
Abstract
The need to develop protein biomanufacturing platforms that can deliver proteins quickly and cost-effectively is ever more pressing. The rapid rate at which genomes can now be sequenced demands efficient protein production platforms for gene function identification. There is a continued need for the biotech industry to deliver new and more effective protein-based drugs to address new diseases. Bacterial production platforms have the advantage of high expression yields, but insoluble expression of many proteins necessitates the development of diverse and optimised refolding-based processes. Strategies employed to eliminate insoluble expression are reviewed, where it is concluded that inclusion bodies are difficult to eliminate for various reasons. Rational design of refolding systems and recipes are therefore needed to expedite production of recombinant proteins. This review article discusses efforts towards rational design of refolding systems and recipes, which can be guided by the development of refolding screening platforms that yield both qualitative and quantitative information on the progression of a given refolding process. The new opportunities presented by light scattering technologies for developing rational protein refolding buffer systems which in turn can be used to develop new process designs armed with better monitoring and controlling functionalities are discussed. The coupling of dynamic and static light scattering methodologies for incorporation into future bioprocess designs to ensure delivery of high-quality refolded proteins at faster rates is also discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Approximately 50% of all new medicines are classified as biopharmaceuticals; the demand for innovative protein-based treatments continues to soar because proteins of the human immune system have shown better efficacy in the fight against disease, compared with chemical-based drugs (Bellott 2010). At the same time, the dawn of the post-genomic era has led to an avalanche of new amino acid sequences waiting to be translated and defined in terms of biological structure–function (Eisenberg et al. 2000; Zhou et al. 2010). Consequently, both industrial and academic research laboratories face increasing pressure to produce pure proteins in sufficiently large quantities at reasonable cost. The recombinant DNA technology platform has been pivotal in allowing proteins to be produced at significant amounts for clinical and structural studies (Shuler and Kargı 2002; Leong and Chen 2008). Escherichia coli is one of the most commercially viable host organisms for protein expression because it offers the advantage of speed, where high growth rates are possible on inexpensive and simple media (Demain and Vaishnav 2009). However, over-expression of heterologous proteins in E. coli often results in the formation of insoluble proteins, more commonly known as inclusion bodies (IBs), due to changes in kinetic competition between folding and aggregation caused by the higher rate of protein synthesis and insufficient supply of chaperones to support correct protein refolding (Vonrhein et al. 1999; Georgiou and Valax 1996). As a matter of fact, only 13% of human-derived proteins expressed in E. coli were found in the soluble form in a previous study (Braun et al. 2002). Therefore, in vitro refolding of these insoluble proteins is necessary to restore biological activity. Although in vitro refolding techniques are well-established, refolding yields are still often sub-optimal due to off-pathway aggregation that competes favourably with the desired unimolecular refolding reaction. Designing a high-yielding refolding-based bioprocess is therefore not a trivial task and can be a time-consuming affair when the search for an optimised refolding environment hinges on trial and error approaches. Therefore, the development of rational approaches for protein refolding that can be robustly applied across a wide range of protein candidates is critically needed. Since aggregate formation in protein refolding is dependent on an additive-modified refolding buffer physicochemical environment, development of refolding analyticals based on in situ monitoring of refolding yield with varying refolding buffer composition will be beneficial to facilitate rapid and rational optimisation of the refolding buffer composition. Until now, the use of trial and error methods to develop refolding recipes prolongs methodology development of in vitro refolding tasks.
In this article, recent efforts to minimise inclusion body formation are first reviewed, followed by a summary on the design rationale of existing protein refolding technologies to increase refolding yield. In the final sections, we discuss how light scattering methodologies can be used for developing rational process designs and recipes for protein refolding. Although light scattering methods are frequently employed by structural biochemists to determine conditions conducive for protein crystallisation, its true potential in protein refolding is yet to be firmly established. Compared to other excellent reviews on protein refolding which have mainly focused on refolding technologies and/or mechanistic studies of buffer additives on protein behaviour, this review presents a systematic investigation of the applicability of light scattering technology in rationally designing refolding buffer compositions and controlling large-scale refolding processes.
Inclusion body formation: can it be avoided?
E. coli is one of the most highly utilized microbial hosts for over-expression of foreign proteins due to its well-characterised genome that eases targeted genetic manipulation to give high protein expression yields. However, despite a three-decade history of use for protein production, there is still a large fraction of proteins that cannot be produced in E. coli in its soluble and hence functional form. There are many hypothesized reasons for insoluble expression, and concerted research efforts to understand protein translation and folding in E. coli have been deployed to address protein insolubility problems. Firstly, over-expression of foreign proteins is believed to overwhelm the bacteria with a high metabolic burden (Hoffmann and Rinas 2004), leading to insufficient supply of chaperones to guide correct folding of the polypeptides. Efforts to co-express endogenous E. coli chaperones such as the DnaK-DnaJ-GrpE chaperones and the GroEL-GroES system during protein expression (Nishihara et al. 1998; Kirschner et al. 2007; de Marco et al. 2007) showed varied levels of success in terms of solubility enhancement for different protein types. The use of chaperones, however, cannot eliminate the occurrence of intracellular proteolytic cleavage, soluble aggregate formation and cellular growth inhibition, which is highly ‘protein sequence’ dependent. Therefore, chaperone co-expression alone cannot guarantee protein solubility (Martínez-Alonso et al. 2010). Secondly, the high proportion of E. coli rare codons in foreign genes can also be problematic. Extensive efforts to optimise rare codon usage in E. coli has helped to improve soluble protein expression (Kleber-Janke and Becker 2000; Burgess-Brown et al. 2008; Chuan et al. 2008b; Zhang et al. 2009), but the success of the codon optimisation strategy is again protein-specific because transcriptional pauses necessary for efficient protein folding in vivo vary with rare codon frequency and composition (Purvis et al. 1987; Krasheninnikov et al. 1988; Marin 2008; Komar 2009). Thirdly, the reducing environment of the bacterial cytoplasm does not promote formation of disulphide bonds necessary for correct folding of the protein. To overcome this problem, new bacterial strains like FA113 and DR473 are engineered to lack thioredoxin and glutathione reductase activities and can now accumulate proteins in the oxidised form (Mansell et al. 2008; Masip et al. 2004).
