
Abstract
A cycloisomaltooligosaccharide (CI; cyclodextran) production system was developed using a Bacillus subtilis expression system for the cycloisomaltooligosaccharide glucanotransferase (CITase) gene. The CITase gene of Bacillus circulans T-3040, along with the α-amylase promoter (PamyQ) and amyQ signal sequence of Bacillus amyloliquefaciens, was cloned into the Bacillus expression vector pUB110 and subsequently expressed in B. subtilis strain 168 and its alkaline (aprE) and neutral (nprE) protease-deficient strains. The recombinant CITase produced by the protease-deficient strains reached 1 U/mL in the culture supernatant within 48 h of cultivation, which was approximately 7.5 times more than that produced by the industrial CITase-producing strain B. circulans G22-10 derived from B. circulans T-3040. When aprE- and nprE-deficient B. subtilis 168 harboring the CITase gene was cultured with 10% dextran 40 for 48 h, 17% of the dextran in the culture was converted to CIs (CI-7 to CI-12), which was approximately three times more than that converted by B. circulans G22-10 under the same dextran concentration. The B. subtilis host–vector system enabled us to produce CIs by direct fermentation of dextran along with high CITase production, which was not possible in B. circulans G22-10 due to growth inhibition by dextran at high concentrations and limited production of CITase.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cyclodextrans are cycloisomaltooligosaccharides (CIs) consisting of 7 to 17 molecules of α-1,6-linked glucose and are represented as CI-7 to CI-17 (Oguma et al. 1993; Funane et al. 2007a, 2008). CIs strongly inhibit glucansucrase activity in mutans streptococci (Kobayashi et al. 1995), suggesting that CIs are antiplaque materials that would be useful in preventing dental caries. Among CIs, cycloisomaltodecaose (CI-10) shows an inclusion ability on par with cyclodextrins, as determined by its ability to stabilize the color of Victoria blue B (Funane et al. 2007a).
CIs are produced from dextran, and this reaction is catalyzed by cycloisomaltooligosaccharide glucanotransferase (CITase; EC 2.4.1.248) (Oguma et al. 1994). Bacillus circulans T-3040 [FERM BP-4132 (NBRC)] simultaneously secretes CITase and CIs into its culture supernatant when grown with dextran (Oguma et al. 1994). However, CITase production by B. circulans strains is very low (0.001 U/mL) and not sufficient for commercial production of CIs. To improve productivity, B. circulans T-3040 was mutagenized. B. circulans strain G22-10, which was obtained by treatment with nitrosoguanidine and streptomycin, has approximately 110 times more CITase activity than the parental strain (Kawabata et al. 2006; Funane et al. 2007b). Thus, B. circulans G22-10 can be used for the efficient commercial production of CIs. However, dextran, the substrate for CI production, inhibits the growth of B. circulans T-3040 and its mutants. B. circulans G22-10 requires a long culture period of at least 4 days to produce sufficiently large amounts of CIs for practical use. Immobilization of Escherichia coli recombinant CITase facilitates CI production, resulting in sufficient yields in 10–16 h (Kawamoto et al. 2001). However, production of CIs by simple fermentation without much equipment and technology is desired for practical use.
Bacillus subtilis has been successfully used as a host for high-level expression of genes for various enzymes (Westers et al. 2004; Inoue et al. 2002; Yamamoto et al. 2005; Fahnestock 1986). Extracellular production of CITase using B. subtilis is a potential strategy to address the problems associated with B. circulans and immobilized CITase and produce CIs by fermentation. In the present study, CITase was produced and secreted by B. subtilis strains carrying the CITase gene, which was under the control of the Bacillus amyloliquefaciens α-amylase promoter (PamyQ). This system successfully produced 7.5 times more extracellular CITase than B. circulans G22-10. Moreover, CIs were simultaneously produced by direct fermentation of dextran at 2%, 5%, and 10% concentrations using this system.
