
Abstract
We report the successful high-yield expression of Candida utilis uricase in Escherichia coli and the establishment of an efficient three-step protein purification protocol. The purity of the recombinant protein, which was confirmed to be C. utilis uricase by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and matrix-assisted laser desorption/ionization time-of-flight mass spectrometer analysis, was >98% and the specific activity was 38.4 IU/mg. Crystals of C. utilis uricase were grown at 18°C using 25% polyethylene glycol 3350 as precipitant. Diffraction by the crystals extends to 1.93 Å resolution, and the crystals belong to the space group P212121 with unit cell parameters a = 69.16 Å, b = 139.31 Å, c = 256.33 Å, and α = β = γ = 90°. The crystal structure of C. utilis uricase shares a high similarity with other reported structures of the homologous uricases from other species in protein database, demonstrating that the three-dimensional structure of the protein defines critically to the catalytic activities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Uricase (also named urate oxidase, EC1.7.3.3), an enzyme involved in the metabolism of purine, catalyzes the oxidative opening of the purine ring of uric acid to form allantoin, which is five to ten times more soluble than uric acid (Chen et al. 2008; Li et al. 2006; Oda et al. 2002; Wu et al. 1989; Pfrimer et al. 2010). Most mammals express functional uricase to yield allantoin as the end product of purine metabolisms. However, in humans and some higher primates, the uricase activity was lost as the result of a few deleterious mutations in the uricase gene. Consequently, the end product of purine metabolism in humans and certain primates is uric acid, rather than allantoin (Wu et al. 1994; Legoux et al. 1992; Koyama et al. 1996).
Uric acid is present in the blood largely as monosodium salt, and both the free acid and the urate salt are relatively insoluble in water. The abnormal accumulation of uric acid in the body usually results in hyperuricemia. In some individuals, however, uric acid can precipitate, leading to symptoms of gout (Pfrimer et al. 2010; Wu et al. 1994; Oda et al. 2002). Gout is a painful disorder characterized by uricemia, recurrent attacks of acute arthritis, deposition of sodium urate in and around joints, and in many cases the formation of uric acid calculi (Legoux et al. 1992). During chemotherapy to treat leukemia or lymphoma, a marked increase in the excretion of uric acid derived from the nucleic acid of malignant cells can obstruct renal tubules, causing acute renal failure (“tumor lysis syndrome”; Jones et al. 1995; Li et al. 2006).
Hyperuricemia has proven difficult to control in some patients and, while several microbial sources of uricase have been proposed for the treatment of this particular clinical syndrome, only Rasburicase (Fasturtec/Elitek) has recently been approved for use both in the USA and Europe for the treatment and prophylaxis of hyperuricemia associated with tumor lysis syndrome in pediatric patients (Li et al. 2006).
Candida utilis uricase shares 49% amino acid sequence identity with Aspergillus flavus uricase, 33% with mammalian uricases, 37% with Drosophila uricase, 33% with Glycine max (soybean) uricase, and 21% with Bacillus uricase (Koyama et al. 1996). C. utilis uricase does not require any cofactor for enzymatic oxidation and is being developed as a therapeutic agent by a number of research groups. Given its potential therapeutic applications, therefore, it is important to investigate the structure–function relationship of the C. utilis enzyme.
The first X-ray crystal structure of a member of the uricase family was determined to 2.2 Å resolution in complex with a competitive inhibitor, 8-azaxanthin, and was further refined to 2.05 Å (Colloc’h et al. 1997). The X-ray structures of uricases from many different species and several uricase-inhibitor complexes have since been resolved (Colloc’h et al. 1997, 2006; Gabison et al. 2008; Juan et al. 2008). C. utilis uricase has been shown to be an attractive candidate for the development of new anti-uric acid therapeutics (Koyama et al. 1996; Chen et al. 2008), yet its three-dimensional structure has not been resolved to date. Here we report the high-yield expression of tag-free C. utilis uricase in Escherichia coli together with an efficient three-step protein purification protocol. We also report on the identification of the recombinant protein and the crystal structure of C. utilis uricase.
Materials and methods
Materials
Enzymes for recombinant DNA technology such as Taq polymerase, T4 DNA ligase, NdeI, and HindIII were purchased from New England Biolabs. Polymerase chain reaction (PCR) amplification kit (including PCR buffer and dNTP mix) was also obtained from New England Biolabs. Plasmid mini kit I and PCR product recovery kit were purchased from Omega Bio-Tek. C. utilis strain CGMCC2.120 was obtained from the China General Microbiological Culture Collection Center.