Manipulation of protein translation rate using low temperature fermentation conditions has been extensively reported, and success has again been highly sequence dependent (Vasina and Baneyx 1997; Fang et al. 1999; Phadtare and Severinov 2005). This outcome is expected considering that protein translation rate decreases in concert with the metabolic rate of the bacteria at reduced temperature, thereby allowing sufficient time for the protein to fold within the bacterial cytoplasm. Fusion protein technology is also commonly used to overcome protein solubility problems and has a higher predictability rate for soluble protein expression than temperature tweaking, especially with the use of fusion tags such as maltose binding protein and thioredoxin A (Bryksa et al. 2006; Pazgier and Lubkowski 2006; Xu et al. 2007; Huang et al. 2008; Huang et al. 2009). However, from a bioprocessing perspective, increased cellular metabolic burden of fusion protein co-expression coupled with inefficient downstream cleavage yields can significantly decrease product yield and increase overall process cost, thus nullifying the effects of increased upstream expression.
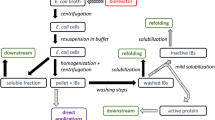
Despite the concerted and intensified efforts to enhance soluble protein expression in E. coli, the ability to “guarantee” soluble protein expression still cannot be achieved. Until now, there seems to be no universal solution to increase protein solubility through any of the abovementioned strategies (Fig. 1). This failure leads naturally to the question of whether tackling the protein insolubility problem in vitro could be the more productive way forward. IB formation can be advantageous in many respects. Apart from the high product yields, resistance towards proteolytic cleavage, minimised product toxicity towards the host cell (Greenshields et al. 2008) and the relative ease of purification of the target protein from other contaminants are strong bioprocessing advantages (Fahnert et al. 2004). Considering that IB formation would continue to feature as an indispensible part of recombinant protein synthesis, the development of rational methods to design protein refolding processes is of critical importance, which forms the basis of this mini-review.

Common strategies to avoid expression of proteins as insoluble IBs and the disadvantages associated with them
Design rationale of refolding methods
To complete the journey from the solubilised (denatured-reduced) state to its native conformation, a protein molecule needs to move down the funnel shaped rugged energy landscape to the lowermost point (Dobson 2003; Radford 2000). At each point within the energy landscape, aggregation-prone protein refolding intermediates remain prone to off-pathway reactions leading to second or higher-ordered aggregation reactions. Hence, the chief objective of any refolding process would be to create an environment that can direct the refolding reactions towards the first-order refolding pathway to form the native product. Unfortunately, since the same physicochemical forces (viz. electrostatic interactions, hydrogen bonding, hydrophobic interactions and disulphide bridging) which are needed to refold a protein are also involved in unproductive inter and intra-molecular protein interactions, protein refolding remains a challenging affair. Intuitively, aggregation can be controlled by manipulating protein concentration during the refolding step and choosing an optimum refolding buffer composition to promote native protein formation (Tsumoto et al. 2003). The design of current refolding strategies has been effective in tackling the concentration problem. Since excellent reviews of the design and operation of different refolding strategies have been published (Middelberg 2002; Jungbauer and Kaar 2007), we will only briefly summarise the rationale of conventional protein refolding strategies in optimising protein concentration for refolding.
Dilution
Refolding by dilution lowers refolding protein concentration to minimise the propensity of inter-molecular interactions of protein refolding intermediates. Despite the low protein concentration, refolding buffer composition still plays an important role in influencing protein refolding kinetics (Leong and Middelberg 2007).
Dialysis
Dialysis is employed in favour of dilution when (a) the starting concentration of the denatured-reduced protein is too low for further dilution into the refolding buffer and/or (b) a complete buffer exchange is required (i.e. no carry-over of denaturants or reducing agents in the refolding buffer). However, dialysis refolding suffers from the disadvantage of protein loss to dialysis membranes, and slow buffer exchange kinetics often can induce aggregation of the refolding intermediates (Leong and Middelberg 2006). The use of multiple step changes in buffer exchange to increase the ‘steepness’ of the folding energy landscape has been successful in improving refolding yields (Tsumoto et al. 2010).
Chromatography
Chromatography-based refolding was developed to improve refolding productivity without compromising refolding yield. Immobilization of protein molecules on chromatographic matrices helps to spatially isolate the proteins from each other (Schmoeger et al. 2010), thereby reducing intermolecular interactions of the protein folding intermediates. Non-native intra-molecular interactions, however, can still promote aggregation of the immobilised protein molecules, but optimal buffer composition can help in maximising the refolding yield (Chen and Leong 2009; Langenhof et al. 2005). The development of chromatographic materials and methods for bioprocess intensification has also been extensively studied (Li et al. 2004; Jungbauer et al. 2004; Kaar et al. 2009; Chen and Leong 2010; Li and Leong 2011; Liu et al. 2009).