Materials and methods
Strains
B. subtilis Marburg 168 (trpC2) and B. amyloliquefaciens NBRC 15535 were used in this study. Alkaline (aprE) and neutral (nprE) protease-deficient strains were produced by transforming wild-type B. subtilis 168 cells with genomic DNA of B. subtilis Marburg 168 (trpC2) strains in which aprE and nprE were disrupted by Spcr and Neor cassettes (Takano Foods Co., Ltd., Omitama, Japan). Transformants were selected on Luria Bertani (LB) agar (Becton, Dickinson and Company, Sparks, MD, USA) plates supplemented with antibiotics (100 μg/mL spectinomycin and 7.5 μg/mL neomycin). Protease activities were assayed as described previously (Amory et al. 1987) with a minor modification using the chromogenic substrate azocasein (Sigma-Aldrich, Saint Louis, MO, USA). Culture supernatants were incubated in 80 mM Tris–HCl (pH 8.0) and 0.4% azocasein at 37°C for 30 min. The reaction was terminated by the addition of 6% trichloroacetic acid. After centrifugation at 10,000×g for 10 min, the supernatants were neutralized with NaOH, and the optical densities (ODs) of the samples were measured at 440 nm. One unit of the enzyme hydrolyzes 1 μg of azocasein per minute.
Construction of CITase gene-carrying vector and transformation of B. subtilis strains
To achieve extracellular expression in B. subtilis, the CITase gene of B. circulans T-3040 (GenBank ID: D61382) was fused with the regulatory region of B. amyloliquefaciens amyQ and the amyQ signal sequence (GenBank ID: V00092). The CITase expression vector pQS-CIT was constructed as follows: first, PamyQ and the amyQ signal peptide region were amplified using primers 1 [5′-AAAGGATCCGCCCCGCACATACGAAAAGACTGG-3′ (BamHI site is underlined)] and 2 [5′- CGATGCCGCCAGAGCCTGAGGCTGATGTTTTTGTAATCG −3′] with chromosomal DNA from B. amyloliquefaciens as the template. Second, the CITase gene without the signal peptide sequence was amplified using primers 3 [5′- TCAGGCTCTGGCGGCATCGAGC-3′] and 4 [5′- AAATCTAGACTAGCTCACATTGATCCCGAAG −3′ (XbaI site is underlined)] with chromosomal DNA from B. circulans T-3040 as the template. Overlap polymerase chain reaction (PCR) was performed using primers 1 and 4 with a mixture of the amplified fragments as a template. The resulting PCR fragment was digested with BamHI and XbaI and ligated into pUB110 at the same restriction sites using the linear form ligation method (Tsuge et al. 2003). The final CITase expression vector pQS-CIT is shown in Fig. 1.

Structure of the CITase expression vector pQS-CIT. pQS-CIT was constructed using pUB110. PamyQ is the promoter region of the α-amylase gene of B. amyloliquefaciens; the amyQ signal sequence is the signal peptide region of the α-amylase gene of B. amyloliquefaciens; CITase is the CITase gene of B. circulans T-3040 without its signal sequence; ori + and palU/ori − are replication origins; Km R is the kanamycin resistance gene; Ph R is the phleomycin resistance gene; repB is the replication initiator protein B; and pUB110 is pUB110 digested with BamHI and XbaI
B. subtilis strains were transformed with pQS-CIT by the method of Anagnostopoulos and Spizizen (1961). Kanamycin- and phleomycin-resistant transformants were screened on blue dextran-containing LB plates [1% peptone, 0.5% yeast extract, 1% NaCl, 0.8% dextran 40 (GE Healthcare, Uppsala, Sweden), 0.2% blue dextran 2000 (GE Healthcare), and 1.5% agar] supplemented with 10 μg/mL of kanamycin and 1 μg/mL of phleomycin. Each CITase-producing clone made a halo around the colony, which was formed by blue dextran 2000 degradation. DNA sequences of the inserted genes in the selected clones were confirmed using an ABI 373A DNA sequencer (Applied Biosystems, Foster City, CA, USA) and an ABI Prism Dye Terminator Cycle Sequencing kit (Applied Biosystems).