Primer design and PCR amplification
The primers were designed according to the nucleotide sequence of C. utilis uricase (GenBank accession no. D32043). In order to facilitate the subsequent cloning, NdeI and HindIII restriction endonuclease sites were attached to the 5′-termini of the upstream and downstream primers, respectively: forward: 5′-GGAATTCCATATGTCAACAACGCTCTCATC-3′; reverse: 5′-CCCAAGCTTACAACTTGGTCTTCTCCT-3′. The polymerase chain reaction was carried out using the genomic DNA of C. utilis as a template. The PCR mixture was subjected to 35 cycles of denaturation (45 s, 94°C), annealing (45 s, 55°C), and extension (60 s, 72°C) using a DNA Thermal Cycler (Eppendorf). The PCR product was separated on a 1% agarose gel, purified using PCR product recovery kit, and digested with NdeI and HindIII overnight.
Cloning, expression, and purification
The digested PCR product was cloned into the prokaryotic expression vector pET-42a (Novagen). The recombinant plasmid was transformed into strain BL21 of E. coli. Protein expression was induced by addition of isopropyl-β-d-thio-galactoside (IPTG; 1 mM final concentration) when the OD600 of the culture reached approximately 0.8. The cultures were allowed to grow for another 7 h at 37°C, and the cells were then harvested by centrifugation at 5,000 rpm for 10 min at 4°C. The bacterial cells were resuspended in lysis buffer (1× PBS, pH 8.0, 1 mM DTT, and 1 mM phenylmethylsulfonyl fluoride) and homogenized by sonication. The cell lysate was centrifuged at 20,000×g for 45 min at 4°C to remove the cell debris completely.
All subsequent purification steps were performed at 16°C. The soluble recombinant uricase in the clear supernatant was salted out by adding (NH4)2SO4 to 60% saturation. The precipitate was dissolved in lysis buffer and centrifuged at 20,000×g for 45 min at 4°C. The supernatant was desalted using a Sephadex G-25 gel filtration column with buffer A (25 mM glycine, pH 10.5). The desalted uricase sample was subjected to further purification on a resource Q column. The fractions containing the target protein were pooled and concentrated using an Ultrafree 5,000 molecular weight cutoff filter unit (Millipore) and further purified using a Superdex-75 (Pharmacia) gel filtration column (using buffer: 25 mM Tris–Cl, pH 8.0). The fractions containing the target protein were pooled and concentrated to 35 mg/ml.
Protein analyses and uricase assay
The purified protein was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometer (MALDI-TOF MS) according to the method described previously (Zhang et al. 2010). The concentration of the protein was determined using the Bradford method (Bradford 1976).
The enzymatic activities of the purified uricase were assayed by following the disappearance of uric acid in the reaction volumes, as indicated by a decrease in absorbance at 292 nm. The reaction mixture contained 5 μl of purified uricase in TEA buffer (7.5 g/l triethanolamine, 0.38 g/l EDTA, pH 8.9) and 0.2 μmol of uric acid in a final volume of 5 ml. The addition of 0.2 ml 20% KOH was used to stop the reactions. One unit of the enzyme activity was defined as the amount of enzyme necessary to transform 1 μmol of uric acid into allantoin in 1 min at 30°C, pH 8.9.
Enzyme characterization
The enzyme activity as a function of pH was tested with 50 mM sodium citrate of pH at 2.0, 3.0, 4.0, and 5.0; 50 mM sodium phosphate of pH at 6.0 and 7.0; and 50 mM Tris–HCl of pH at 8.0, 8.5, 9.0, and 10.0. The optimum temperature was evaluated by measuring the uricase activities at different temperatures at 10°C, 20°C, 30°C, 35°C, 37°C, 40°C, 50°C, 60°C, 70°C, and 80 °C. The thermostability of the enzyme was evaluated by incubating the enzyme (0.3 mg/ml enzyme in 0.1 M Tris, pH 8.0) at −20°C, 4°C, or 37°C for 0, 12, 24, 48, and 72 h. Each aliquot of the enzyme subjected to thermostability tests was assayed at 25°C for catalytic activities. The activity of the enzyme was represented as E t/E o (observed activity at a given time/initial activity).
