
Abstract
In the present work, Bacillus subtilis was engineered as the cell factory for isobutanol production due to its high tolerance to isobutanol. Initially, an efficient heterologous Ehrlich pathway controlled by the promoter P43 was introduced into B. subtilis for the isobutanol biosynthesis. Further, investigation of acetolactate synthase of B. subtilis, ketol-acid reductoisomerase, and dihydroxy-acid dehydratase of Corynebacterium glutamicum responsible for 2-ketoisovalerate precursor biosynthesis showed that acetolactate synthase played an important role in isobutanol biosynthesis. The overexpression of acetolactate synthase led to a 2.8-fold isobutanol production compared with the control. Apart from isobutanol, alcoholic profile analysis also confirmed the existence of 1.21 g/L ethanol, 1.06 g/L 2-phenylethanol, as well as traces of 2-methyl-1-butanol and 3-methyl-1-butanol in the fermentation broth. Under microaerobic condition, the engineered B. subtilis produced up to 2.62 g/L isobutanol in shake-flask fed-batch fermentation, which was 21.3% higher than that in batch fermentation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Isobutanol has been approved as a platform compound in food industry as well as in pharmaceutical and chemical industry for a long time (Karabektas and Hosoz 2009). Nowadays, it gains much interest as an ideal supplement or a sustainable replacement of gasoline due to its distinguished advantages, such as higher energy density and lower hygroscopicity compared with traditional biofuels (Atsumi et al. 2008; Nielsen et al. 2009).
The current demand for isobutanol is fulfilled by chemical synthesis from syngas, which requires high reaction temperature (350–500°C) and expensive catalyst such as palladium or rhodium (Carlini et al. 2003). Due to general environmental and economic issues, it is desirable to develop biosynthesis process to skip such energy-consumed and uneconomical steps. Vary from ethanol and butanol, which can be naturally produced by Saccharomyces cerevisiae and Clostridia, isobutanol biosynthesis is hampered owing to the lack of an economic native producer. Clostridial species, though they can produce n-butanol, are not ideal because of the relative lack of genetic tools to manipulate their metabolism, their slow growth, their sensitive to oxygen, and so on (Inui et al. 2008; Steen et al. 2008). In recent years, various synthetic metabolic pathways toward target compounds have been devised based on the progress of synthetic biology and metabolic engineering (Liu et al. 2010; Martin et al. 2003; Mukherji and van Oudenaarden 2009), which is holding great hopes for providing solutions to the unmet needs for humankind. In particular, the implementation of isobutanol biosynthesis in Escherichia coli is an excellent exemplar for new energy production by synthetic biological methodology (Atsumi et al. 2008; Zhanga et al. 2008).
However, further enhancement of isobutanol production by E. coli is impeded owing to its lower isobutanol-tolerant threshold (Smith et al. 2010). A more competent host is capable of enduring isobutanol at a high concentration; therefore, it is of great necessity in the long run. Bacillus subtilis is regarded as a great potential workhorse for isobutanol production because of its strong solvent-tolerant capacity (Fischer et al. 2008; Nielsen et al. 2009). Furthermore, it is also advantageous because of its native bio-safety, non-bias in codon usage, clear genetic background, and mature fermentative technology (Kunst et al. 1997; Wong 1995). So far to our knowledge, this organism has never been engineered for isobutanol production, and here in our present work, B. subtilis was engineered for isobutanol production as an attempt.
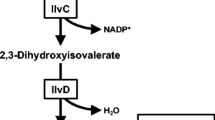
The production of desired chemicals can always be enhanced by overexpressing the precursor pathway, and this has been confirmed in many cases such as in riboflavin and antibiotic biosynthesis (Duan et al. 2010; Thykaer et al. 2010). The 2-ketoisovalerate (KIV) is the important precursor for isobutanol production, which is also shared by valine biosynthesis. KIV biosynthesis begins with the condensation of two pyruvate molecules to 2-acetolactate, which is catalyzed by acetolactate synthase (AlsS; EC 2.2.1.6). And then, KIV is produced through the consecutively catalysis by nicotinamide adenine dinucleotide phosphate (NADPH)-dependent ketol-acid reductoisomerase (IlvC; EC 1.1.86) and dihydroxy-acid dehydratase (IlvD; EC 4.2.1.9; Fig. 1). Previous researches have demonstrated that AlsS of Corynebacterium glutamicum is the key enzyme in KIV biosynthesis pathway because of feed-back inhibition (Leyval et al. 2003). However, the literature on AlsS of B. subtilis is rather limited to its enzymatic properties and regulatory factors (Atsumi et al. 2009; Holtzclaw and Chapman 1975; Renna et al. 1993), rarely mentioned its role in KIV biosynthesis pathway. Therefore, it raises our interest to investigate the position of AlsS in precursor and isobutanol biosynthesis pathways of the engineered B. subtilis.

Schematic representation of heterologous pathway leading to isobutanol formation in B. subtilis (gene names are shown in italics). Red arrows and bold gene names represent the reconstructed isobutanol biosynthesis pathway and the enzymes involved, respectively. PEP phosphoenolpyruvate, PYR pyruvate, OAA oxaloacetate, ACLAC acetolactate, DHVAL 2,3-dihydroxy-3-methylbutanoate, KIV 2-ketoisovalerate, KIC 2-ketoisocaproate, BUT butanol, 2MB 2-methyl-1-butanol, 3MB 3-methyl-1-butanol, PHPYR phenylpyruvate, PHETA 2-phenylethanol, NADPH nicotinamide adenine dinucleotide phosphate (reduced form), PPP pentose phosphate pathway, TCA citrate cycle, BPP biosynthetic 2-ketoisovalerate precursor pathway. Genes ldh, eutD, ydaP, adhB, alsS, ilvC, ilvD, kivd, and adh2 encode lactate dehydrogenase, phosphate acetyltransferase, pyruvate oxidase, alcohol dehydrogenase, acetolactate synthase, ketol-acid reductoisomerase, dihydroxy-acid dehydratase, keto-acid decarboxylase, and alcohol dehydrogenase
In this work, a recombinant B. subtilis for isobutanol biosynthesis was first constructed. Additionally, the important role of AlsS on isobutanol biosynthesis was elucidated, and strain improvement was performed by overexpressing a de novo biosynthetic KIV precursor pathway. Genetic stability, metabolic profile, and fermentation strategy of the engineered B. subtilis were also investigated.