Cultivation of pQS-CIT-carrying B. subtilis strains and preparation of extracellular and intracellular fractions
B. subtilis strains harboring pQS-CIT were precultured using a Bio Shaker BR 40 L (Taitec Co. Ltd., Koshigaya, Japan) with shaking at 200 rpm at 37°C for 24 h in 2 mL of mineral salts-supplemented P4M2 medium [4% peptone, 2% maltose, and 1% yeast extract containing 0.001% of NaCl, MgSO4·7H2O, FeSO4·7H2O, MnCl2·4H2O, and 0.005% CaCl2·2H2O (pH 7.2)] with 10 μg/mL of kanamycin and 1 μg/mL of phleomycin. The cells were grown in 14-mL tubes until late log phase. Each preculture was inoculated into 5.5 mL of the same medium containing 1 μg/mL phleomycin in L-shaped tubes. The initial absorbance at 600 nm was adjusted to an OD of 0.04. The cells were cultured with a TN-1506 biophotorecorder (Advantec MFS, Inc., Dublin, CA, USA) with shaking at 70 rpm at 37°C. The culture was centrifuged at 10,000×g for 15 min, and the supernatants were pooled as an extracellular fraction. Precipitated cells were washed two times with 20 mM Tris–HCl buffer (pH 7.0), suspended in the same buffer containing lysozyme (10 mg/mL), and incubated at 37°C for 30 min with gentle mixing. Subsequently, 100 μL of BugBuster (Merck KGaA, Darmstadt, Germany) and 1 μL of DNase I (Takara Bio Inc., Otsu, Japan) were added to the cell suspension and mixed. The cell lysate was centrifuged at 10,000×g for 15 min to remove cell debris and pooled as an intracellular fraction.
Cultivation of the B. circulans G22-10 strain and preparation of the extracellular fraction
B. circulans G22-10 was precultured with shaking at 200 rpm at 30°C for 2 days in 2 mL of LB broth (Becton, Dickinson and Company) containing 2% dextran 40 as described above. The preculture was inoculated into 5.5 mL of the same medium in L-shaped tubes. The initial absorbance at 600 nm was adjusted to an OD of 0.04 and cultured with shaking at 70 rpm at 30°C with the TN-1506 biophotorecorder. The culture was centrifuged at 10,000×g for 10 min, the precipitants were discarded, and the supernatants were pooled as an extracellular fraction.
Purification of CITase
B. subtilis 168 transformed with pQS-CIT and B. circulans G22-10 were cultured in 1 L mineral salts-supplemented P4M2 medium and 1 L LB broth containing 2% dextran 40, respectively, as described in the “Materials and methods” section, in 5 L baffled-bottom Erlenmeyer flasks with a shaker (TB-128R; Takasaki Scientific Instruments Corp., Kawaguchi, Japan) at 160 rpm. The growing conditions were 37°C for 2 days for B. subtilis and 30°C for 3 days for B. circulans. Culture supernatants were collected by centrifugation (5,000×g for 10 min), brought to 20–80% saturation with ammonium sulfate, and centrifuged at 10,000×g for 20 min. The resulting pellets were suspended in an appropriate volume of 50 mM sodium phosphate buffer (pH 8.0) and dialyzed against the same buffer. The supernatants were collected by centrifugation (10,000×g for 20 min) and loaded onto a Resource Q column (6 mL; GE Healthcare) equilibrated with 50 mM sodium phosphate buffer (pH 8.0). The column was washed with the same buffer, and the enzyme was eluted with a 0–0.6 M linear NaCl gradient. The eluted fraction was loaded onto a gel filtration column (Superose 12, 1.0 × 30 cm; GE Healthcare) equilibrated with 50 mM sodium phosphate buffer (pH 7.0) containing 0.15 M NaCl. Purified enzyme produced by B. circulans G22-10 strain was obtained at this step. For the enzyme produced by pQS-CIT-carrying B. subtilis 168, four times the fraction volume of 50 mM sodium phosphate buffer (pH 8.0) was added to the eluted fraction from the Superose 12 column. It was loaded onto a Mono Q column (1 mL; GE Healthcare) equilibrated with 50 mM sodium phosphate buffer (pH 8.0), washed with the same buffer, and the enzyme was eluted with a 0–0.6 M linear NaCl gradient to obtain purified enzyme. Protein concentration was determined using the bicinchoninic acid protein assay kit (Pierce, Rockford, IL, USA) with bovine serum albumin used as a standard.