To determine the apparent K m for the decomposition of uric acid, the assay mixtures contained 100 mM Tris (pH 8.0), 0.5 ml uricase sample, and a variable concentration (25.00, 16.67, 12.50, 10.00, 8.33, and 6.25 mM, respectively) of uric acid at 37°C. Nonlinear regression was used for the fitting to the Michaelis–Menten equation for the determination of the kinetic constants. To determine the cation specificities, a series of assays were performed with 2 mM MgCl2, CaCl2, CuCl2, MnCl2, KCl, or NaCl.
Crystallization, data collection, and structure determination
The protein sample was centrifuged at 20,000×g for 45 min to clarify the solution prior to crystallization trials. Initial screening was performed at 18°C in 24-well plates by the hanging-drop vapor-diffusion method using sparse-matrix screen kits from Hampton Research (Crystal Screen reagent kits I and II), followed by a refinement of the conditions through the variation of precipitants, pH, protein concentrations, and additives. Typically, drops consisting of 1 μl protein solution and the equivalent volume of reservoir solution were equilibrated against 200 μl of reservoir solution.
X-ray diffraction data sets from the crystals were collected on Beamline BL17U of the Shanghai Synchrotron Radiation Facility with an X-ray wavelength of 0.9794 Å. All data were collected on a MAR-CCD245 image plate detector.
Prior to data collection, the crystals were immersed in a cryoprotectant solution (reservoir solution supplemented with 12% glycerol) for 5–10 s, picked up with a loop, and then flash-cooled in a nitrogen gas stream at −173°C. A full data set of 720 frames was collected with an exposure time per frame of 0.8 s, a detector distance of 150 mm, and an oscillation range per frame of 0.5°. All intensity data were indexed, integrated, and scaled using the HKL2000 software suite (Otwinowski and Minor 1997).
The PHASER program (McCoy et al. 2007) was used to find the correct solution via the molecular replacement method, using the crystal structure of A. flavus uricase (protein database (PDB) code 3BJP) as the initial searching model. Manual model building and refinement were performed with COOT (Emsley and Cowtan 2004) and PHENIX (Adams et al. 2002). During the later stages of positional refinement, restraints were relaxed, and a bulk solvent correction was applied under the guidance of R free. Model geometry was verified using the program PROCHECK (Laskowski et al. 1993). Solvent molecules were located from stereochemically reasonable peaks in the σA-weighted F o − F c difference electron density map. Structural figures were drawn with the program PyMOL (DeLano 2002).
Results
Cloning, expression, and purification
A 912-bp DNA fragment encoding C. utilis uricase was obtained by PCR amplification and cloned into pET-42a. The bacteria containing the recombinant plasmid were identified by PCR and plasmid digestion and confirmed by DNA sequencing.
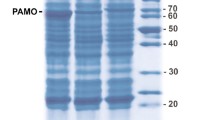
C. utilis uricase was expressed in E. coli with high yield. The recombinant uricase accumulated intracellularly in E. coli in a totally soluble form (Fig. 1a). After a three-step purification procedure (Table 1), the protein was determined to be >98% pure by SDS-PAGE stained with Coomassie brilliant blue (Fig. 1b). The molecular weight of the purified protein was 34.16 kDa as determined by MALDI-TOF MS (Fig. 2a), and the peptide mass fingerprinting analysis of the trypsin-digested purified protein confirmed that the expressed protein was indeed C. utilis uricase (Fig. 2b).

a Expression of C. utilis uricase in E. coli. 1 Marker, 2 before induction, 3 after induction with IPTG. b SDS-PAGE of the purified C. utilis uricase. 1 Marker, 2 the purified C. utilis uricase used for crystallization

MALDI-TOF MS spectrum of C. utilis uricase. a Determination of molecular weight. b Peptide mass fingerprinting
Enzyme activity assay and characterization
The specific activity of the purified uricase was 38.4 IU/mg (see Table 1). The effects of pH and temperature on enzyme activity were shown in Fig. 3a and b, respectively. The purified uricase showed a maximum activity at pH 8.5 and 37°C. As shown in Fig. 4, the residual activity after 72 h incubation at −20°C or 4°C was practically constant but was significantly affected by incubation at 37°C.

Effect of pH and temperature on enzyme activity. Purified uricase was incubated under different pH (a) and temperatures (b) and assayed as described in “Materials and methods” section

Thermal stability. Purified enzyme was pre-incubated at −20°C, 4°C, and 37°C for the indicated times and assayed at 37°C. Residual activity corresponds to the percentage of activity determined for the nonincubated samples
The correlation of enzyme velocity versus substrate concentration was determined by nonlinear regression fitting for the apparent kinetic constants (Fig. 5). The K m value was measured as 33.7 μM. Among the cations tested for specificities, Cu2+ was shown to inactivate the enzyme completely, whereas Mn2+ increased the uricase activity substantially. Other cations have little influence on the activity of uricase (Fig. 6).

