrer and Domdey 1991). Poly(A) + RNA was purified from total RNA using OligotexTM-dT (Super) mRNA purification kit (Takara Bio, Shiga, Japan). After reverse transcription, the full-length cDNA sequence of URA5 was determined by 5′- and 3′-RACE (rapid amplification of cDNA ends) methods using 5′- and 3′-Full RACE Core Sets (Takara Bio, Shiga, Japan), respectively.
The genomic sequences of the upstream and downstream regions of URA5 were obtained by inverted PCR (Triglia et al. 1988). The genomic DNA was isolated by gene trapping using a Liquid Extraction kit (Takara Bio, Shiga, Japan) and digested by PstI. Using self-ligated genomic DNA as a template, inverted PCR was performed with the following primers: sense primer, 5′-AACCAAATCGACGGTTCAGG-3′, and antisense primer, 5′-GGCACTCGAGATGACACAAG-3′. Amplified fragments were cloned into pGEM-T vectors and sequenced. Genomic DNA of U-5 was also isolated. The DNA fragment containing URA5 was amplified by PCR, and its sequence was analyzed along with a wild-type strain.
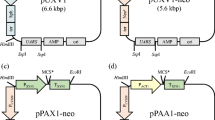
The entire URA5 gene of S-2 including parts of the 5′ and 3′ UTRs was amplified by PCR using genomic DNA as template and sense and antisense primers AACCTGCAGGTGCTTCGAGTGCTTGGTACC and AAACCCGGGCTGATGGATTGCAGGTCTGC, which include Sse8387I and SmaI sites (underlined). The product was digested by Sse8387I and SmaI and cloned into the multicloning site of pUC19 to construct the plasmid pCsURA5 (Fig. 1a).

Schematic representation of the plasmids. a The plasmid pCsURA5 containing URA5 of Cryptococcus sp. strain S-2. The 5′ flanking region (777 bp) and the 3′ flanking region (481 bp) are represented as thick lines. b The plasmid pCsUCLE for multiple integration of CLE1. CLE1, including 1,003 bp of 5′ flanking (CLE-pro) and 111 bp of 3′ flanking (CLE-ter) regions, is cloned into pCsURA5. c The plasmid pCsUX containing xylanase promoter (Xyl-pro; 837 bp) and terminator (Xyl-ter; 277 bp) regions. d The plasmid pCsUA containing α-amylase promoter (Amy-pro; 940 bp) and terminator (Amy-ter; 297 bp) regions
Transformations
To transform the uracil auxotroph U-5, cells were cultured overnight in 100 ml of YM broth in a 500-ml baffled Erlenmeyer flask at 25 °C with vigorous aeration in a shaker at 130 rpm. The culture was harvested at an optical density of 0.8–1.0 at 660 nm (OD660 nm) by centrifugation at 2,300 × g and 4 °C for 3 min. The harvested cells were washed twice in a wash solution and resuspended in 0.5 ml electroporation solution.
Cells were transformed by three different electroporation methods as previously described for S. cerevisiae (Becker and Guarente 1991) (method 1) and C. neoformans (Varma et al. 1992) with some modifications (methods 2 and 3). In method 1, the wash and electroporation solutions were 1 M sorbitol. The cell suspension (50 μl) in the electroporation solution was transferred to 0.2-cm-gap disposable electroporation cuvettes (Bio-Rad, Hercules, USA). After the addition of plasmid DNA (2 μg), the cells were subjected to an electric pulse from a Gene Pulser (Bio-Rad). The pulse setting was 1.5 kV, 25 μF, and 200 Ω. In method 2, the wash solution contained 270 mM sucrose, 1 mM MgCl2, 10 mM Tris–HCl (pH 7.6), and 4 mM DTT and the electroporation solution contained 270 mM sucrose, 1 mM MgCl2, and 10 mM Tris–HCl (pH 7.6). The pulse setting was 470 kV, 25 μF, and 1,000 Ω. Method 3 was identical to method 2 except the pulse setting was 470 kV, 25 μF, and ∞.