Assay of CITase activity and CI production
CITase activity was determined by measuring the amounts of CI-7, CI-8, and CI-9 produced from dextran as described previously (Yamamoto et al. 2006). The CITase assays were performed in 40 mM sodium acetate buffer (pH 5.5) containing 10 mM CaCl2 and 2% dextran 40 at 40°C. CIs in the reaction mixtures were measured by high-performance liquid chromatography (HPLC; CLASS-VP with Evaporative Light Scattering Detector System; Shimadzu, Kyoto, Japan) using a TSK gel Amide-80 column (4.6 × 250 mm; Tosoh, Tokyo, Japan). The mobile phase was acetonitrile–water (55:45, v/v), and the flow rate was 1 ml/min. One unit of CITase activity was defined as the amount of enzyme that produced 1 μmol of the sum of CI-7, CI-8, and CI-9 per minute.
The amount of CI (CI-7 to CI-12) produced in the culture supernatants was measured using HPLC as described above. The standard CIs were purchased from C-I Bio, Ltd. (Tomigusuku, Okinawa, Japan).
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and zymography
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was performed using an 8% polyacrylamide gel following the method of Laemmli (1970). Protein bands were stained with Coomassie Brilliant Blue R-250 (CBB). Enzyme zymography was performed according to the method of Igarashi et al. (1992) with a slight modification. Proteins were separated on an 8% SDS-PAGE gel containing 0.5% blue dextran 2000. Subsequently, the proteins in the gels were renatured by washing the gel two times with water and then soaking it in 50 mM sodium acetate buffer (pH 5.5) at 37°C until active CITase was detected by the formation of a clear band on a blue background.
Results
Expression of the CITase gene from B. circulans T-3040 in the B. subtilis 168 strain
To express the CITase gene from B. circulans T-3040 in B. subtilis and produce extracellular CITase, an expression vector (pQS-CIT) was constructed containing PamyQ and the amyQ signal sequence of B. amyloliquefaciens situated upstream of the CITase gene (Fig. 1). The B. subtilis Marburg 168 strain carrying pQS-CIT mostly produced CITase in the extracellular fraction after 48 h of cultivation, with only a trace amount of CITase activity detected in the intracellular fraction. The B. subtilis 168 strain without pQS-CIT and the strain harboring only pUB110 showed no CITase activity in either the extracellular or intracellular fraction.
The 168 strain grew to an OD of approximately 30 at an absorbance of 600 nm (Fig. 2a) and exhibited CITase activity of 0.682 ± 0.057 U/mL in the culture medium after 36 h of cultivation (Fig. 2b). This value was 15 times higher than that of B. circulans G22-10 after 36 h of cultivation (0.0408 ± 0.017 U/mL) and approximately 5 times higher than that of the same strain after 48 h of cultivation (0.139 ± 0.045 U/mL). Purified CITase produced by the G22-10 strain and B. subtilis recombinant CITase possessed almost the same specific activities (Table 1). Higher CITase activity in the pQS-CIT-carrying B. subtilis 168 culture supernatants could be due to the production of large amounts of enzyme protein. However, the CITase activity of the 168 strain decreased after 48 h of cultivation.