Materials and methods
Regents
All enzymes were purchased from Fermentas Co., Ltd (Glen Burnie, MD, USA). All antibiotics were purchased from Sigma-Aldrich (St. Louis, MO, USA). Oligonucleotides were ordered from Invitrogen Biotechnology Co., Ltd (Carlsbad, CA, USA), and DNA sequencing was served by BGI (Beijing, China).
Bacterial strains, plasmids, and genetic manipulation
The strains and plasmids used in this work are listed in Table 1. E. coli JM109 was used to propagate all plasmids. All Bacillus strains used were derived from B. subtilis 168, which was used as the wild type (WT). All DNA manipulations were carried out using standard techniques (Sambrook and Russell 2001). The transformation of E. coli and B. subtilis were performed by heat shock (Sambrook and Russell 2001) and the competent cell method (Anagnostopoulos and Spizizen 1961), respectively. Primers used for plasmid construction are listed in Table 2.
To engineer isobutanol biosynthesis recombinants, plasmids pHPL(01–04) were constructed. First of all, plasmid pHPL01 was created by blunting the large BsmI-NcoI fragment of pHP13 and then auto-ligated. P43 promoter (GenBank GeneID, 140119566) and alsS gene (GenBank GeneID, 936852) were amplified with two pairs of primers P43-F/P43-R and alsS-F/alsS-R by using B. subtilis genomic DNA (GenBank accession no. NC_000964) as PCR template. The P43 PCR product digested with HindIII and PstI was cloned into pHPL01 plasmid cut with the same enzymes to obtain pHPL02. pHPL03 was constructed by amplifying adh2 (GenBank GeneID, 171020) of S. cerevisiae from its genomic DNA (GenBank accession no. NC_001140) with adh2-F/adh2-R, digesting the PCR product with BamHI and SmaI and ligating into pHPL02 cut with the same enzymes. Similarly, pHPL04 was constructed by amplifying kivd (GenBank GeneID, 51870501) of Lactococcus lactics from its genomic DNA with kivd-F/kivd-R, digesting the PCR product with BamHI and PstI and ligating into pHPL03 cut with the same enzymes.
Plasmid pDGMPKA was used to construct the isobutanol production mutant B. subtilis BSUL02. Before obtaining pDGMPKA, a multiple cloning site (MCS) cut from pUCMCS with HindIII and EcoRI was cloned into the plasmid pDG1730 cut with the same enzymes, creating pDGM. Then PKA (P43::kivd-adh2) was amplified with a pair of primers PKA-F and PKA-R using pHPL04 as a PCR template. The PCR product was digested with SphI and SalI and ligated into pDGM cut with the same enzymes to obtain pDGMPKA.
Plasmids pHPL(05–09) were used to investigate the biosynthetic precursor pathway for isobutanol production. The alsS PCR product digested with XmaI and PvuI was cloned into pHPL02 and pHPL04 cut with the same enzymes, respectively, creating pHPL05 and pHPL06. To clone ilvC (GenBank GeneID, 1019254) and ilvD (GenBank GeneID, 1019249), C. glutamicum genomic DNA (GenBank accession no. NC_006958) was used as a template with two pairs of primers ilvC-F/ilvC-R and ilvD-F/ilvD-R, respectively. The ilvD PCR product was digested with KpnI and PstI and cloned into pHPL02 cut with the same enzymes to obtain pHPL07. Similarly, pHPL08 was obtained by digesting the ilvC PCR product with BglII and PvuI and ligating into pHPL07 cut with the same enzymes. Finally, pHPL09 was obtained by digesting alsS PCR product with AfeI and XhoI and ligating into pHPL08 cut with the same enzymes.
The B. subtilis recombinants were selected by antibiotic screening. The mutants were further confirmed by observing amylase halo, PCR verification, and DNA sequencing with a pair of primers amyE-F and amyE-R.
Medium and cultivation
Unless stated otherwise, all E. coli and B. subtilis strains were cultured in Luria–Bertani (LB) at 37°C. For stepwise construction of isobutanol and biosynthetic precursor pathways, the cultures were grown in 5 mL LB medium in 16 × 200 mm test tubes. To prepare seed cultures, stains stored at −80°C were incubated on LB agar plate for 18 h, and one colony of recombinants was selected and cultivated at 240 rpm. To study the isobutanol biosynthesis behavior and metabolic profile, the seed cultures were diluted 1:100 into 20 mL culture medium (LBGSM-I or LBGSM-II) and cultivated under microaerobic condition (cultured in the screw-cap flask at 240 rpm). LBGSM-I medium was composed of LB medium with 20 g/L glucose, 100 mM potassium phosphate buffer (pH 7.0), and 1,000 dilution of Trace Metal Mix A5 (Atsumi et al. 2008); LBGSM-II medium was prepared by adding another 20 g/L glucose into LBGSM-I medium. For isobutanol fermentation, experiments were carried out in the 1-L flasks under microaerobic condition with a 200-mL work volume and 1% inoculation. LBGSM-II medium was used for batch fermentation, while fed-batch fermentation was initiated with LBGSM-I medium and followed by a glucose addition after 18 h cultivation in order to keep the total glucose concentration equal to that in LBGSM-II medium. The pH values were adjusted to 7.0 by adding 10 M NaOH at 18 h. Antibiotics were added appropriately (erythromycin 0.5 μg/mL, ampicillin 100 μg/mL, and spectinomycin 100 μg/mL).
Isobutanol toxicity tolerance assays
To assay isobutanol toxicity tolerance, B. subtilis and E. coli were cultivated in LB medium at 37°C, while C. glutamicum was cultured in GCIII medium at 30°C (Menkel et al. 1989). When the optical density at 600 nm (OD600) reached to 0.4~0.6, some of the cultures were properly diluted and spread evenly onto corresponding agar plates with varied isobutanol concentration (0%, 0.5%, 1.0%, 1.5%, and 2.0%) for the viable rate calculation after 24 h incubation, and some of the cultures were directly inoculated into the corresponding liquid medium to cultivate for the first 2 h, followed by adding isobutanol at a cascading concentration stated above and kept on cultivating for another 22 h to measure OD600 for the relative growth calculation. The viable rate and the relative growth were defined as below:
Enzymatic activity assays in crude extracts
The crude cell extract was prepared as described previously (Leyval et al. 2003). Quantification of ADH activity was performed by using the method developed by Smith et al. (2010). Total protein concentrations were measured by Bradford (1976) assay.