Under all conditions, circular and linear plasmids were applied to transformations. EcoRI digested at a multicloning site located at the outside of the 3′ flanking region of URA5. Eco52I digested pCsURA5 at an internal region of URA5 ORF. The amount of plasmid was also changed from 0.1 to 5 μg for method 3. After electroporation, cells were plated on an SC(−ura) plate, which was incubated at 25°C until colonies appeared. The colonies on the SC(−ura) plates were counted to estimate transformation efficiency under each condition.
Introduction of the CLE gene (CLE1) to construct multicopy transformants
Plasmid pCsURA5 was digested by SmaI (blunt end digestion) and EcoRI and purified using a gel extraction kit (Qiagen, Hilden, Germany). CLE1 including 1,003 bp of the 5′ flanking region and 111 bp of the 3′ flanking region was amplified by PCR. EcoRI cleaved only the 3′ region of the PCR product. This fragment, digested by EcoRI, was purified and cloned into pCsURA5. The resulting plasmid, pCsUCLE (Fig. 1b), was linearized by digestion with EcoRI and transferred into U-5 by electroporation as described above. Ten transformants were randomly selected and inoculated into 50 ml of CLE production medium (1% yeast extract, 0.5% lactose, 1% KH2PO4, 0.1% MgSO47H2O, 1% triolein, pH 5.5) in a 200-ml baffled Erlenmeyer flask and cultured at 25°C with vigorous aeration in a shaker at 140 rpm for 7 days (Kamini et al. 2000). The culture supernatant was assayed for CLE activity using p-nitrophenyl butyrate (PNPB) as a substrate. An aliquot (10 μl) of supernatant was used for one well of sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Following electrophoresis, gels were stained by Coomassie Brilliant Blue using CBB Stain One (Nacalai Tesque, Kyoto, Japan).
CLE activity
CLE activity was assayed by measuring the amount of p-nitrophenol released from PNPB. The reaction mixture contained 2.5 mM PNPB and 50 mM sodium acetate (pH 5.5) and 2% (w/v) of Triton X-100. The reaction was initiated by adding 50 μl of the enzyme solution to 950 μl of the reaction mixture. The reaction was terminated by adding 2 ml of acetone. The concentration of p-nitrophenol was determined spectrophotometrically at 410 nm using an extinction coefficient of 0.62 mM−1 cm−1. One unit of hydrolysis activity was defined as the hydrolysis of 1 μmol of the substrate per minute, at 37 °C.
Construction of plasmids containing inducible promoters for recombinant protein expression
Plasmid pCsURA5 was digested by SmaI and EcoRI and purified using a gel extraction kit (Qiagen, Hilden, Germany). The xylanase terminator region (277 bp) was amplified by PCR and also digested by SmaI and EcoRI. The two restriction enzyme-treated fragments were ligated. The resulting plasmid was digested by EcoRI and used directly in the next step. The xylanase promoter region (837 bp) was amplified by PCR, digested by EcoRI, and cloned into the constructed plasmid to generate pCsUX (Fig. 1c).
The α-amylase terminator region (297 bp) was also amplified by PCR and digested by EcoRI and SmaI and cloned into the plasmid pCsURA5, which had previously been digested with EcoRI and SmaI. The resulting constructed plasmid was digested by EcoRI and used directly in the next step. The α-amylase promoter region (940 bp) was amplified by PCR and digested by EcoRI and cloned into the constructed plasmid to generate pCsUA (Fig. 1d).
Cloning the GFP gene into pCsUX and pCsUA
The GFP gene (GFP) was amplified from a pEGFP DNA template by PCR using a forward primer that contained an MluI site and a reverse primer that contained an SpeI site. The amplified fragment was digested by MluI and SpeI, and the resulting fragment was cloned into pCsUX at the same sites to generate plasmid pCsUX.GFP. The amplified GFP with SpeI sites at the 5′ and 3′ ends was digested by SpeI and cloned into pCsUA at the SpeI site to produce plasmid pCsUA.GFP.