Growth (a) and CITase production (b) by the B. circulans G22-10 and pQS-CIT-carrying B. subtilis strains. Growth was assessed as culture turbidity by measuring absorbance at 600 nm. CITase activity was determined as described in the “Materials and methods” section. White bar, 12-h culture; light gray bar, 24-h culture; dark gray bar, 36-h culture; and black bar, 48-h culture. The error bars indicate the standard deviation of triplicate experiments. G22-10 is B. circulans G-22-10; Wild is B. subtilis 168; ΔaprE is B. subtilis 168 ΔaprE; and ΔaprEΔnprE is B. subtilis 168 ΔaprEΔnprE
Expression of the CITase gene in protease-deficient B. subtilis 168 strains
We hypothesized that the reduction in CITase activity in the later stage of cultivation was caused by proteolysis by the host proteases secreted into the medium. Kawamura and Doi (1984) reported that extracellular protease activity was reduced by 65% in the aprE-deficient (ΔaprE) mutant, and the aprE and nprE double-deficient (ΔaprEΔnprE) mutant exhibited 96% decrease. Thus, to prevent CITase degradation, aprE- and/or nprE-deficient mutants were used as host strains. In this study, the ΔaprE and ΔaprEΔnprE mutants obtained from the B. subtilis 168 strain also exhibited protease activities reduced by approximately 65% and 90%, respectively. Furthermore, the CITase activities of ΔaprE and ΔaprEΔnprE mutants reached 0.980 ± 0.070 U/mL and 1.062 ± 0.059 U/mL, respectively, approximately two times higher than the parental strain, and the activities did not decrease until after 48 h of cultivation (Fig. 2b).
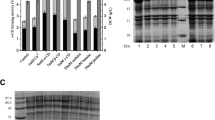
Zymography analysis showed that the protein bands of the protease-deficient mutants were thicker than those of the parental strain (Fig. 3a). The molecular size of CITase is predicted to be 103 kDa, which is in agreement with that of the protein band in the CBB-stained gel (Fig. 3a) and the halos observed by zymography (Fig. 3b). Two smaller bands with a halo at 96 and 88 kDa were also observed by zymography.

SDS-PAGE (a) and zymography (b) of the culture supernatant of pQS-CIT-carrying B. subtilis 168 and its protease-deficient strains. The B. subtilis transformants were grown in mineral salts-supplemented P4M2 medium at 37°C for 48 h. Each culture supernatant (10 μL) was loaded onto an 8% (w/v) polyacrylamide gel with or without 0.5% blue dextran 2000. After electrophoresis, the gel without blue dextran was stained with CBB. The blue gel was washed two times with water with gentle shaking for 30 min and subsequently incubated in 50 mM sodium acetate (pH 5.5) at 37°C for 3 h. Lanes Marker, Wild, ΔaprE, and ΔaprEΔnprE: Amersham high molecular weight calibration kit for SDS electrophoresis (GE Healthcare), B. subtilis 168, B. subtilis 168 ΔaprE, and B. subtilis 168 ΔaprEΔnprE, respectively
The CITase activity of the B. subtilis ΔaprEΔnprE culture supernatant was 7.5 times greater than that of the B. circulans G22-10 culture supernatant, and the specific activity of the B. subtilis ΔaprEΔnprE culture supernatant was almost twice that of the B. circulans G22-10 culture supernatant (Table 1). Both CITases were purified as 103 kDa proteins, and their specific activities were approximately equal, with 14.6 U/mL for B. subtilis ΔaprEΔnprE recombinant CITase and 14.2 U/mL for B. circulans G22-10 CITase (Table 1).
Production of CIs using the pQS-CIT-carrying protease-deficient ΔaprEΔnprE mutant
CI (CI-7 to CI-12) production was measured in the culture supernatant of pQS-CIT-carrying B. subtilis 168 ΔaprEΔnprE grown in mineral salts-supplemented P4M2 medium with 2%, 5%, and 10% dextran 40. CI production was compared with that of B. circulans G22-10 strains grown in 2%, 5%, and 10% dextran 40-supplemented LB broth. As shown in Fig. 4a, dextran 40 inhibited the growth of the G22-10 strain, and thus, its CI production level was not enhanced by an increasing dextran 40 concentration (Fig. 4b). The G22-10 strain grown with 2%, 5%, and 10% dextran 40 for 48 h produced CIs (sum of CI-7 to CI-12) at yields of 6.02 ± 0.69 mg/mL, 5.30 ± 0.41 mg/mL, and 5.43 ± 0.27 mg/mL, respectively.