Real-time PCR analysis of alsS transcription in recombinant B. subtilis
The relative mRNA abundance of alsS of different recombinants was measured by real-time PCR (RT-PCR; Bio-Rad, USA) to further validate its key role in isobutanol biosynthesis pathway. Total RNA was extracted by Trizol reagent (Qiagen, Germany) and then treated with DNase I (Fermentas, USA). Gene gapA (GenBank Gene ID, 938627) was used as internal control. Primers used were prefixed with RT and listed in Table 2. Data analysis was performed according to the comparative C T method (Livak and Schmittgen 2001).
Genetic stability assays
Genetic stability and assessment were performed as described (Bi et al. 2009). The strain retaining both erythromycin and spectinomycin resistance was cultured for ten generations to evaluate the stability of isobutanol biosynthesis.
Analytical methods
Metabolite identification by gas chromatography–mass spectrum
Alcohol compounds produced by our strains were identified by gas chromatography (GC)–mass spectrum (6890N GC System and 5975C mass selective detector, Agilent Technologies, USA) with a DB-5ms capillary column (30 m, 0.25-mm internal diameter, 0.25-μm film thickness, Agilent Technologies, USA). The supernatant was harvested by centrifugation at 10,000×g for 10 min, and alcohols were extracted by vortex mixing 500 μL supernatant with 300 μL GC standard grade chloroform for 2 min and then centrifuged for 5 min to obtain the sample-containing chloroform for analysis. A 1-μL sample was injected for analysis. The operating conditions were performed as described previously (Atsumi et al. 2008).
Metabolite quantified by gas chromatography–flame ionization detector and high-performance liquid chromatography
The alcohol compounds were quantified by gas chromatography–flame ionization detector (7890A G3440A, Agilent Technologies, USA) equipped with PEG-20M capillary column (30 m, 0.32-mm internal diameter, 0.25-μm film thickness, Shanghai Kechung Chromatograph Instruments Co., Ltd, China). The oven temperature was programmed to maintain at 60°C for 1 min, then increased at a rate of 10°C/min to 110°C, further at a rate of 30°C/min to 230°C, and maintained for 2 min. Injector and detector temperatures were both kept on 230°C. Other fermentation products were determined by high-performance liquid chromatography (1200, Agilent Technologies, USA) equipped with a Zorbax SB-C18 column (250 × 4.6 mm, Agilent Technologies, USA) and a UV detector (210 nm). The mobile phase of 5 mM H2SO4 at a 0.3-mL/min flow rate was adopted. The working temperature of the column was kept on 30°C.
OD600, cell dry weight, and glucose concentration measurement
The OD600 was measured using a microplate reader (Model 550, Bio-Rad, USA). Cell dry weight (CDW) was measured by filtering the cell suspension with a filter and drying the filter paper and cells to a constant weight for 24 h at 105°C. The linear relationship between CDW and OD600 was obtained with a conversion factor of 0.325. Glucose concentration in culture broth was determined enzymatically by a bioanalyzer (SBA-40C, Shandong, China).
Results
Isobutanol toxicity tolerance assay
B. subtilis can withstand 1-butanol very well (Fischer et al. 2008), while little was known about its isobutanol tolerance. In the present study, the isobutanol toxicity tolerance of B. subtilis was first evaluated by exposing the organism to this solvent at different concentrations. Additionally, the isobutanol production host E. coli (Atsumi et al. 2008) and the latest reported C. glutamicum (Smith et al. 2010) were selected as control. For agar plate cultivation, both B. subtilis and C. glutamicum grew well when isobutanol concentration was less than 1%, and the viable rate was 56.0% and 55.7%, respectively, higher than 30.4% of E. coli. B. subtilis exhibited the highest toxicity tolerance under high isobutanol concentration (2%), and the viable rate was 2.5-fold of C. glutamicum and 20-fold of E. coli, respectively. The similar results were observed in liquid cultivation. Though C. glutamicum and E. coli showed a little higher relative growth (94.6% and 89.5%, respectively) than B. subtilis (82.5%) at 0.5% isobutanol concentration, B. subtilis exhibited a higher relative growth value in the presence of 2% isobutanol, which was 1.9-fold of C. glutamicum and 3.8-fold of E. coli, respectively (Fig. 2). These results showed a beneficial characteristic of B. subtilis of being a cell factory for isobutanol production.

Comparison of isobutanol toxicity tolerance of B. subtilis, C. glutamicum, and E. coli by exposure to isobutanol. Data were expressed as average values and standard deviations of three parallel studies
Engineering B. subtilis for isobutanol production by constructing an efficient heterologous Ehrlich pathway
For the initial attempt to engineer the B. subtilis cell factory for isobutanol production, the pathway capable of converting KIV to isobutanol should be constructed first. Atsumi et al. (2009) showed that the E. coli strain overexpressing alsS of B. subtilis without keto-acid decarboxylase (KDC; EC 4.1.1.72) was still able to produce isobutanol, demonstrating that AlsS functions as both 2-ketoisovalerate decarboxylase (Kdc activity) and AlsS (Als activity). Additionally, the enzyme activity of native alcohol dehydrogenase (ADH; EC 1.1.1.1) to isobutyraldehyde was 54.50 ± 2.44 U/mg in WT in the present work. However, the fact that isobutanol could not be detected in WT might ascribe to the inadequate ADH activity for isobutanol biosynthesis. Therefore, adh2 from S. cerevisiae was expressed heterologously under the control of a strong promoter P43 of B. subtilis to ensure the efficiency. The ADH activity of the resulted recombinant was 94.54 ± 1.98 U/mg, approximately doubled than that of WT, confirming that both P43 and heterogeneous ADH function well. Unfortunately, the fact that isobutanol could not be detected in this recombinant probably ascribed to the insufficient Kdc activity of native AlsS. This assumption was confirmed by further overexpression of kivd from L. lactics. As we expected, the resulted strain BSUL01-2 could accumulate 0.69 ± 0.05 g/L isobutanol (Table 3).