These constructed plasmids were transferred into U-5 by electroporation as described above. Integrations of GFP expression cassettes in the transformants were confirmed by colony PCR using primers located in the promoter region and the GFP gene.
Transformants resulting from integration of pCsUX.GFP or pCsUA.GFP were cultured in yeast nitrogen base medium (0.67% yeast nitrogen base without amino acids, 2% carbon source) at 25°C for 24 h. Glucose, xylose, maltose, dextrin, and xylan were used as carbon source. After the cells were harvested by centrifugation and washed with water, expression levels of GFP were observed by fluorescence microscopy (ECLIPSE E600 microscope, Nikon, Tokyo, Japan), using the appropriate filter set.
Cloning CLE1 into pCsUX and construction of recombinant CLE production yeast
CLE1 from genomic DNA of S-2 was amplified by PCR using a forward and a reverse primer that contained an MluI site before the start and stop codons. The amplified fragment was digested by MluI, and the resulting fragment was cloned into pCsUX at these same sites to produce plasmid pCsUX.CLE. The constructed plasmid was digested by Sse8387I and transferred into U-5 by electroporation as described above. After electroporation, cells were plated on an SC(−ura) plate, which was incubated at 25°C until colonies appeared. Forty transformants were cultured in 5 ml SC(−ura) medium containing 2% xylose as the sole carbon source at 25°C for 3 days. The transformant showing the highest CLE activity was selected and used for subsequent CLE expression. The selected transformant was inoculated into YM medium and cultured for 2 days as a preculture. The preculture (2.5 ml) was inoculated into 50 ml of expression medium consisting of 6% xylose and 1% yeast extract in a 200-ml baffled flask and cultured at 25°C and 140 rpm. The activity of the supernatant from the culture was measured each day for 6 days.
Accession numbers
The accession number of the cDNA sequence of URA5 is AB470526 in DDBJ. Accession numbers of α-amylase promoter, xylanase promoter, and CLE1 are D83540, D63381, and AB671329, respectively.
Results
Isolation of the uracil auxotroph of S-2
Spontaneous 5-FOA-resistant mutants were isolated from medium containing 5-FOA. One of them was found to be a uracil auxotroph. 5-FOA-resistant uracil auxotrophs are usually deficient in OPRTase and/or orotidine-5′-phosphate decarboxylase (OMPdecase, EC 4.1.1.23) because these enzymes catalyze the last two steps in the biosynthesis of pyrimidine (Bai et al. 1999). This isolated mutant could grow in a medium containing orotidine, but not in a medium containing orotic acid. Therefore, the isolated mutant was deficient in OPRTase because orotic acid is converted into orotidine-5′-phosphate by OPRTase. The uracil auxotroph, named U-5, was subsequently used as a host strain for the following host–vector system construction.
Isolation of URA5 of S-2
An approximately 400-bp fragment of URA5 encoding OPRTase of S-2 was amplified by degenerate PCR using four degenerate primers, which were designed from highly conserved regions of URA5 from other microbes. Using the sequence of an isolated genomic DNA fragment, the full cDNA sequence of URA5 was obtained by 3′ and 5′ RACE. The upstream and downstream sequences of the URA5 ORF (1,126 and 522 bp, respectively) were derived by inverted PCR. The URA5 ORF region of genomic DNA contains three exons and two introns. The OPRTase of S-2 consists of 235 amino acids and has a molecular weight of 25 kDa. Its deduced amino acid sequence exhibits 71% and 43% homology with the OPRTases of C. neoformans and S. cerevisiae, respectively. One base substitution was detected in URA5 of U-5, which resulted in a Ser-to-Leu substitution at amino acid position 48.
Transformation by electroporation
URA5 including 777 bp of the 5′ flanking region and 481 bp of the 3′ flanking region was cloned into the multicloning site between the SmaI and Sse8387I sites of plasmid pUC19. The resulting plasmid, named pCsURA5 (Fig. 1a), was used to transform U-5 by electroporation. Three forms were prepared: a circular plasmid, a linear plasmid digested by EcoRI outside the URA5 region, and a linear plasmid digested by Eco52I within the URA5 ORF. Three methods that differed in the preparation of competent cells and the electric pulse were compared using each of the three plasmid forms. The highest transformation efficiency was obtained with method 3 using linear DNA digested by EcoRI (Table 1).