The substrate-dependent activity of uricase. Michaelis–Menten plot of reaction velocity as a function of concentration uric acid was used to derive the kinetic parameter. The solid line represents the nonlinear least-squares best fit of the data to the Michaelis–Menten equation. The R 2 value was indicated

The cation specificity of C. utilis uricase; n number of independent experiments
Crystallization data collection and structure determination
Small C. utilis uricase crystals were observed after about 5 days from one condition of the Crystal Screen kit (Hampton Research) containing PEG3350 as precipitant. The conditions were further optimized by varying the precipitants, buffer pH, and protein concentrations. Larger crystals were obtained at 18°C using the hanging-drop vapor-diffusion method by mixing 1 μl protein with 1 μl reservoir solution (0.1 M Tris, pH 8.0, 25% polyethylene glycol 3350) and equilibrating against 200 μl of reservoir solution. The larger crystals were suitable for X-ray diffraction. An X-ray diffraction data set was collected from a single crystal. Data collection statistics and final refinement statistics are summarized in Table 2.
The crystal structure of the 34-kDa, full-length C. utilis uricase was solved by the molecular replacement method and refined to 1.9 Å resolution, resulting in a final R work value of 18.3% (R free = 23.9%). The crystal belongs to the space group P212121, with unit cell parameters a = 69.2 Å, b = 139.3 Å, c = 256.3 Å, and α = β = γ = 90°. A Matthews coefficient of 2.26 Å3/Da suggested the presence of eight uricase molecules in the asymmetric unit composed of two homotetramers, corresponding to a solvent content of 45.53% (Fig. 7a, b). Gel filtration and analytical ultracentrifugation (data not shown) indicated that C. utilis uricase exists as a homotetramer in solution, which was consistent with the crystallographic result. The C. utilis uricase displays a compact, barrel-shaped structure consisting of two relatively separated domains: the α-helical domain and a β-stranded domain with an identical substrate-binding site at the connection region between Asn261-Gly289 and Lys15-Leu189 (Fig. 7c). Unexpectedly, the high degree of structural similarity of C. utilis uricase with A. flavus uricase was beyond their similarity in amino acid sequence. The α-helical domain consists of six canonical α-helices, while the β-stranded domains are mainly anti-parallel β-stands. The connection region between Asn261-Gly289 and Lys15-Leu189, which twists to form C. utilis uricase homotetramer, contains the identical substrate-binding sites as characterized by the structure of A. flavus uricase (Fig. 7c, d).

Structure of C. utilis uricase. a, b Overall structure of the C. utilis uricase homotetramer. The four monomers are colored cyan, magenta, green, and white, respectively; a and b are two orthogonal views of the tetramer. c Superposition of structures of uricases from C. utilis and A. flavus (PDB: 3BJP). The structure of C. utilis uricase is colored in cyan and the other one in yellow. d Stereoview of the active-site region. Relevant residues involved are shown in sticks, with nitrogen atoms and oxygen atoms colored in blue and red, respectively. The uric acid molecule is indicated in magenta
Discussion
The choice of expression vector
Up to now, the only commercial product Rasburicase was produced by expressing A. flavus uricase gene in Saccharomyces cerevisiae. Compared with S. cerevisiae or other fungal expression system, E. coli has more advantages, such as less expense and time needed and easy to operate, especially in industrial scale. Generally, the fermentation of E. coli could be finished within 10 h, while the fermentation of fungi, such as S. cerevisiae, Hansenula polymorpha, or Pichia pastoris, takes a much longer time, usually more than 4 days. Certainly, some biological and biochemical disadvantages also exist, such as the lack of posttranslational modifications, the residues of bacterial lipopolysaccharides in the purified target protein, etc. In this study, the pET-42a vector was selected for expression of C. utilis uricase because it has an NdeI site near the RBS, thus enabling us to clone the DNA sequence encoding C. utilis uricase near the RBS, immediately downstream of the TATA box. As a result, the C. utilis uricase can be expressed in its native form without any additional amino acid residues. The recombinant C. utilis uricase we expressed has no tag and is therefore suitable for development as a potential novel therapeutics.