To increase the genetic stability of recombinant, the heterologous Ehrlich pathway was then inserted into the chromosome of WT (Fig. 3). By using the integration vector pDGMPKA (Table 1) with a neutral site amyE for the aim of eliminating the polar effect caused by gene disruption and insertion, the resulted strain BSUL02-1 produced 0.57 ± 0.06 g/L isobutanol (Table 3).

Schematic diagram of the construction of recombinant BSUL02-1. The heterologous Ehrlich pathway (P43::kivd-adh2) with a spectinomycin cassette was inserted into amyE locus of B. subtilis chromosome
BSUL02-1 with single copy of Ehrlich pathway (integration into the chromosome) produced less isobutanol than BSUL01-2 with multi-copy (without integration; Table 3). Simultaneously, BSUL02-1 showed a longer exponential phase and lower specific growth rate (Fig. 4). That was probably due to the imbalance of KDC, ADH, and AlsS accumulation in vivo, as KDC and ADH were expressed constitutively while the synthesis of AlsS was regulated by many factors (Renna et al. 1993). For that speculation, alsS was constitutively overexpressed and the resulted recombinants BSUL02-2 and BSUL01-3 produced almost equivalent isobutanol (Table 3).

Comparison of the isobutanol biosynthesis and cell growth between BSUL01-2 and BSUL02-1 in LBGSM-I medium cultured for 30 h. The solid line, dash line, and dot line represent cell growth (OD600), isobutanol production, and glucose concentration, respectively. Data were expressed as average values and standard deviations of three parallel studies
Strain improvement by overexpressing a de novo biosynthetic KIV precursor pathway
It was observed that isobutanol production was unexpectedly enhanced after overexpression of AlsS (Table 3). AlsS of C. glutamicum has been identified as the key enzyme in valine biosynthesis (Leyval et al. 2003), whereas in B. subtilis, little was reported about its role in isobutanol biosynthesis that shard the same KIV precursor with valine biosynthesis. For this limitation, AlsS, IlvC, and IlvD involved in the biosynthetic precursor pathway were investigated. Compared with the parental strain BSUL02-1, BSUL02-3 with IlvD and IlvC overexpression increased isobutanol production to 1.1-fold, while BSUL02-2 with only AlsS overexpression elevated the production to 2.2-fold. BSUL03 harboring the de novo biosynthetic precursor pathway (P43::ilvD-ilvC-alsS) accumulated 1.58 ± 0.21 g/L isobutanol, which was 2.8-fold of BSUL02-1 and 2.5-fold of BSUL02-3, respectively, whereas only 1.3-fold of BSUL02-2 (Table 3). These results implied that AlsS was an important enzyme for KIV precursor biosynthesis. Then it was validated by analyzing the relative mRNA abundance of alsS with RT-PCR. Taking gapA gene as internal standard, it was clearly observed that the transcription levels of alsS in BSUL02-2 and BSUL03 were much higher than those in BSUL02-1 and BSUL02-3 (Fig. 5) and presented an approximately equal level. These results further confirmed that it was AlsS rather than IlvC or IlvD that played a more significant role in isobutanol biosynthesis.

Effect alsS in biosynthetic precursor pathway on isobutanol production and its transcriptional analysis. Both isobutanol production and the relative mRNA abundance of alsS were represented by column. Plus symbols denote overexpression of the indicated gene(s) or pathway(s) in recombinant B. subtilis. Data were expressed as average values and standard deviations of three parallel studies
Genetic stability analysis of the engineered BSUL03
Genetic stability is important for engineered strains in industrial production. Analysis showed that plasmid pHPL08 transformant was quite stable in BSUL03. The percentages of cells cultivated without antibiotic selection pressure retained erythromycin resistance were as follows: 100 ± 2.2% after five generations and 97.5 ± 9.4% after ten generations. In addition, all the progeny still retained spectinomycin resistance with the percentage of 100 ± 3.2% after ten generations indicated a stable homologous recombination in BSUL03. Strain with both erythromycin and spectinomycin resistance was cultured for ten generations, and a stable isobutanol production of 1.73 ± 0.11 g/L was observed. These observations suggested that BSUL03 was genetic stable for isobutanol production.
Isobutanol biosynthesis and metabolic profile analysis of recombinant B. subtilis
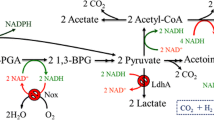
To investigate the isobutanol biosynthesis and metabolic profile, WT and recombinants were cultured under microaerobic condition. All the recombinants showed less biomass than WT after 40 h growth, especially the strains with the Ehrlich pathway genes encoded in multi-copy plasmid. Recombinants with only alsS overexpression (BSUL01-3 and BSUL02-2) enhanced isobutanol production by 95.7% and 132.5% than their parental strains. BSUL03 harboring the de novo biosynthetic precursor pathway produced the highest isobutanol production of 2.02 ± 0.26 g/L (Table 4). In addition to isobutanol, other branched-chain higher alcohols were analyzed. 2-Phenylethanol (PHETA) existed in all recombinants, while 2-methyl-1-butanol (2MB) and 3-methyl-1-butanol (3MB) were only detected with trace amount in alsS overexpressed recombinants. Acetate, lactate, and ethanol were detected as major by-products in fermentation broth. In contrast to the stable production of ethanol in all strains, the production of acetate and lactate varied from strains with alsS overexpression or not. For BSUL01-2 and BSUL01-3, the decrease of acetate and lactate were 109% and 14%, respectively, while the values were 34% and 24% for BSUL02-1 and BSUL02-2. Therefore, acetate and lactate could be regarded as potential reconstruction targets for further strain improvement (Ji et al. 2008).
Isobutanol fermentation of BSUL03 by different fermentation strategies
To select a suitable fermentation process for the best isobutanol producer BSUL03, both batch and fed-batch fermentation strategies were investigated. The experiments were carried out in 1 L shake flasks under microaerobic condition. In fed-batch fermentation, though the cell grew with a slower specific rate of 0.16 h−1, isobutanol was produced at a faster production rate of 0.086 g/L/h; comparatively, the values were 0.27 h−1 and 0.073 g/L/h in batch fermentation, respectively. Besides, the biomass of fed-batch fermentation was 14.8% higher than that of batch fermentation. At the end of fermentation, the maximal isobutanol titer was up to 2.62 ± 0.19 g/L at 48 h, which was 21.30% higher than that in batch fermentation at 42 h (Fig. 6). In addition, 8.12 ± 1.11 g/L residual glucose, 5.32 g/L acetate, and 6.35 g/L lactate were detected in batch fermentation broth, which was 1.3-fold, 1.7-fold, and 1.3-fold of those in fed-batch fermentation, respectively (Fig. 6). The higher concentration of residual glucose, acetate, and lactate might explain the lower isobutanol production rate and final titer due to the lower conversion ratio in batch fermentation.