The optimal concentration of plasmid for transformation by method 3 was also studied. Both circular and linear (EcoRI digested) plasmids (0.1–5 μg) were used for this evaluation. The transformation efficiency was higher for the linear plasmid under all concentrations. Although the transformation efficiency at low concentration (0.1 μg) was apparently greater than that at higher concentrations (1–5 μg plasmid), only a few colonies appeared at low concentration (0.1 μg). Therefore, 2 μg of plasmid was used for the following studies with method 3, which exhibited the highest transformation efficiency using 1–5 μg plasmid.
Introduction of CLE1 to construct multicopy transformants
We attempted to introduce multiple CLE1 genes containing its promoter region into U-5 to construct a multicopy transformant. CLE1 containing 1,003 bp of 5′ flanking region and 111 bp of 3′ flanking region was cloned into the multicloning site between SmaI and EcoRI sites of plasmid pCsURA5 to form pCsUCLE (Fig. 1b). Linearized plasmid was introduced into the uracil auxotroph by electroporation and transformants that appeared on an SC(−ura) plate were isolated. Eight of 10 randomly selected transformants showed higher CLE activity than the host strain, U-5 (Fig. 2a), indicating that most of the transformants contained URA5 and CLE1. Growth of each transformant, U-5 as well as the wild type, was the same (Fig. 2b). Four transformants exhibited approximately six to nine times higher CLE activities than the U-5, and transformants 4 and 6 exhibited approximately 3.5 times higher activity than the wild type. SDS-PAGE of the culture supernatant revealed a strong 21-kDa band of CLE in the transformants (Fig. 2c, lanes 4, 5, 6, and 8). This result was consistent with an enzymatic assay (Fig. 2a). Furthermore, CLE was a major protein in the culture supernatant of multicopy transformants, and other extracellular proteins were present in very low amounts. Multiple integration of CLE1 was confirmed by genomic Southern analysis in four transformants with higher CLE activity (nos. 4, 5, 6, and 8) (Supplemental Fig. S1). Each transformant showed a different pattern of multiple bands of CLE1 in the Southern analysis. This result indicated that the transformation resulted in ectopic integration of the gene.

CLE expression from multicopy transformants. Transformants (nos. 1–10) were randomly selected. a CLE activities of transformant cultures. WT and U-5 represent CLE activities of supernatants of the wild-type yeast and uracil auxotroph, respectively. b Growth of transformants. c SDS-PAGE of supernatants (10 μl) of transformant cultures
Heterologous recombinant protein expression vectors including inducible promoters
Two different expression plasmids were constructed for the expression of heterologous proteins in S-2 as shown in Fig. 1c (pCsUX) and d (pCsUA). Plasmid pCsUX contained a xylanase promoter, which is induced by xylan (Iefuji et al. 1996a) and xylose, and the corresponding terminator. Plasmid pCsUA contained an α-amylase promoter, which is induced by maltose (Iefuji et al. 1996b), and the corresponding terminator. Then, a DNA fragment of the desired protein was integrated between the promoter and terminator. GFP expression was investigated with both plasmids. Induction and expression of GFP were observed by fluorescence microscopy. When the xylanase promoter was used, GFP expression was strongly induced by xylose or xylan, but not by glucose or maltose (Fig. 3a). When the α-amylase promoter was used, GFP expression was strongly induced by maltose or dextrin (Fig. 3b). When glucose or xylose was added, only weak fluorescence of GFP was observed.