Growth (a) and CI production (b) by B. circulans G22-10 and pQS-CIT-carrying B. subtilis 168 ΔaprEΔnprE strains. Growth was measured as described in the legend of Fig. 2. The CI produced (mg/mL) is indicated as the sum of CI-7 to CI-12 per milliliter of culture broth. White bar, 12-h culture; light gray bar, 24-h culture; dark gray bar, 36-h culture; and black bar, 48-h culture. The error bars indicate the standard deviation of triplicate experiments. % dex 40 is dextran 40 concentration (percent) in culture broth
In contrast, the growth of the ΔaprEΔnprE mutant was apparently unaffected by dextran 40, and no reduction in cell density was observed after 48 h of growth with 10% dextran in the medium (Fig. 4a). In addition, CI production by the ΔaprEΔnprE mutant was enhanced with increasing dextran 40 concentration (Fig. 4b). CIs (CI-7 to CI-12) were produced after 48 h of cultivation from 2%, 5%, and 10% dextran 40 in the amounts of 8.59 ± 0.32 mg/mL, 12.27 ± 0.22 mg/mL, and 16.62 ± 1.14 mg/mL, respectively. The yields of CIs from the conversion of the substrate dextran were 43%, 25%, and 17%, respectively.
Discussion
Using PamyQ and the amyQ signal sequence, the CITase gene was successfully expressed extracellularly by B. subtilis. In previous studies, the CITase gene has been expressed in E. coli BL21 (DE3) using pET systems (Yamamoto et al. 2006; Funane et al. 2011). However, there are disadvantages in the use of E. coli expression systems for CITase and CI production. For example, a low temperature must be maintained during expression; 18°C in Yamamoto et al. (2006) or 25°C in Funane et al. (2011). At higher temperatures, the CITase gene is barely expressed in E. coli and the recombinant CITase becomes inactive inclusion bodies. Another problem is that the enzyme accumulates inside the E. coli cells, and the cells must be disrupted to use the enzyme. To produce CIs efficiently using E. coli recombinant CITase, the enzyme was immobilized on Chitopearl BCW-3505 (Kawamoto et al. 2001), resulting in an enzyme activity of 1.75 U/ml carrier. They calculated the yield of CIs as the sum of CI-7, CI-8, and CI-9 produced versus the amount of substrate dextran and found that the maximum conversion yield of CIs (CI-7 to CI-9) in batch reactions for 2% and 10% dextran was 24% and 12%, respectively. We have shown that a pQS-CIT-carrying B. subtilis 168 ΔaprEΔnprE strain yields CIs (CI-7 to CI-9) from 2% and 10% dextran at 24% and 11%, respectively, after 48 h of cultivation. The CI production level of this strain is nearly identical to that of the immobilized CITase system.
B. circulans T-3040 and U-155 are known as CI- and CITase-producing strains (Oguma et al. 1994; Oguma and Kawamoto 2003). The optimum growth temperature of these strains is 30°C, and they do not grow well at higher temperatures. CITase activity in B. circulans T-3040 is only 0.001 U/mL, whereas the high CITase-producing mutant B. circulans G22-10 has 110 times more activity. However, it is still low at 0.110 U/mL after 3 days of culturing (Kawabata et al. 2006; Funane et al. 2007b). CITase is an inducible enzyme, and dextran is required for enzyme induction in B. circulans strains. However, dextran inhibits cell growth at high concentrations (Fig. 4).