Isobutanol biosynthesis and metabolic profile of BSUL03 by two different fermentation strategies. Black circle batch fermentation, white circle fed-batch fermentation; a black line CDW, dash line isobutanol, blue line glucose; b black line lactate, dash line acetate. Data were expressed as average values and standard deviations of three parallel studies
Discussion
Isobutanol attracts intensive interest as a promising biofuel for its notable advantages and has recently been biosynthesized in the engineered E. coli (Atsumi et al. 2008). However, B. subtilis with a much higher solvent-tolerant capacity can be considered as a preferable cell factory for isobutanol production.
The isobutanol toxicity tolerance of B. subtilis, E. coli, and C. glutamicum was compared in the present work, and results showed that B. subtilis exhibits a higher tolerance to isobutanol than the others (Fig. 2). The reasons might be as follows: On the one hand, the thick cell wall of B. subtilis protects the cell against the external stresses such as pressure and hyperosmotic shock (Hayhurst et al. 2008), which is important for a cell to keep alive; on the other hand, a large group of nonspecific stress proteins, which are mediated by the general stress σ B factor and induced together by environmental stress, possess an essential protective function on cell survival (Marles-Wright et al. 2008; Petersohn et al. 2001). These protective mechanisms are crucial for B. subtilis to stand against high isobutanol concentration, which will be a distinct advantage of B. subtilis once isobutanol titer reaches the threshold of the present producers.
Previous and our present work demonstrated the existence of ADH encoded by adhB in WT (Kunst et al. 1997) and its enzyme activity to isobutyraldehyde (54.50 ± 2.44 U/mg), respectively. Hence, isobutanol can be biosynthesized theoretically since the native AlsS possesses Kdc activity to convert KIV to isobutyraldehyde (Atsumi et al. 2009). However, the failure of isobutanol detection in WT and recombinant BSUL01-1 with adh2 overexpression (Table 3) might ultimately ascribe to the low affinity of the native AlsS to isobutyraldehyde, whose K m value is 300 mM in vitro (Atsumi et al. 2009), approximately 158 times of that of L. lactic (1.9 mM; de la Plaza et al. 2004). Therefore, the heterologous Ehrlich pathway was constructed by putting kivd and adh2 in series under the control of P43, which is a well-characterized strong promoter of B. subtilis and has been used to strengthen the gene expression (Wang and Doi 1984). By introducing this heterologous Ehrlich pathway into B. subtilis, isobutanol could be detected in all recombinants. Simultaneously, it was interesting to find that the strains with single copy of the Ehrlich pathway showed a longer exponential phase but lower isobutanol production in comparison with those with multi-copy (Table 3). This is plausible as AlsS responsible for KIV formation is synthesized in detectable quantities only in stationary-phase cultures (Renna et al. 1993), and it was observed that 80–90% of isobutanol was accumulated in this period in the present work (Fig. 4), whereas the discrepancies between strains just mentioned were vanished after overexpressing the alsS gene constitutively (Table 3), further indicating a concernful influence of AlsS on isobutanol biosynthesis. Additionally, it was worth noting that isobutanol production was greatly enhanced beyond our expectation in alsS overexpressed recombinants, which dropped the hint of the significant role of AlsS for isobutanol biosynthesis.
In view of the potential importance of AlsS stated above, it was attractive to investigate the AlsS and the other enzymes involved in KIV precursor pathway. Overexpressing alsS, ilvC, and ilvD individually and together showed that IlvC and IlvD had little influence on isobutanol production (Table 3), which highlighted the importance of AlsS. Moreover, the obvious dependence of isobutanol production on alsS expression level showed by transcriptional analysis further validated the dominated role of AlsS in the KIV precursor biosynthesis pathway of B. subtilis (Fig. 5), which consisted with the results elucidated in several microorganisms (Eggeling et al. 1987; Leyval et al. 2003). To enhance isobutanol production, a de novo biosynthetic KIV precursor pathway was constructed. It was important to retain the native AlsS because of its high affinity to pyruvate (Gollop et al. 1990). Additionally, IlvC and IlvD from C. glutamicum were also selected as they exhibit the highest affinities for their respective substrate (the K m values of both of them are around 1 mM) among all the bacteria so far as we known at pH 7.3 and pH 8.0, respectively, which are close to the optimal pH for native AlsS (Chang et al. 2009; Leyval et al. 2003). However, it has to point out that the present work did not investigate the enzyme activities of native IlvC and IlvD. Recombinant BSUL03 harboring the de novo biosynthetic KIV precursor pathway produced 2.8-fold isobutanol of its parent BSUL02-1 (Table 3), which demonstrated the great importance of precursor supply on production enhancement of the desired chemicals (Thykaer et al. 2010). Inspired by these results, other approaches such as promoting the enzyme activity by directed evolution can be adopted to enhance precursor biosynthesis and further improve isobutanol production.