High hydrostatic pressure technology
Another way to promote native refolding reactions is to create a refolding environment that can destabilise off-pathway aggregation. High hydrostatic pressure technology was developed with this strategy in mind, where hydrostatic pressure is applied to ‘ensure’ that only proteins refolded to the native form and hence having the lowest specific volumes are stabilised (Malavasi et al. 2011; Seefeldt et al. 2009). Application of pressure can also help to solubilise the IBs directly and refold the target protein spontaneously. However, modulation of hydrogen bonds and disulphide bridges needed for solubilising the IBs and obtaining the correctly refolded product can only be achieved by an optimised buffer system (Qoronfleh et al. 2007).
As evident from Table 1, the design of these refolding methods is focused mainly on controlling refolding concentration to obtain high refolding yields, but optimisation of the physicochemical environment still needs to be independently performed for each of these methods. In recent years, high throughput screening of refolding buffers for protein refolding based on protein solubility has been reported (Vincentelli et al. 2004). Some of these screening kits have been commercialized; for example, Refold Master™ developed by Novexin Ltd (Cambridge, UK) comprises a matrix of different buffer compositions which allows screening of protein foldability in 96-well plate formats. These screening kits, however, do not allow in situ monitoring of the refolding reaction, which can provide important insights into the influence of different refolding additives on refolding yields. The major hurdle for such monitoring is because different proteins require different detection strategies (Table 2) and a generic screening platform is not practically possible. It would, therefore, be considerably more advantageous if new screening methods could be developed to simultaneously provide quantitative information on aggregation kinetics under different refolding environments, thus providing a better correlation between physicochemical parameters and refolding yield for a given protein. In the following sections, we investigate how light scattering methods can be used not only as valuable tools for measuring such data but also to develop rational process designs and recipes for protein refolding.
Light scattering methods: a rational approach to designing protein refolding recipes
The random movement of protein molecules during refolding is influenced by the inter-particle and hydrodynamic interactions amongst them. This random motion leads to fluctuation in the light intensities scattered from the protein molecules in solution when irradiated by an incident light wave. The use of static light scattering to study the behaviour of protein molecules in solution was first reported in the 1990s, where it was concluded that that successful crystallisation takes place only when the second virial coefficient (SVC) of the solutions lie within a specific value (George and Wilson 1994). The ability to quantitatively predict environments conducive for protein crystallisation indicates that the light scattering platform can also be instrumental for studying protein–protein interaction within a refolding system by providing thermodynamic information on protein phase behaviour. Before discussing the applications of light scattering in the rational design of refolding recipes, we will first briefly review the principles of static and dynamic light scattering to set the context of our discussion.
Static light scattering
Static light scattering (SLS) is primarily used to measure the SVC of protein solutions. SVC is a thermodynamic parameter which originates from the virial expansion of solution osmotic pressure (Neal et al. 1998). According to statistical mechanics, the SVC (B 2) can be expressed as an integration of the potential of mean force, w 2(r), between two molecules (Eq. 1):
where N A stands for the Avogadro’s constant, M w is the molar mass, k is the Boltzmann constant and r is the centre-to-centre distance between two molecules (Pan and Glatz 2003). Therefore, SVC takes into account all the interactions that regulate protein phase behaviour such as electrostatic, van der Waals, excluded volume, hydration forces and hydrophobic interactions (Valente et al. 2005). SVC values of protein solutions are determined using a Debye plot according to Eq. 2 (Stockmayer 1950):
where R(θ) is the excess Rayleigh scattering of the protein solution, K is the light-scattering optical constant and c is the protein mass concentration. K depends on the solution’s scattering properties and is defined as:
where λ is the wavelength of the incident light, n is the refractive index of the protein solution and n s is the refractive index of the solvent. Qualitatively, SVC measures two-body interactions in dilute solution conditions (i.e. protein–protein interaction in the case of protein refolding). A positive value indicates a repulsive interaction amongst the protein molecules, implying that protein–solvent interactions are favoured over protein–protein interactions while a negative value indicates the reverse (George and Wilson 1994). Experimentally determined SVC values have thus been established to correlate well with protein solubility (Guo et al. 1999; Ruppert et al. 2001). Since refolding yield is dependent on the extent of aggregation that occurs during refolding and the latter is a function of the physicochemical parameters of the refolding buffer, optimisation of the refolding buffer based on SVC measurements can help in developing rational strategies for designing the optimum refolding buffer recipes.
Dynamic light scattering
Whilst inter-particle interactions can be determined from SVC measurements, hydrodynamic interactions on the other hand can be measured by dynamic light scattering (DLS). Hydrodynamic interactions arise because a moving molecule induces solvent flow and hence exerts viscous forces on diffusing protein molecules nearby (Liu et al. 2005). Thus, when a second protein molecule comes in contact within the same flow field, it experiences attraction or repulsion due to the hydrodynamic interactions. The net interacting forces amongst the protein molecules can be quantitated by DLS using Eq. 4, which indicates that the mutual diffusion coefficient (D m) of solute molecules under a given solvent condition is dependent on protein concentration (Meechai et al. 1999):
where dρ/dc is the ‘protein concentration’-dependent increase of the solution osmotic pressure, indicating the thermodynamic driving force for diffusion, while f m is the concentration-dependent friction factor derived from the resistance to collective translation of particles due to the potential and hydrodynamic interactions amongst them. Expressing the protein solution concentration in terms of volume fraction ϕ, Eq. 4 takes the form shown below (Liu et al. 2005)
where D 0 is the infinite dilution diffusion coefficient while k m is the interaction parameter between the solute particles, obtained from the slope of the normalised diffusivities (D m/D 0) versus volume fraction plots (Grigsby et al. 2000; Mirarefi and Zukoski 2004; Rubin et al. 2010; Liu et al. 2005). Just like the SVC values, attractive or repulsive inter-particle interactions are reflected by negative or positive k m values, respectively. This interaction term is given by Eq. 6:
where k d and k h represent the interaction potential and hydrodynamic interactions between particles, respectively. A detailed interpretation of k d and k h is presented elsewhere (Muschol and Rosenberger 1995), but for the purpose of this review, it is reasonable to consider that k d can be obtained from the following equation (Liu et al. 2005):
where v is the partial specific volume of the protein (i.e. ratio of the volume fraction to the protein concentration). Diffusivity values obtained from the DLS data can also provide information on the hydrodynamic diameter of the refolding protein molecules using the Stoke–Einstein equation (Liu et al. 2005), which gives indications of aggregation kinetics at a given time.