Induction and expression of GFP observed by fluorescence microscopy. Transformants were cultured in media containing the indicated carbon source at 25°C, 24 h. Left: differential interference contrast microscopy. Right: fluorescence microscopy. a Microscope observations of transformants with pCsUX.GFP. Glucose, xylose, maltose, and xylan were used as the carbon source. b Transformants with pCsUA.GFP. Glucose, xylose, maltose, and dextrin were used as the carbon source
Recombinant CLE production under inducible conditions of the xylanase promoter
The inducible plasmid for recombinant CLE production was constructed by cloning CLE1 into pCsUX. The constructed plasmid (pCsUX.CLE) was introduced into U-5 and transformants were screened for CLE production. Most transformants demonstrated higher CLE activities in the culture medium than the wild type. Of 40 transformants examined, the one with the highest CLE was selected. The selected transformant was inoculated into a medium containing 6% xylose to induce the xylanase promoter. CLE activity of the culture medium increased, reaching about 220 U/ml after 6 days (Fig. 4), which was twice the activity of the multicopy transformant in Fig. 2. In the presence of xylose, the wild type produced approximately 15 U/ml of CLE, whereas the transformant produced approximately 15 times this amount.

Recombinant CLE production under the inducible condition of the xylanase promoter
Discussion
We have constructed a host–vector system of the basidiomycetous yeast Cryptococcus sp. strain S-2 cells, using a plasmid that contains the URA5 and a uracil auxotroph. This system was useful for recombinant CLE expression. In general, yeast expression systems have some advantages for recombinant protein production. For example, a yeast can secrete an own protein in appropriate conformation directly into the medium. Moreover, fewer native proteins are secreted in the culture medium as shown in Fig. 2c, which is also preferred for these applications.
URA5 was used as a marker for the constructed system. In the case of S. cerevisiae, 5-FOA-resistant isolates are predominantly defective in URA3, which encodes OMPdecase (Boeke et al. 1984), because S. cerevisiae has a minor OPRTase isozyme (URA10) (De Montigny et al. 1990). However, URA5 has been used as a marker for some yeast host–vector systems. In the case of C. neoformans, most of the spontaneous 5-FOA-resistant mutants were OPRTase deficient, i.e., ura5 cells (Edman and Kwon-Chung 1990; Kwon-Chung et al. 1992). In S-2, URA5 is probably the only gene encoding OPRTase. In U-5, the OPRTase has a Ser48 to Leu substitution, which could be the cause of OPRTase deficiency because Ser48 was found to be conserved in 10 microbial OPRTases described in “Materials and methods”.
Method 3 in Table 1, which is optimized for C. neoformans (Varma et al. 1992), was most effective for S-2. In addition, using a linearized plasmid increased the transformation frequency, as was also found to be the case with C. neoformans (Edman and Kwon-Chung 1990). The optimal transformation efficiency (obtained with method 3; Table 1) was comparable to that obtained with C. neoformans (Varma et al. 1992). The plasmid linearized by EcoRI was more effective for transformation than the circular plasmid. Although the plasmid digested by Eco52I was also linear, its transformation efficiency was not higher than that of the circular plasmid. Because Eco52I was used to digest the internal region of URA5 ORF to use it for transformation, developing a nonmutated URA5 by homologous recombination at the ura5 locus in the chromosome of the uracil auxotroph was necessary. On the other hand, EcoRI did not digest the URA5 including its promoter and terminator regions. Therefore, the introduced plasmid digested by EcoRI could directly produce an active OPRTase.
Multicopy transformants incorporating CLE1 were generated under the optimized transformation conditions. In general, multicopy transformants express higher quantity of protein than single-copy transformants (Read et al. 2007; Werten et al. 1999). Because the wild-type strain has one copy of the intrinsic CLE1, it is expected that most of the transformants have two or more copies of this gene in its genome. Four of 10 randomly selected transformants clearly contained more than two copies of CLE1 as determined by genomic Southern analysis (Supplemental Fig. S1). The transformants that exhibited nine times higher CLE activities than the host strain were isolated during the screening process that contained only 10 transformants. High-frequency integration of CLE1 into the genome may result in multiple integrations. Although the mechanism of multiple integrations has not yet been elucidated, this feature of S-2 is desirable in the host strain for recombinant protein production.