The metabolic profiles of recombinants were investigated under microaerobic condition. In addition to isobutanol, both 2MB and 3MB were detected with trace amount, whereas to our surprise, at least 1.06 g/L PHETA was existed in every recombinant (Table 4). PHETA is widely used in food and cosmetic industry, and its biosynthesis mainly relies on yeast at present (Etschmann and Schrader 2006). Since other PHETA producers are less productive, such as E. coli, which produces at most 0.89 g/L PHETA by adding 8 g/L phenylpyruvate precursor (Atsumi et al. 2008), and C. glutamicum possesses a weak biosynthesis ability with the production of 0.11 g/L PHETA (Smith et al. 2010), B. subtilis is considered as a promising PHETA producer succeeding to yeast. By employing B. subtilis, PHETA can be biosynthesized as the main product after further strain development, or be produced as a major byproduct that can be easily separated from isobutanol. It was also observed that ethanol production in all B. subtilis strains was stable, a litter higher than PHETA (Table 4). Previous work demonstrated that ethanol, which is formed by acetyl-CoA come from pyruvate, was another product in addition to the major anaerobic fermentation products acetate and lactate (Cruz-Ramos et al. 2000; Nakano et al. 1997). These results indicated that during isobutanol fermentation, B. subtilis underwent the mixed acid–butanediol fermentation style. Acetate and lactate were produced at a high concentration in fermentation broth under microaerobic condition, which was not only a waste of carbon source but also a hindrance for cell growth. Usually, it is preferred to increase the conversion of carbon substrates to desired products by deleting the mixed acid biosynthesis pathway (Jian et al. 2010; Unrean et al. 2010). In B. subtilis, it was reported that 80% acetate could be reduced by alsS overexpression with the desired product increased by 50% (Zhu et al. 2007). Lactate was also demonstrated as an important competitor with acetolactate to pyruvate available (Romer-Garcia et al. 2009). In accordance with those facts, our results (Table 4) indicated a redistribution of carbon flux by alsS overexpression, which led to an increase of pyruvate availability to KIV biosynthesis. This strategy might also provide a positive signal for menp1 promoter and coordinate the activation of TCA cycle and formation of the respiratory chain (Qin and Taber 1996) and further influenced the primarily energy metabolism that was important for isobutanol biosynthesis because of its dependence on adenosine triphosphate and NADP(H) supply. It was observed that lactate did not decrease so sharply as acetate in the present work, which probably owing to the discrepancy of K m value of different enzymes to pyruvate: 3.0 mM of lactate dehydrogenase (Garvie 1980) and 13.6 mM of AlsS (Atsumi et al. 2009). Therefore, acetolactate was greatly competed by lactate for pyruvate consumption, and it was helpful to increase isobutanol production by inactivating lactate biosynthetic pathway. For instance, isobutanol production was increased by ~25% to 4.9 g/L by constructing an isobutanol-producing C. glutamicum in a ΔpycΔldh background (Smith et al. 2010).
The suitable fermentation process was also surveyed by employing different strategies in this work. Compared with batch fermentation, the isobutanol production was increased by 21.3% and up to 2.62 g/L in fed-batch fermentation, while a 36.6% and 23.9% reduction of acetate and lactate were observed simultaneously. A slower decrease of pH value was observed in fed-batch fermentation than that in batch fermentation (data not shown, the pH value at 18 h was 6.17 in fed-batch fermentation while 4.82 in batch fermentation), as by intermittent substrate feeding, the cell growth could be well regulated to avoid the broth overacidification. Therefore, the possible reasons for the higher isobutanol production were: (1) the avoidance of overacidification kept a relative favorable pH value for the enzymes responsible for isobutanol production and therefore raised the efficiency of bio-catalysis and (2) the reduction of mixed acid resulted in the increase of pyruvate availability to KIV and therefore enhanced isobutanol production. Further, isobutanol production can be effectively improved by different fed-batch operational strategies such as constant substrate feeding rate and pH control (Gordillo et al. 1998; Mu et al. 2009).
Despite some achievements have been made in engineering B. subtilis as a promising cell factory for isobutanol production, there are still many difficulties to be dealt with, such as how to devise an efficient biosynthesis pathway and how to direct the carbon flux. On the one hand, it is important to explore and screen enzymes with high catalytic activity to further increase the isobutanol biosynthesis efficiency. For instance, adh2 from S. cerevisiae is used to encode ADH for branched-chain higher alcohols production (Atsumi et al. 2008; Connor and Liao 2008), while Atsumi et al. (2010) confirmed that ADH encoded by yqhD or adhA from E. coli is better than adh2 for isobutanol production. On the other hand, it is necessary to decrease the by-products as much as possible, which can prevent the inhibition to cell growth and direct the carbon flux toward the target effectively. By blocking ethanol formation, Ji et al. (2010) improved 2,3-butanediol to 130 g/L in Klebsiella oxytoca, and this was also worked for isobutanol production improvement (Atsumi et al. 2008). At present, with the development of metabolic engineering and computational biology (Hädicke and Klamt 2010; Leonard et al. 2009), strains can be improved by rational design, which is based on the genome-scale network that contains comprehensive relationships of genes, proteins, and reactions. Over the last few years, a number of approaches have been proposed for the phenotype simulation of microorganisms under different environmental and genetic conditions. An in silico aided metabolic engineering strategy is popular because of its accurate identification of target genes that lead to production enhancement, which saves lots of time to obtain an ideal strain. Strain improvement by in silico design based on genome-scale network has been well applied to E. coli (Feist et al. 2010) and S. cerevisiae (Brochado et al. 2010), which sets a good example to further optimize the B. subtilis cell factory for isobutanol production.