The combination of static and dynamic light scattering data is therefore highly effective in identifying the type of interactions prevalent during protein refolding reactions. As summarised in Fig. 2, SLS data can provide SVC values, which in turn can be used to determine k d (using Eq. 7) for any given system at any given point of time (Fig. 2a). Similarly, DLS data can be used to determine the normalised diffusivities (D m/D 0) to obtain k m values using Eq. 5 (Fig. 2b). Thus, by knowing the values of k d and k m for any given time point, one can easily calculate the corresponding k h values using Eq. 6 (Fig. 2c). The combination of DLS and SLS data can thus provide a vivid picture of the types of interaction encountered by protein refolding intermediates at any given time point, thereby providing vital information necessary that will guide the control of a protein refolding process. In the next section, we discuss how light scattering experiments can be used to design smarter protein refolding processes in the future.

Strategy for quantifying the type of interacting forces amongst the protein refolding intermediates using light scattering data: a slopes from the Debye plots obtained from the SLS experiments, under different environmental conditions, can be used to determine the SVC for the protein solutions (from Velev et al. 1998); b slopes from the diffusivity plots, obtained from DLS experiments, can be used to obtain the k m values for the different protein intermediates at different conditions; c combination of DLS and SLS data helps to determine the hydrodynamic and interaction-potential of the interacting moieties at any given point of time within the protein refolding system (from Liu et al. 2005)
Future directions: using light scattering tools for rational design of refolding processes
Over the past decade, SVC and protein diffusivities have been increasingly used to obtain better insights into protein behaviour under different environmental conditions. Experimentally determined SVC values can shed light on methods to manipulate protein thermodynamic properties and phase behaviour (Curtis and Lue 2006; Rosenbaum and Zukoski 1996) and can even provide partial structural information (Neal et al. 1998). SVC values have thus been used as an unbiased estimator for protein-specific behavioural patterns to guide determination of optimal crystallisation conditions for proteins (Tessier and Lenhoff 2003; Velev et al. 1998). In view of protein refolding applications, SVC values can accurately correlate the relationship between refolding additives and protein interactions (Kulkarni et al. 2000; Kulkarni and Zukoski 2001; Curtis et al. 2002; Liu et al. 2004) and has been exploited to predict (a) the propensity for aggregation during protein refolding (Ho et al. 2003) and (b) potential refolding yields of inclusion body proteins (Ho and Middelberg 2004). Therefore, SVC measurements of refolding mixtures carried out at statistically determined design points can rapidly provide quantitative correlations between the choice and concentration of additives on the interactive behaviour of proteins (Valente et al. 2005; Basu et al. 2011). In addition to SLS, other methods have also been used to determine SVC (Tessier and Lenhoff 2003), amongst which static interaction chromatography has gained considerable popularity both in the conventional chromatography (Valente et al. 2005) and microfluidic device-based platforms (Deshpande et al. 2009; Martin and Lenhoff 2011). However, with the advent of the avalanche photodiode for recording scattered intensity signals, modern light scattering instruments allow fast determination of accurate SLS and DLS data within a single setup (Yadav et al. 2011). By monitoring protein interactions under varying pH, temperature and ionic strengths through diffusivity measurements (Grigsby et al. 2000), it is possible to determine equilibrium association constants (Kuehner et al. 1997), which can be used to predict protein stability within a given refolding environment. Therefore, modern light scattering instruments can be exploited to obtain combined DLS and SLS data to monitor protein inter-particle and hydrodynamic interactions in order to pinpoint the type of forces involved amongst interacting protein molecules during refolding (Prinsen and Odijk 2007).
Therefore, it is reasonable to say that the stage is set to exploit light scattering technologies for better monitoring and controlling of the refolding processes. Although there are a few studies on monitoring protein refolding reactions using SLS or DLS data (Crisman and Randolph 2009; Gast et al. 1997), development of an analytical to control refolding processes using this strategy is yet to be realised. Since DLS has been successfully used to develop titration based strategies for crystallisation of a target protein (Mirarefi and Zukoski 2004), there remains little doubt about the potential of light scattering methods for controlling protein refolding processes as well. The schematic shown in Fig. 3 provides a glimpse of a potential solution for designing a rational protein refolding process, where data from an online light-scattering detector can be fed into a controller that can keep the refolding process under a desired environment (i.e. maintain the critical thermodynamic parameters like the SVC and k m within a specified range). The controller in turn can be loaded with information regarding changes in protein behaviour under different environmental conditions, quantified off-line by monitoring the vital thermodynamic parameters at statistically determined design points. For better quantitative interpretation of light scattering data, it is essential to segregate the different aggregating species within the analysed samples (George and Wilson 1994), and therefore, the presence of a suitable separating system prior to the light-scattering detector can prove beneficial. In this regard, field-flow techniques like the asymmetric flow field-flow fractionation (AF4) which has the capability to segregate molecules based on their diffusivities (Roda et al. 2009) can be relevant. Applicability of AF4 over a wide size range up to 500 nm (Cao et al. 2009) has already made it a potential fractionating methodology for protein bioprocessing (Chuan et al. 2008a; Luo et al. 2006).