We investigated two inducible promoters for construction of an expression vector. The xylanase promoter of S-2 was induced by xylan as well as xylose. Xylose is produced from xylan and xylooligosaccharides by the xylanase of S-2 (Iefuji et al. 1996a). In another yeast, Cryptococcus albidus, endo-1,4-β-xylanase induction by xylose was quite limited, while xylan or xylobiose strongly induced production of the enzyme (Biely et al. 1980; Biely and Petrakova 1984). Xylanase from the basidiomycete Sclerotium rolfsii was induced by xylan and xylobiose as well as a broad variety of substrates (Sachslehner et al. 1998). The xylanase promoter in the constructed expression vector was tightly controlled. Glucose and maltose did not induce expression of GFP, whose gene was cloned downstream of the xylanase promoter. On the other hand, the regulation of the α-amylase promoter seemed less constrained than that of the xylanase promoter. The α-amylase promoter was strongly induced by maltose or dextrin. Unexpectedly, xylose also induced the α-amylase promoter but at a lower level than did glucose.
Next, we used the xylanase promoter for recombinant CLE production because this promoter was more effective than the α-amylase promoter, whose efficiency we evaluated. Under inducible conditions of the xylanase promoter, the isolated transformant produced about 220 U/ml of CLE (Fig. 4). Under the same condition (6% xylose and 1% yeast extract), approximately 15 U/ml of CLE was produced by the wild-type yeast, although this condition was not optimal for intrinsic CLE production.
Kamini et al. (2000) determined the optimal conditions for CLE production by the S-2 wild type. As shown in Fig. 2a, the wild-type yeast produced approximately 30 U/ml of CLE under the optimum condition for intrinsic CLE production. Until this study, maximal production has been 30 U/ml. In this study, the production of CLE was highest under the control of the xylanase promoter and was more than seven times higher than the maximum productivity of the wild-type yeast. We have indicated that CLE can effectively degrade plastic (Kawai et al. 2011; Masaki et al. 2005) and the transesterification of triglycerides to fatty acid methyl esters for biodiesel fuel (Kamini and Iefuji 2001). The ability to achieve a higher level of CLE production enhances its overall potential for these and other applications.
We constructed a yeast with an enhanced ability to produce CLE. The basidiomycetous yeast S-2 produces certain unique extracellular enzymes under different conditions. The S-2 system can also be used to increase the production of these enzymes by multicopy transformants and/or by using a xylanase promoter. Because the marker and the promoters, which are the main parts of this system, are derived from the host yeast, constructing a self-cloning strain that does not contain a heterologous gene and produces desirable homologous enzymes of S-2 is possible.
References
Avis TJ, Anguenot R, Neveu B, Bolduc S, Zhao Y, Cheng Y, Labbé C, Belzile F, Bélanger RR (2005) The potential of Pseudozyma yeastlike epiphytes for the production of heterologous recombinant proteins. Appl Microbiol Biotechnol 69:304–311
Avis TJ, Record E, Lomascolo A, Scholtmeijer K, Asther M, Wessels JGH, Wösten MAB (2008) Usefulness of heterologous promoters in the Pseudozyma flocculosa gene expression system. Biosci Biotechnol Biochem 72:456–462
Bai X, Larsen M, Meinhardt F (1999) The URA5 gene encoding orotate-phosphoribosyl transferase of the yeast Kluyveromyces lactis: cloning, sequencing and use as a selectable marker. Yeast 15:1393–1398
Beach D, Nurse P (1981) High-frequency transformation of the fission yeast Schizosaccharomyces pombe. Nature 290:140–142