References
Anagnostopoulos C, Spizizen J (1961) Requirements for transformation in Bacillus subtilis. J Bacteriol 81:741–746
Atsumi S, Hanai T, Liao JC (2008) Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451:86–89
Atsumi S, Li Z, Liao JC (2009) Acetolactate synthase from Bacillus subtilis serves as a 2-ketoisovalerate decarboxylase for isobutanol biosynthesis in Escherichia coli. Appl Environ Microbiol 75:6306–6311
Atsumi S, Wu T-Y, Eckl E-M, Hawkins SD, Buelter T, Liao JC (2010) Engineering the isobutanol biosynthetic pathway in Escherichia coli by comparison of three aldehyde reductase/alcohol dehydrogenase genes. Appl Microbiol Biotechnol 85:651–657
Bi CH, Zhang XL, Inogram LO, Prestson JF (2009) Genetic engineering of Enterobacter asburiae strain JDR-1 for efficient production of ethanol from hemicellulose hydrolysates. Appl Environ Microbiol 75:5743–5749
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brochado AR, Matos C, MØller BL, Hansen J, Mortensen UH, Patil KR (2010) Improved vanillin production in baker’s yeast through in silico design. Microb Cell Fact 9:84. doi:10.1186/1475-2859-9-84
Carlini C, Macinai A, Marchionna M, Noviello M, Galletti AMR, Sbrana G (2003) Selective synthesis of isobutanol by means of the Guerbet reaction: part 3: methanol/n-propanol condensation by using bifunctional catalytic systems based on nickel, rhodium and ruthenium species with basic components. J Mol Catal A Chem 206:409–418
Chang A, Scheer M, Grote A, Schomburg I, Schomburg D (2009) BRENDA, AMENDA and FRENDA the enzyme information system: new content and tools in 2009. Nucleic Acid Res 37:588–592
Connor MR, Liao JC (2008) Engineering of an Escherichia coli strain for the production of 3-methyl-1-butanol. Appl Environ Microbiol 74:5769–5775
Cruz-Ramos H, Hoffmann T, Marino M, Nedjari H, Presecan-Siedel E, Dreesen O, Glaser P, Jahn D (2000) Fermentative metabolism of Bacillus subtilis: physiology and regulation of gene expression. J Bacteriol 182:3072–3080
de la Plaza M, Fernández de Palencia P, Peláez C, Requena T (2004) Biochemical and molecular characterization of alpha-ketoisovalerate decarboxylase, an enzyme involved in the formation of aldehydes from amino acid by Lactococcus lactis. FEMS Microbiol Lett 238:367–374
Duan YX, Chen T, Chen X, Zhao XM (2010) Overexpression of glucose-6-phosphate dehydrogenase enhances riboflavin production in Bacillus subtilis. Appl Microbiol Biotechnol 85:1907–1914
Eggeling I, Cordes C, Eggeling L, Sahm H (1987) Regulation of acetohydroxy acid synthase in Corynebacterium glutamicum during fermentation of α-ketobutyrate to L-isoleucine. Appl Microbiol Biotechnol 25:346–351
Etschmann MMW, Schrader J (2006) An aqueous-organic two-phase bioprocess for efficient production of the natural aroma chemicals 2-phenylethanol and 2-phenylethylacetate with yeast. Appl Microbiol Biotehcnol 71:440–443
Feist AM, Zielinski DC, Orth JD, Schellenberger J, Herrgard MJ, Palsson BØ (2010) Model-driven evaluation of the production potential for growth-coupled products of Escherichia coli. Metab Eng 12:173–186
Fischer CR, Klein-Marcuschamer D, Stephanopoulos G (2008) Selection and optimization of microbial hosts for biofuels production. Metab Eng 10:295–304
Garvie EI (1980) Bacterial lactate dehydrogenase. Microbiol Rev 44:106–139
Gollop N, Damri B, Chipman DM, Barak Z (1990) Physiological implications of the substrate specificities of acetohydroxy acid synthases from varied organisms. J Bacteriol 172:3444–3449
Gordillo MA, Sanz A, Sánchez A, Valero F, Montesinos JL, Lafuente J, Solà C (1998) Enhancement of Candida rugosa lipase production by using different control fed-batch operational strategies. Biotechnol Bioeng 60:156–168
Guérout-Fleury AM, Frandsen N, Stragier P (1996) Plasmids for ectopic integration in Bacillus subtilis. Gene 180:57–61
Hädicke O, Klamt S (2010) CASOP: a computational approach for strain optimization aiming at high productivity. J Biotechnol 147:88–101
Haima P, Bron S, Venema G (1987) The effect of restriction on shotgun cloning and plasmid stability in Bacillus subtilis Marburg. Mol Gen Genet 209:335–342
Hayhurst EJ, Kallas L, Hobbs JK, Foster SJ (2008) Cell wall peptidoglycan architecture in Bacillus subtilis. P Natl Acad Sci USA 105:14603–14608
Holtzclaw WD, Chapman LF (1975) Degradative acetolactate synthase of Bacillus subtilis: purification and properties. J Bacteriol 121:917–922
Inui M, Suda M, Kimura S, Yasuda K, Suzuki H, Toda H, Yamamoto S, Okino S, Suzuki N, Yukawa H (2008) Expression of Clostridium acetobutylicum butanol synthetic genes in Escherichia coli. Appl Microbiol Biotechnol 77:1305–1316
Ji XJ, Huang H, Li S, Du J, Lian M (2008) Enhanced 2,3-butanediol production by altering the mixed acid fermentation pathway in Klebsiella oxytoca. Biotechnol Lett 30:731–734
Ji XJ, Huang H, Zhu JG, Ren LJ, Nie ZK, Du J, Li S (2010) Engineering Klebsiella oxytoca for efficient 2,3-butanediol production through insertional inactivation of acetaldehyde dehydrogenase gene. Appl Microbiol Biotechnol 85:1751–1758
Jian J, Zhang SQ, Shi ZY, Wang W, Chen GQ, Wu Q (2010) Production of polyhydroxyalkanoates by Escherichia coli mutants with defected mixed acid fermentation pathways. Appl Microbiol Biotechnol 87:2247–2256
Karabektas M, Hosoz M (2009) Performance and emission characteristics of a diesel engine using isobutanol–diesel fuel blends. Renew Energy 34:1554–1559
Karmazyn-Campelli C, Fluss L, Leighton T, Stragier P (1992) The spoIIN279(ts) mutation affects the FtsA protein of Bacillus subtilis. Biochimie 74:689–694
Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero G, Bessières P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Nm C, Choi SK, Codani JJ, Connerton IF, Cummings NJ, Daniel RA, Denizot F, Devine KM, Düsterhöft A, Ehrlich SD, Emmerson PT, Entian KD, Errington J, Fabret C, Ferrari E, Foulger D, Fritz C, Fujita M, Fujita Y, Fuma S, Galizzi A, Galleron N, Ghim SY, Glaser P, Goffeau A, Golightly EJ, Grandi G, Guy BJ, Haga K, Haiech J, Harwood CR, Hénaut A, Hilbert H, Holsappel S, Hosono S, Hullo MF, Itaya M, Jones L, Joris B, Karamata D, Kasahara Y, Klaerr-Blanchard M, Klein C, Kobayashi Y, Koetter P, Koningstein G, Krogh S, Kumano M, Kurita K, Lapidus A, Lardinois S, Lauber J, Lazarevic V, Lee SM, Levine A, Liu H, Masuda S, Mauël C, Médigue C, Medina N, Rp M, Mizuno M, Moestl D, Nakai S, Noback M, Noone D, O’Reilly M, Ogawa K, Ogiwara A, Oudega B, Park SH, Parro V, Pohi TM, Portetelle D, Porwollik S, Prescott AM, Presecan E, Pujic P, Purnelle B, Rapoport G, Rey M, Reynolds S, Rieger M, Rivolta C, Rocha E, Roche B, Rose M, Sadaie Y, Sato T, Scanlan E, Schleich S, Schroeter R, Scoffone F, Sekiguchi J, Sekowska A, Seror SJ, Serror P, Shin BS, Soldo B, Sorokin A, Tacconi E, Takagi T, Takahashi H, Takemaru K, Takeuchi M, Tamakoshi A, Tanaka T, Terpstra P, Tognoni A, Tosato V, Uchiyama S, Vandenbol M, Vannier F, Vassarotti A, Viari A, Wambutt R, Wedler E, Wedler H, Weitzenegger T, Winters P, Wipat A, Yamamoto H, Yamane K, Yasumoto K, Yata K, Yoshida K, Yoshikawa HF, Zumstein E, Yoshikawa H, Danchin A (1997) The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390:249–256
Leonard E, Runguphan W, O’Connor S, Prather KJ (2009) Opportunities in metabolic engineering to facilitate scalable alkaloid production. Nat Chem Biol 5:292–300
Leyval D, Uy D, Delaunay S, Goergen JL, Engasser JM (2003) Characterization of the enzyme activities involved in the valine biosynthetic pathway in a valine-producing strain of Corynebacterium glutamicum. J Biotechnol 104:241–252
Liu X, Brune D, Vermaas W, Curtiss R (2010) Production and secretion of fatty acids in genetically engineered cyanobacteria. P Natl Acad Sci USA. doi:10.1073/pnas.1001946107
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:4020–4408
Marles-Wright J, Grant T, Delumeau O, Delumeau O, van Duinen G, Firbank SJ, Lewis PJ, Murray JW, Newman JA, Quin MB, Race PR, Rohou A, Tichelaar W, van Heel M, Lewis RJ (2008) Molecular architecture of the “Stressosome”, a signal integration and transduction hub. Science 322:92–96
Martin VJJ, Pitera DJ, Withers ST, Newman JD, Keasling JD (2003) Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol 21:796–802
Menkel E, Thierbach G, Eggeling L, Sahm H (1989) Influence of increased aspartate availability on lysine formation by a recombinant strain of Corynebacterium glutamicum and utilization of fumarate. Appl Environ Microbiol 55:684–688
Mu WM, Liu FL, Jia JH, Chen C, Zhang T, Jiang B (2009) 3-Phenyllactic acid production by substrate feeding and pH-control in fed-batch fermentation of Lactobacillus sp. SK007. Bioresour Technol 100:5226–5229
Mukherji S, van Oudenaarden A (2009) Synthetic biology: understanding biological design from synthetic circuits. Nat Rev Genet 10:859–871
Nakano MM, Dailly YP, Zuber P, Clark DP (1997) Characterization of anaerobic fermentative growth of Bacillus subtilis: identification of fermentation end products and genes required for growth. J Bacteriol 179:6749–6755
Nielsen DR, Leonard E, Yoon SH, Tseng HC, Yuan C, Prather KLJ (2009) Engineering alternative butanol production platforms in heterologous bacteria. Metab Eng 11:262–273
Petersohn A, Brigulla M, Haas S, Hoheisel JD, Völker U, Hecker M (2001) Global analysis of the general stress response of Bacillus subtilis. J Bacteriol 183:5617–5631
Qin X, Taber HW (1996) Transcriptional regulation of the Bacillus subtilis menp1 promoter. J Bacteriol 178:705–713
Renna MC, Najimudin N, Winik LR, Zahler SA (1993) Regulation of the Bacillus subtilis alsS, alsD, and alsR genes involved in post-exponential-phase production of acetoin. J Bacteriol 175:3863–3875
Romer-Garcia S, Hernández-Bustos C, Merino E, Gosset G, Martinez A (2009) Homolactic fermentation from glucose and cellobiose using Bacillus subtilis. Microb Cell Fact 8:23. doi:10.1186/1475-2859-8-23
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor
Schleifer KH, Kraus J, Dvorak C, Kilpper-Balz R, Collins MD, Fischer W (1985) Transfer of Streptococcus lactis and related streptococci to the genus Lactococcus gen. nov. Syst Appl Microbiol 6:183–195
Smith KM, Cho KM, Liao JC (2010) Engineering Corynebacterium glutamicum for isobutanol production. Appl Microbiol Biotechnol 87:1045–1055
Steen EJ, Chan R, Prasad N, Myers S, Petzold CJ, Redding A, Ouellet M, Keasling JD (2008) Metabolic engineering of Saccharomyces cerevisiae for the production of n-butanol. Microb Cell Fact 7:36. doi:10.1186/1475-2859-7-36
Thykaer J, Nielsen J, Wohlleben W, Weber T, Gutknecht M, Lantz AE, Stegmann E (2010) Increased glycopeptide production after overexpression of shikimate pathway genes being part of the balhimycin biosynthetic gene cluster. Metab Eng 12:455–461
Unrean P, Trinh CT, Srienc F (2010) Rational design and construction of an efficient E. coli for production of diapolycopendioic acid. Metab Eng 12:112–122
Wang PZ, Doi RH (1984) Overlapping promoters transcribed by Bacillus subtilis sigma 55 and sigma 37 RNA polymerase holoenzymes during growth and stationary phases. J Biol Chem 259:8619–8625
Wong SL (1995) Advances in the use of Bacillus subtilis for the expression and secretion of heterologous proteins. Curr Opin Biotechnol 6:215–223
Zhanga K, Sawaya MR, Eisenberg DS, Liao JC (2008) Expanding metabolism for biosynthesis of nonnatural alcohols. P Natl Acad Sci USA 105:20653–22065
Zhu YB, Chen X, Chen T, Zhao XM (2007) Enhancement of riboflavin production by overexpression of acetolactate synthase in a pta mutant of Bacillus subtilis. FEMS Microbiol Lett 266:224–230
Acknowledgments
The authors appreciate the kind donation of strain B. subtilis 168 and plasmid pHP13, pDG364 and pDG1730 from Dr. Danier R. Zeigler and the Bacillus Genetic Stock Center, The Ohio State University. This research was financially supported by the National 973 Project of China (No. 2007CB714302), the Key Program of National Natural Science Foundation of China (Grant No. 20936002), and National Nature Science Foundation of China (No. 20976124 and No. 20906070).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, S., Wen, J. & Jia, X. Engineering Bacillus subtilis for isobutanol production by heterologous Ehrlich pathway construction and the biosynthetic 2-ketoisovalerate precursor pathway overexpression. Appl Microbiol Biotechnol 91, 577–589 (2011). https://doi.org/10.1007/s00253-011-3280-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3280-9