A schematic representation of a process design which can allow better controlling of a protein refolding process. Samples from the protein refolding reactor can be fed directly into a light-scattering detector via a suitable separating unit. The data from the detector can be fed directly into a suitable controller that can help to maintain the desired thermodynamic parameters of the ongoing reaction within a specific range
In summary, the process design schematically depicted in Fig. 3 can help to enhance our understanding and control of any refolding process, thereby qualifying it as a potential candidate within the arsenal of process analytical technology (PAT) tools used for protein refolding processes (Read et al. 2010). Therefore, in addition to the dissolved oxygen, currently being used as a PAT tool for this process (Pizarro et al. 2009), SVC and diffusivity data can be highly beneficial for rational design of protein refolding recipes and platforms.
Concluding remarks
To maximise refolding yields, two factors must be optimised: (a) protein concentration and (b) physicochemical environment of the refolding buffer. The design rationale for existing refolding platforms has been mainly targeted at optimising the former factor, while little progress has been made in tool development to rapidly optimise the latter. Huge amounts of information on the impact of different types of additives on the refolding reactions are now available from many earlier studies, from which different refolding buffer systems can be designed. However, the lack of quantitative data on aggregation kinetics to complement solubility data which is often used to infer protein foldability in existing refolding screening methods increases the difficulty in design and scale-up of refolding processes. The use of ‘light scattering’-based analyticals in a high throughput format has the advantage of qualitative screening for solubility as well as quantitative determination of aggregation behaviour based on protein–protein interaction, leading to reduced time to design optimum refolding recipes. The successful development of such tools will underpin protein bioprocessing by significantly reducing the time and cost for developing high-yielding refolding processes and expedite delivery of proteins for a wide range of applications.
References
Anselment B, Baerend D, Mey E, Buchner J, Weuster-Botz D, Haslbeck M (2010) Experimental optimization of protein refolding with a genetic algorithm. Protein Sci 19(11):2085–2095
Basu A, Chen WN, Leong SSJ (2011) A rational design for hepatitis B virus X protein refolding and bioprocess development guided by second virial coefficient studies. Appl Microbiol Biot 90(1):181–191
Bellott E (2010) The rising tide of biotech. Eur Biopharm Rev (Autumn):92–97
Benoit I, Coutard B, Oubelaid R, Asther M, Bignon C (2007) Expression in Escherichia coli, refolding and crystallization of Aspergillus niger feruloyl esterase A using a serial factorial approach. Protein Expres Purif 55(1):166–174
Braun P, Hu Y, Shen B, Halleck A, Koundinya M, Harlow E, LaBaer J (2002) Proteome-scale purification of human proteins from bacteria. Proc Natl Acad Sci USA 99(5):2654–2659
Bryksa BC, MacDonald LD, Patrzykat A, Douglas SE, Mattatall NR (2006) A C-terminal glycine suppresses production of pleurocidin as a fusion peptide in Escherichia coli. Protein Expres Purif 45(1):88–98
Burgess-Brown NA, Sharma S, Sobott F, Loenarz C, Oppermann U, Gileadi O (2008) Codon optimization can improve expression of human genes in Escherichia coli: a multi-gene study. Protein Expres Purif 59(1):94–102
Cao S, Pollastrini J, Jiang Y (2009) Separation and characterization of protein aggregates and particles by field flow fractionation. Curr Pharm Biotechnol 10(4):382–390
Chen Y, Leong SSJ (2009) Adsorptive refolding of a highly disulfide-bonded inclusion body protein using anion-exchange chromatography. J Chromatogr A 1216(24):4877–4886
Chen Y, Leong SSJ (2010) High productivity refolding of an inclusion body protein using pulsed-fed size exclusion chromatography. Process Biochem 45(9):1570–1576
Chuan YP, Fan YY, Lua L, Middelberg APJ (2008a) Quantitative analysis of virus-like particle size and distribution by field-flow fractionation. Biotechnol Bioeng 99(6):1425–1433
Chuan YP, Lua LHL, Middelberg APJ (2008b) High-level expression of soluble viral structural protein in Escherichia coli. J Biotechnol 134(1–2):64–71
Cowan RH, Davies RA, Pinheiro TTJ (2008) A screening system for the identification of refolding conditions for a model protein kinase, p38α. Anal Biochem 376(1):25–38
Crisman RL, Randolph TW (2009) Refolding of proteins from inclusion bodies is favored by a diminished hydrophobic effect at elevated pressures. Biotechnol Bioeng 102(2):483–492
Curtis RA, Lue L (2006) A molecular approach to bioseparations: protein–protein and protein–salt interactions. Chem Eng Sci 61(3):907–923
Curtis RA, Ulrich J, Montaser A, Prausnitz JM, Blanch HW (2002) Protein–protein interactions in concentrated electrolyte solutions. Biotechnol Bioeng 79(4):367–380. doi:https://doi.org/10.1002/bit.10342