Becker DM, Guarente L (1991) High-efficiency transformation of yeast by electroporation. Methods Enzymol 194:182–187
Biely P, Petrakova E (1984) Novel inducers of the xylan-degrading enzyme system of Cryptococcus albidus. J Bacteriol 160:408–412
Biely P, Krátký Z, Vrsanská M, Urmanicová D (1980) Induction and inducers of endo-1,4-β-xylanase in the yeast Cryptococcus albidus. Eur J Biochem 108:323–329
Boeke JD, La Croute F, Fink GR (1984) A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197:345–346
Böer E, Steinborn G, Kunze G, Gellissen G (2007) Yeast expression platforms. Appl Microbiol Biotechnol 77:513–523
Buckholz RG, Gleeson MAG (1991) Yeast systems for the commercial production of heterologous proteins. Bio/Technology 9:1067–1072
Cheng Y, Avis TJ, Bolduc S, Zhao Y, Anguenot R, Neveu B, Labbé C, Belzile F, Bélanger RR (2008) Recombinant protein secretion in Pseudozyma flocculosa and Pseudozyma antarctica with a novel signal peptide. Biosci Biotechnol Biochem 72:3158–3166
Cregg JM, Barringer KJ, Hessler AY, Madden KR (1985) Pichia pastoris as a host system for transformations. Mol Cell Biol 5:3376–3385
Das S, Hollenberg CP (1982) A high-frequency transformation system for the yeast Kluyveromyces lactis. Curr Genet 6:123–128
De Montigny J, Kern L, Hubert J, Lacroute F (1990) Cloning and sequencing of URA10, a second gene encoding orotate phosphoribosyl transferase in Saccharomyces cerevisiae. Curr Genet 17:105–111
Domînguez Á, Ferminân E, Sânchez M, Gonzalez FJ, Pérez-Campo FM, Garcia S, Herrero AB, Vicente AS, Cabello J, Prado M, Iglesias FJ, Choupina A, Burguillo FJ, Fernândcz-Lago L, Carmen López M (1998) Non-conventional yeasts as hosts for heterologous protein production. Internat Microbiol 1:131–142
Edman JC, Kwon-Chung KJ (1990) Isolation of the URA5 gene from Cryptococcus neoformans var. neoformans and its use as a selective marker for transformation. Mol Cell Biol 10:4538–4544
Fonseca Á, Boekhout T, Fell JW (2011) Cryptococcus Vuillemin (1901). In: Kurtzman CP, Fell JW, Boekhout T (eds) The yeasts, a taxonomic study, 5th edn. Elsevier, Amsterdam, pp 1661–1737
Gasser B, Sauer M, Maurer M, Stadlmayr G, Mattanovich D (2007) Transcriptomics-based identification of novel factors enhancing heterologous protein secretion in yeasts. Appl Environ Microbiol 73:6499–6507
Hinnen A, Hicks JB, Fink GR (1978) Transformation of yeast. Proc Natl Acad Sci USA 75:1929–1933
Iefuji H, Iimura Y, Obata T (1994) Isolation and characterization of a yeast Cryptococcus sp. S-2 that produces raw starch-digesting α-amylase, xylanase, and polygalacturonase. Biosci Biotechnol Biochem 58:2261–2262
Iefuji H, Chino M, Kato M, Iimura Y (1996a) Acid xylanase from yeast Cryptococcus sp. S-2: purification, characterization, cloning, and sequencing. Biosci Biotechnol Biochem 60:1331–1338
Iefuji H, Chino M, Kato M, Iimura Y (1996b) Raw-starch-digesting and thermostable α-amylase from the yeast Cryptococcus sp. S-2: purification, characterization, cloning and sequencing. Biochem J 318:989–996
Kamini NR, Iefuji H (2001) Lipase catalyzed methanolysis of vegetable oils in aqueous medium by Cryptococcus spp. S-2. Process Biochem 37:405–410
Kamini NR, Fujii T, Kurosu T, Iefuji H (2000) Production, purification and characterization of an extracellular lipase from the yeast, Cryptococcus sp. S-2. Process Biochem 36:317–324
Kawai F, Nakadai K, Nishioka E, Nakajima H, Ohara H, Masaki K, Iefuji H (2011) Different enantioselectivity of two types of poly(lactic acid) depolymerases toward poly(L-lactic acid) and poly(D-lactic acid). Polym Degrad Stab 96:1342–1348