Our study revealed that the B. subtilis 168 ΔaprEΔnprE mutant strain produces 1.062 ± 0.0590 U/mL CITase in a 48-h culture (Fig. 2b), and its specific activity is as strong as B. circulans G22-10 CITase (Table 1). CITase activity in the culture supernatant was not decreased using the protease-deficient strains as host cells (Figs. 2 and 3), which suggests that the decrease in CITase activity in the protease-positive host cells was due to proteolysis by AprE and NprE. The 96- and 88-kDa protein bands in the zymogram were observed in the samples from both the protease-positive- and -negative hosts. A previous deletion study of T-3040 CITase revealed that when the enzyme lacks the 234-amino acid C-terminal variable region (78 kDa), it retains its CI-producing activity with the same k cat values as the wild-type enzyme, but further C-terminal deletion completely abolishes the enzyme activity (Funane et al. 2011). The 96- and 88-kDa proteins in the zymogram were larger than the 78-kDa deletion mutant protein. These proteins may be CITases with truncated C-terminal regions that were partially digested by remnant proteases secreted by the host cells. When the wild-type B. subtilis 168 strain was used as a host, its proteases, including AprE and NprE, probably digested CITase until it was no longer active, causing the decrease in CITase activity after 48 h of cultivation (Fig. 2b).
Currently, CIs are commercially produced from dextran using CITase obtained by culturing B. circulans G22-10. However, production of sufficiently large amounts of CITase from the G22-10 strain for practical use is time consuming. The growth of the CI-producing B. circulans is inhibited by the substrate dextran, limiting CI production by direct CI fermentation with B. circulans (Fig. 4). In this study, we demonstrated a simple method for producing CI by cultivating a strain of B. subtilis with dextran. As dextran concentration increased, increased CI production was observed with this system. Notably, since CITase produces larger molecules of CIs up to CI-17 (Funane et al. 2008), the conversion efficiency to CIs may be larger than the yield calculated from the production of CI-7 to CI-12. Based on our findings, this system could be used for the industrial production of CIs along with CITase by direct fermentation.
References
Amory A, Kunst F, Aubert E, Klier A, Rapoport G (1987) Characterization of the sacQ genes from Bacillus licheniformis and Bacillus subtilis. J Bacteriol 169:324–333
Anagnostopoulos C, Spizizen J (1961) Requirements for transformation in Bacillus subtilis. J Bacteriol 81:741–746
Fahnestock SR (1986) Expression of the staphylococcal protein A gene in Bacillus subtilis by gene fusions utilizing the promoter from a Bacillus amyloliquefaciens α-amylase gene. J Bacteriol 165:796–804
Funane K, Terasawa K, Mizuno Y, Ono H, Miyagi T, Gibu S, Tokashiki T, Kawabata Y, Kim YM, Kimura A, Kobayashi M (2007a) A novel cyclic isomaltooligosaccharide (cycloisomaltodecaose, CI-10) produced by Bacillus circulans T-3040 displays remarkable inclusion ability compared with cyclodextrins. J Biotechnol 130:188–192. doi:10.1016/j.jbiotec.2007.03.009
Funane K, Tokashiki T, Gibu S, Kawabata Y, Oguma T, Ito H, Nakachi M, Miyagi S, Kobayashi M (2007b) Finding of cyclodextrans and attempts of their industrialization for cariostatic oligosaccharides. J Appl Glycosci 54:103–107
Funane K, Terasawa K, Mizuno Y, Ono H, Gibu S, Tokashiki T, Kawabata Y, Kim YM, Kimura A, Kobayashi M (2008) Isolation of Bacillus and Paenibacillus bacterial strains that produce large molecules of cyclic isomaltooligosaccharides. Biosci Biotechnol Biochem 72:3277–3280. doi:10.1271/bbb.80384