Dechavanne V, Barrillat N, Borlat F, Hermant A, Magnenat L, Paquet M, Antonsson B, Chevalet L (2011) A high-throughput protein refolding screen in 96-well format combined with design of experiments to optimize the refolding conditions. Protein Expres Purif 75(2):192–203
de Marco A, Deuerling E, Mogk A, Tomoyasu T, Bukau B (2007) Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol 7:32
Demain AL, Vaishnav P (2009) Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv 27(3):297–306
Deshpande K, Ahamed T, Van Der Wielen LAM, Ter Horst JH, Jansens PJ, Ottens M (2009) Protein self-interaction chromatography on a microchip. Lab Chip—Miniaturisation for Chemistry and Biology 9(4):600–605
Dobson CM (2003) Protein folding and misfolding. Nature 426(6968):884–890
Eisenberg D, Marcotte EM, Xenarios I, Yeates TO (2000) Protein function the post-genomic era. Nature 405(6788):823–826
Ejima D, Ono K, Tsumoto K, Arakawa T, Eto Y (2006) A novel “reverse screening” to identify refolding additives for activin-A. Protein Expres Purif 47(1):45–51
Fahnert B, Lilie H, Neubauer P (2004) Inclusion bodies: formation and utilisation. Adv Biochem Eng/Biotechnol 89:93–142
Fang L, Xia B, Inouye M (1999) Transcription of cpsA, the gene for the major cold-shock protein of Escherichia coli, is negatively regulated at 37°C by the 5′-untranslated region of its mRNA. FEMS Microbiol Lett 176(1):39–43
Gast K, Nöppert A, Müller-Frohne M, Zirwer D, Damaschun G (1997) Stopped-flow dynamic light scattering as a method to monitor compaction during protein folding. Eur Biophys J 25(3):211–219. doi:https://doi.org/10.1007/s002490050033
George A, Wilson WW (1994) Predicting protein crystallization from a dilute solution property. Acta Crystallogr D 50:361–365
Georgiou G, Valax P (1996) Expression of correctly folded proteins in Escherichia coli. Curr Opin Biotech 7(2):190–197
Greenshields AL, Knickle LC, Syvitski R, Douglas SE (2008) Strategies for recombinant expression of small, highly disulphide-bonded, cationic antimicrobial peptides. Protein Peptide Lett 15(9):985–994
Grigsby JJ, Blanch HW, Prausnitz JM (2000) Diffusivities of lysozyme in aqueous MgCl2 solutions from dynamic light-scattering data: effect of protein and salt concentrations. J Phys Chem B 104(15):3645–3650
Guo B, Kao S, McDonald H, Asanov A, Combs LL, Wilson WW (1999) Correlation of second virial coefficients and solubilities useful in protein crystal growth. J Cryst Growth 196(2–4):424–433
Ho JGS, Middelberg APJ (2004) Estimating the potential refolding yield of recombinant proteins expressed as inclusion bodies. Biotechnol Bioeng 87(5):584–592
Ho JGS, Middelberg APJ, Ramage P, Kocher HP (2003) The likelihood of aggregation during protein renaturation can be assessed using the second virial coefficient. Protein Sci 12(4):708–716
Hoffmann F, Rinas U (2004) Stress induced by recombinant protein production in Escherichia coli. Adv Biochem Eng/Biotechnol 89:73–92
Huang L, Ching CB, Jiang R, Leong SSJ (2008) Production of bioactive human beta-defensin 5 and 6 in Escherichia coli by soluble fusion expression. Protein Expres Purif 61(2):168–174
Huang L, Leong SSJ, Jiang R (2009) Soluble fusion expression and characterization of bioactive human beta-defensin 26 and 27. Appl Microbiol Biot 84(2):301–308
Jungbauer A, Kaar W (2007) Current status of technical protein refolding. J Biotechnol 128(3):587–596
Jungbauer A, Kaar W, Schlegl R (2004) Folding and refolding of proteins in chromatographic beds. Curr Opin Biotech 15(5):487–494
Kaar W, Hartmann BM, Fan Y, Zeng B, Lua LHL, Dexter AF, Falconer RJ, Middelberg APJ (2009) Microbial bio-production of a recombinant stimuli-responsive biosurfactant. Biotechnol Bioeng 102(1):176–187
Kirschner A, Altenbuchner J, Bornscheuer UT (2007) Cloning, expression, and characterization of a Baeyer–Villiger monooxygenase from Pseudomonas fluorescens DSM 50106 in E. coli. Appl Microbiol Biot 73(5):1065–1072
Kleber-Janke T, Becker WM (2000) Use of modified BL21(DE3) Escherichia coli cells for high-level expression of recombinant peanut allergens affected by poor codon usage. Protein Expres Purif 19(3):419–424
Komar AA (2009) A pause for thought along the co-translational folding pathway. Trends Biochem Sci 34(1):16–24
Krasheninnikov IA, Komar AA, Adzhubeǐ IA (1988) Rol’ klasterov redkikh kodonov v opredenelenii granits uchastkov polipeptidnoǐ tsepi s odnotipnoǐ vtoprichnoǐ strukturoǐ v protsesse ko-transliatsionnogo svorachivaniia belka [Role of the rare codon clusters in defining the boundaries of polypeptide chain regions with identical secondary structures in the process of co-translational folding of proteins]. Dokl Akad Nauk SSSR 303(4):995–999
Kuehner DE, Heyer C, Rämsch C, Fornefeld UM, Blanch HW, Prausnitz JM (1997) Interactions of lysozyme in concentrated electrolyte solutions from dynamic light-scattering measurements. Biophys J 73(6):3211–3224
Kulkarni A, Zukoski C (2001) Depletion interactions and protein crystallization. J Cryst Growth 232(1–4):156–164
Kulkarni AM, Chatterjee AP, Schweizer KS, Zukoski CF (2000) Effects of polyethylene glycol on protein interactions. J Chem Phys 113(21):9863–9873