Kodama Y, Masaki K, Kondo H, Suzuki M, Tsuda S, Nagura T, Shimba N, Suzuki E, Iefuji H (2009) Crystal structure and enhanced activity of a cutinase-like enzyme from Cryptococcus sp. strain S-2. Proteins 77:710–717
Köhrer K, Domdey H (1991) Preparation of high molecular weight RNA. Methods Enzymol 194:398–405
Kurtzman CP, Fell JW, Boekhout T (2011) The yeasts, a taxonomic study, 5th edn. Elsevier, Amsterdam
Kwon-Chung KJ, Varma A, Edman JC, Bennett JE (1992) Selection of ura5 and ura3 mutants from the two varieties of Cryptococcus neoformans on 5-fluoroorotic acid medium. J Med Vet Mycol 30:61–69
Masaki K, Kamini NR, Ikeda H, Iefuji H (2005) Cutinase-like enzyme from the yeast Cryptococcus sp. strain S-2 hydrolyzes polylactic acid and other biodegradable plastics. Appl Environ Microbiol 71:7548–7550
Neveu B, Michaud M, Belzile F, Bélanger RR (2007) The Pseudozyma flocculosa actin promoter allows the strong expression of a recombinant protein in the Pseudozyma species. Appl Microbiol Biotechnol 74:1300–1307
Read JD, Colussi PA, Ganatra MB, Taron CH (2007) Acetamide selection of Kluyveromyces lactis cells transformed with an integrative vector leads to high-frequency formation of multicopy strains. Appl Environ Microbiol 73:5088–5096
Sachslehner A, Nidetzky B, Kulbe KD, Haltrich D (1998) Induction of mannanase, xylanase, and endoglucanase activities in Sclerotium rolfsii. Appl Environ Microbiol 64:594–600
Takagi M, Kawai S, Chang MC (1986) Construction of a host–vector system in Candida maltosa by using an ARS site isolated from its genome. J Bacteriol 167:551–555
Takashima M, Nakase T (1999) Molecular phylogeny of the genus Cryptococcus and related species based on the sequences of 18S rDNA and internal transcribed spacer regions. Microbiol Cult Coll 15:35–47
Thirunavukarasu K, Edwinoliver NG, Anbarasan SD, Gowthaman MK, Iefuji H, Kamini NR (2008) Removal of triglyceride soil from fabrics by a novel lipase from Cryptococcus sp. S-2. Process Biochem 43:701–706
Thongekkaew J, Ikeda H, Masaki K, Iefuji H (2008) An acidic and thermostable carboxymethyl cellulase from the yeast Cryptococcus sp. S-2: purification, characterization and improvement of its recombinant enzyme production by high cell-density fermentation of Pichia pastoris. Protein Expr Purif 60:140–146
Triglia T, Peterson MG, Kemp DJ (1988) A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences. Nucleic Acids Res 16:8186
Valkonen M, Ward M, Wang H, Penttilä M, Saloheimo M (2003) Improvement of foreign-protein production in Aspergillus niger var. awamori by constitutive induction of the unfolded-protein response. Appl Environ Microbiol 69:6979–6986
Varma A, Edman JC, Kwon-Chung KJ (1992) Molecular and genetic analysis of URA5 transformants of Cryptococcus neoformans. Infect Immun 60:1101–1108
Werten MWT, Van Den Bosch TJ, Wind RD, Mooibroek H, De Wolf FA (1999) High-yield secretion of recombinant gelatins by Pichia pastoris. Yeast 15:1087–1096
Acknowledgments
We thank Dr. Masako Takashima (Japan Collection of Microorganisms, RIKEN BioResource Center) for discussions about the taxonomy of the genus Cryptococcus and Profs. Hideyo Yamaguchi and Katsuhisa Uchida (Teikyo University Institute of Medical Mycology) for information on the safety test of Cryptococcus sp. strain S-2 in mice.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
ESM 1
(PDF 173 kb)
Rights and permissions
About this article
Cite this article
Masaki, K., Tsuchioka, H., Hirano, T. et al. Construction of a new recombinant protein expression system in the basidiomycetous yeast Cryptococcus sp. strain S-2 and enhancement of the production of a cutinase-like enzyme. Appl Microbiol Biotechnol 93, 1627–1636 (2012). https://doi.org/10.1007/s00253-011-3680-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3680-x