Funane K, Kawabata Y, Suzuki R, Kim YM, Kang HK, Suzuki N, Fujimoto Z, Kimura A, Kobayashi M (2011) Deletion analysis of regions at the C-terminal part of cycloisomaltooligosaccharide glucanotransferase from Bacillus circulans T-3040. Biochim Biophys Acta 1814:428–434. doi:10.1016/j.bbapap.2010.12.009
Igarashi T, Yamamoto A, Goto N (1992) Characterization of the dextranase purified from Streptococcus mutans Ingbritt. Microbiol Immunol 36:969–976
Inoue Y, Yasutake N, Oshima Y, Yamamoto Y, Tomita T, Miyoshi S, Yatake T (2002) Cloning of the maltose phosphorylase gene from Bacillus sp. strain RK-1 and efficient production of the cloned gene and the trehalose phosphorylase gene from Bacillus stearothermophilus SK-1 in Bacillus subtilis. Biosci Biotechnol Biochem 66:2594–2599. doi:10.1271/bbb.66.2594
Kawabata Y, Kitao S, Funane K, Tokashiki T, Gibu S, Miyagi S (2006) Strain improvement of cyclomaltoligosaccharide glucanotransferase (CITase) production in Bacillus circulans by nitrosoguanidine and streptomycin-resistance mutanogenesis. Food Clin Nutr 1:43–48 (in Japanese)
Kawamoto H, Oguma T, Sekine H, Kobayashi M (2001) Immmobilization of cycloisomaltooligosaccharide glucanotransferase for the production of cycloisomaltooligosaccharide from dextran. Enzyme Microb Technol 28:515–521. doi:10.1016/S0141-0229(01)00304-0
Kawamura F, Doi RH (1984) Construction of a Bacillus subtilis double mutant deficient in extracellular alkaline and neutral proteases. J Bacteriol 160:442–444
Kobayashi M, Funane K, Oguma T (1995) Inhibition of dextran and mutan synthesis by cycloisomaltooligosaccharides. Biosci Biotechnol Biochem 59:1861–1865
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi:10.1038/227680a0
Oguma T, Kawamoto H (2003) Production of cyclodextran and its application. Trends Glycosci Glycotechnol 15:91–99
Oguma T, Horiuchi T, Kobayashi M (1993) Novel cyclic dextrins, cycloisomaltooligosaccharides, from Bacillus sp. T-3040 culture. Biosci Biotech Biochem 57:1225–1227
Oguma T, Tobe K, Kobayashi M (1994) Purification and properties of a novel enzyme from Bacillus spp. T-3040, which catalyzes the conversion of dextran to cyclic isomaltooligosaccharides. FEBS Lett 345:135–138. doi:10.1016/0014-5793(94)00418-8
Tsuge K, Matsui M, Itaya M (2003) One step assembly of multiple DNA fragments with a designed order and orientation in Bacillus subtilis plasmid. Nucleic Acids Res 31:e133. doi:10.1093/nar/gng133
Westers L, Westers H, Quax WJ (2004) Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochem Biophys Acta 1694:299–310. doi:10.1016/j.bbamcr.2004.02.011
Yamamoto T, Mukai K, Maruta K, Watanabe H, Yamashita H, Nishimoto T, Kubota M, Chaen H, Fukuda S (2005) Hyper expression of kojibiose phosphorylase gene and trehalose phosphorylase gene from Thermoanaerobacter brockii ATCC35047 in Bacillus subtilis and selaginose synthesis utilizing two phosphorylases. J Biosci Bioeng 100:343–346. doi:10.1263/jbb.100.343
Yamamoto T, Terasawa K, Kim YM, Kimura A, Kitamura Y, Kobayashi M, Funane K (2006) Identification of catalytic amino acids of cyclodextran glucanotransferase from Bacillus circulans T-3040. Biosci Biotechnol Biochem 70:1947–1953. doi:10.1271/bbb.60105
Acknowledgments
This study was supported in part by the project “Collaboration of Industry, University, and Government” (Okinawa Prefecture, Japan) and by a Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (BRAIN, Japan).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kawabata, Y., Kimura, K. & Funane, K. Extracellular production of cycloisomaltooligosaccharide glucanotransferase and cyclodextran by a protease-deficient Bacillus subtilis host–vector system. Appl Microbiol Biotechnol 93, 1877–1884 (2012). https://doi.org/10.1007/s00253-011-3671-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3671-y