Langenhof M, Leong SSJ, Pattenden LK, Middelberg APJ (2005) Controlled oxidative protein refolding using an ion-exchange column. J Chromatogr A 1069(2):195–201
Leong SSJ, Chen WN (2008) Preparing recombinant single chain antibodies. Chem Eng Sci 63(6):1401–1414
Leong SSJ, Middelberg APJ (2006) Dilution versus dialysis: a quantitative study of the oxidative refolding of recombinant human alpha-fetoprotein. Food Bioprod Process 84(1 C):9–17
Leong SSJ, Middelberg APJ (2007) A simplified bioprocess for human alpha-fetoprotein production from inclusion bodies. Biotechnol Bioeng 97(1):99–117
Li X, Leong SSJ (2011) A chromatography-focused bioprocess that eliminates soluble aggregation for bioactive production of a new antimicrobial peptide candidate. J Chromatogr A 1218(23):3654–3659
Li M, Su ZG, Janson JC (2004) In vitro protein refolding by chromatographic procedures. Protein Expres Purif 33(1):1–10
Lin L, Seehra J, Stahl ML (2006) High-throughput identification of refolding conditions for LXRβ without a functional assay. Protein Expres Purif 47(2):355–366
Liu W, Bratko D, Prausnitz JM, Blanch HW (2004) Effect of alcohols on aqueous lysozyme–lysozyme interactions from static light-scattering measurements. Biophys Chem 107(3):289–298
Liu W, Cellmer T, Keerl D, Prausnitz JM, Blanch HW (2005) Interactions of lysozyme in guanidinium chloride solutions from static and dynamic light-scattering measurements. Biotechnol Bioeng 90(4):482–490
Liu X, Du Y, Guo Z, Gunasekaran S, Ching CB, Chen Y, Leong SSJ, Yang Y (2009) Monodispersed MCM-41 large particles by modified pseudomorphic transformation: direct diamine functionalization and application in protein bioseparation. Micropor Mesopor Mat 122(1–3):114–120
Luo J, Leeman M, Ballagi A, Elfwing A, Su Z, Janson JC, Wahlund KG (2006) Size characterization of green fluorescent protein inclusion bodies in E. coli using asymmetrical flow field-flow fractionation-multi-angle light scattering. J Chromatogr A 1120(1–2):158–164
Malavasi NV, Foguel D, Bonafe CFS, Braga CACA, Chura-Chambi RM, Vieira JM, Morganti L (2011) Protein refolding at high pressure: optimization using eGFP as a model. Process Biochem 46(2):512–518
Mansell TJ, Fisher AC, DeLisa MP (2008) Engineering the protein folding landscape in gram-negative bacteria. Curr Protein Pept Sci 9(2):138–149
Marin M (2008) Folding at the rhythm of the rare codon beat. Biotechnol J 3(8):1047–1057
Mark JK, Smith S, Hefford MA (2007) Over-expression and refolding of MAP kinase phosphatase 3. Protein Expres Purif 54(2):253–260
Martin C, Lenhoff AM (2011) Self-interaction chromatography of proteins on a microfluidic monolith. Biochem Eng J 53(2):216–222
Martínez-Alonso M, García-Fruitós E, Ferrer-Miralles N, Rinas U, Villaverde A (2010) Side effects of chaperone gene co-expression in recombinant protein production. Microb Cell Fact 9:64
Masip L, Pan JL, Haldar S, Penner-Hahn JE, DeLisa MP, Georgiou G, Bardwell JCA, Collet JF (2004) An engineered pathway for the formation of protein disulfide bonds. Science 303(5661):1185–1189
Meechai N, Jamieson AM, Blackwell J (1999) Translational diffusion coefficients of bovine serum albumin in aqueous solution at high ionic strength. J Colloid Interf Sci 218(1):167–175
Middelberg APJ (2002) Preparative protein refolding. Trends Biotechnol 20(10):437–443
Mirarefi AY, Zukoski CF (2004) Gradient diffusion and protein solubility: use of dynamic light scattering to localize crystallization conditions. J Cryst Growth 265(1–2):274–283
Muschol M, Rosenberger F (1995) Interactions in undersaturated and supersaturated lysozyme solutions: static and dynamic light scattering results. J Chem Phys 103(24):10424–10432
Neal BL, Asthagiri D, Lenhoff AM (1998) Molecular origins of osmotic second virial coefficients of proteins. Biophys J 75(5):2469–2477
Nishihara K, Kanemori M, Kitagawa M, Yanagi H, Yura T (1998) Chaperone coexpression plasmids: differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl Environ Microb 64(5):1694–1699
Pan X, Glatz CE (2003) Solvent effects on the second virial coefficient of subtilisin and solubility. Crystal Growth and Design 3(2):203–207
Pazgier M, Lubkowski J (2006) Expression and purification of recombinant human α-defensins in Escherichia coli. Protein Expres Purif 49(1):1–8
Phadtare S, Severinov K (2005) Extended-10 motif is critical for activity of the cspA promoter but does not contribute to low-temperature transcription. J Bacteriol 187(18):6584–6589
Pizarro SA, Dinges R, Adams R, Sanchez A, Winter C (2009) Biomanufacturing process analytical technology (PAT) application for downstream processing: using dissolved oxygen as an indicator of product quality for a protein refolding reaction. Biotechnol Bioeng 104(2):340–351
Prinsen P, Odijk T (2007) Collective diffusion coefficient of proteins with hydrodynamic, electrostatic, and adhesive interactions. J Chem Phys 127(11)
Purvis IJ, Bettany AJE, Santiago TC, Coggins JR, Duncan K, Eason R, Brown AJP (1987) The efficiency of folding of some proteins is increased by controlled rates of translation in vivo. A hypothesis. J Mol Biol 193(2):413–417
Qoronfleh MW, Hesterberg LK, Seefeldt MB (2007) Confronting high-throughput protein refolding using high pressure and solution screens. Protein Expres Purif 55(2):209–224
Radford SE (2000) Protein folding: progress made and promises ahead. Trends Biochem Sci 25(12):611–618
Read EK, Park JT, Shah RB, Riley BS, Brorson KA, Rathore AS (2010) Process analytical technology (PAT) for biopharmaceutical products: part I. Concepts and applications. Biotechnol Bioeng 105(2):276–284
Roda B, Zattoni A, Reschiglian P, Moon MH, Mirasoli M, Michelini E, Roda A (2009) Field-flow fractionation in bioanalysis: a review of recent trends. Anal Chim Acta 635(2):132–143
Rosenbaum DF, Zukoski CF (1996) Protein interactions and crystallization. J Cryst Growth 169(4):752–758
Rubin J, Miguel AS, Bommarius AS, Behrens SH (2010) Correlating aggregation kinetics and stationary diffusion in protein-sodium salt systems observed with dynamic light scattering. J Phys Chem B 114(12):4383–4387
Ruppert S, Sandler SI, Lenhoff AM (2001) Correlation between the osmotic second virial coefficient and the solubility of proteins. Biotechnol Prog 17(1):182–187
Scheich C, Niesen FH, Seckler R, Büssow K (2004) An automated in vitro protein folding screen applied to a human dynactin subunit. Protein Sci 13(2):370–380
Schmoeger E, Wellhoefer M, Dürauer A, Jungbauer A, Hahn R (2010) Matrix-assisted refolding of autoprotease fusion proteins on an ion exchange column: a kinetic investigation. J Chromatogr A 1217(38):5950–5956. doi:https://doi.org/10.1016/j.chroma.2010.07.053
Seefeldt MB, Rosendahl MS, Cleland JL, Hesterberg LK (2009) Application of high hydrostatic pressure to dissociate aggregates and refold proteins. Curr Pharm Biotechnol 10(4):447–455
Shuler ML, Kargı F (2002) Bioprocess engineering: basic concepts. Prentice-Hall international series in the physical and Chem Eng Scis, 2nd edn. Prentice Hall, Upper Saddle River
Stockmayer WH (1950) Light scattering in multi-component systems. J Chem Phys 18(1):58–61
Tessier PM, Lenhoff AM (2003) Measurements of protein self-association as a guide to crystallization. Curr Opin Biotech 14(5):512–516
Trésaugues L, Collinet B, Minard P, Henckes G, Aufrère R, Blondeau K, Liger D, Zhou CZ, Janin J, Van Tilbeurgh H, Quevillon-Cheruel S (2004) Refolding strategies from inclusion bodies in a structural genomics project. J Struct Funct Genomics 5(3):195–204
Tsumoto K, Ejima D, Kumagai I, Arakawa T (2003) Practical considerations in refolding proteins from inclusion bodies. Protein Expres Purif 28(1):1–8
Tsumoto K, Arakawa T, Chen L (2010) Step-wise refolding of recombinant proteins. Curr Pharm Biotechnol 11(3):285–288
Valente JJ, Payne RW, Manning MC, Wilson WW, Henry CS (2005) Colloidal behavior of proteins: effects of the second virial coefficient on solubility, crystallization and aggregation of proteins in aqueous solution. Curr Pharm Biotechnol 6(6):427–436
Vasina JA, Baneyx F (1997) Expression of aggregation-prone recombinant proteins at low temperatures: a comparative study of the Escherichia coli cspA and tac promoter systems. Protein Expres Purif 9(2):211–218
Velev OD, Kaler EW, Lenhoff AM (1998) Protein interactions in solution characterized by light and neutron scattering: comparison of lysozyme and chymotrypsinogen. Biophys J75(6):2682–2697
Vincentelli R, Canaan S, Campanacci V, Valencia C, Maurin D, Frassinetti F, Scappucini-Calvo L, Bourne Y, Cambillau C, Bignon C (2004) High-throughput automated refolding screening of inclusion bodies. Protein Sci 13(10):2782–2792
Vonrhein C, Schmidt U, Ziegler GA, Schweiger S, Hanukoglu I, Schulz GE (1999) Chaperone-assisted expression of authentic bovine adrenodoxin reductase in Escherichia coli. FEBS Lett 443(2):167–169
Willis MS, Hogan JK, Prabhakar P, Liu X, Tsai K, Wei Y, Fox T (2005) Investigation of protein refolding using a fractional factorial screen: a study of reagent effects and interactions. Protein Sci 14 (7):1818–1826
Xu X, Jin F, Yu X, Ji S, Wang J, Cheng H, Wang C, Zhang W (2007) Expression and purification of a recombinant antibacterial peptide, cecropin, from Escherichia coli. Protein Expres Purif 53(2):293–301
Yadav S, Scherer TM, Shire SJ, Kalonia DS (2011) Use of dynamic light scattering to determine second virial coefficient in a semidilute concentration regime. Anal Biochem 411(2):292–296
Zhang SM, Fan R, Yang TY, Sun Y, Li JY, Xu QZ, Zhou PK (2009) An improved strategy for efficient expression and purification of soluble HIV-1 tat protein in E. coli. Virol Sin 24(6):518–528
Zhou C, Qi X, Li P, Chen WN, Mouad L, Chang MW, Leong SSJ, Chan-Park MB (2010) High potency and broad-spectrum antimicrobial peptides synthesized via ring-opening polymerization of α-aminoacid-N-carboxyanhydrides. Biomacromolecules 11(1):60–67
Acknowledgements
The authors would like to thank Professor Anton P. Middelberg (University of Queensland) for evaluating the content of the paper and also providing valuable suggestions for its betterment.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Basu, A., Li, X. & Leong, S.S.J. Refolding of proteins from inclusion bodies: rational design and recipes. Appl Microbiol Biotechnol 92, 241–251 (2011). https://doi.org/10.1007/s00253-011-3513-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3513-y