Activity of the recombinant C. utilis uricase
It was reported that expression of the uricase genes from C. utilis, A. flavus, Nilaparvata lugens, and soybean in E. coli produced recombinant uricases that were as active as their natural counterparts (Suzuki and Verma 1991; Legoux et al. 1992; Koyama et al. 1996; Hongoh et al. 2000). C. utilis uricase shares 49% and 35% amino acid sequence identity with uricases from A. flavus and Arthrobacter globiformis, respectively (Fig. 8). The specific activity of the recombinant C. utilis uricase we purified is 38.4 IU/mg protein, which is higher than those reported for the commercial product Rasburicase (Li et al. 2006) and previously reported recombinant uricases from C. utilis (Koyama et al. 1996; Nishimura et al. 1982; Chen et al. 2008). The uricase activity of our recombinant protein is also higher than for the uricases from A. flavus (Legoux et al. 1992; Li et al. 2006; Conley and Priest 1980), N. lugens (Hongoh et al. 2000), soybean (Suzuki and Verma 1991), fish (Kinsella et al. 1985), and camel (Osman et al. 1989), but lower than the activity of the uricase from Bacillus subtilis expressed in E. coli (Pfrimer et al. 2010).

Sequence alignment of uricases from C. utilis, A. flavus, and A. globiformis. The sequence alignment was made using Clustal W2
Structure determination
Although C. utilis uricase shares a maximum of 49% amino acid sequence identity with homologous uricases from other species, the uricase crystal structures determined to date are very similar, implying that the three-dimensional structure of protein plays a very important role in its function. Compared with structure of A. flavus uricase, the highly conserved Phe162, Arg179, Gln235, and Asn261 and several other residues of C. utilis uricase appeared to be directly involved in substrate binding. Although our attempt to get the crystal structure of C. utilis uricase in complex with the substrate has not been successful yet, our data sufficiently demonstrated that C. utilis uricase possessed very similar, if not the same, substrate-binding sites and enzymatic profile to those of A. flavus uricase. The high-resolution three-dimensional structure of C. utilis uricase helped us to understand its specific functions better. Furthermore, the structure of C. utilis uricase should provide a structural basis for the modification of the native enzyme to construct mutants with higher biological activity and may also be useful in the rational design of new drugs.
References
Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) Acta Crystallogr D Biol Crystallogr 58:1948–1954
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chen Z, Wang Z, He X, Guo X, Li W, Zhang B (2008) Uricase production by a recombinant Hansenula polymorpha strain harboring Candida utilis uricase gene. Appl Microbiol Biotechnol 79:545–554
Colloc'h N, El Hajji M, Bachet B, L'Hermite G, Schiltz M, Prange T, Castro B, Mornon JP (1997) Crystal structure of the protein drug urate oxidase-inhibitor complex at 2.05 A resolution. Nat Struct Biol 4:947–952
Colloc'h N, Girard E, Dhaussy AC, Kahn R, Ascone I, Mezouar M, Fourme R (2006) High pressure macromolecular crystallography: the 140-MPa crystal structure at 2.3 a resolution of urate oxidase, a 135-kDa tetrameric assembly. Biochim Biophys Acta 1764:391–397
Conley TG, Priest DG (1980) Non-classical inhibition of uricase by cyanide. Biochem J 187:733–738
DeLano WL (2002) The PyMOL molecular graphics system. DeLano Scientific, San Carlos
Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132
Gabison L, Prange T, Colloc’h N, El Hajji M, Castro B, Chiadmi M (2008) Structural analysis of urate oxidase in complex with its natural substrate inhibited by cyanide: mechanistic implications. BMC Struct Biol 8:32
Hongoh Y, Sasaki T, Ishikawa H (2000) Cloning, sequence analysis and expression in Escherichia coli of the gene encoding a uricase from the yeast-like symbiont of the brown planthopper, Nilaparvata lugens. Insect Biochem Mol Biol 30:173–182
Jones DP, Mahmoud H, Chesney RW (1995) Tumor lysis syndrome: pathogenesis and management. Pediatr Nephrol 9:206–212
Juan EC, Hoque MM, Shimizu S, Hossain MT, Yamamoto T, Imamura S, Suzuki K, Tsunoda M, Amano H, Sekiguchi T, Takenaka A (2008) Structures of Arthrobacter globiformis urate oxidase–ligand complexes. Acta Crystallogr D Biol Crystallogr D64:815–822
Kinsella JE, German B, Shetty J (1985) Uricase from fish liver: isolation and some properties. Comp Biochem Physiol B 82:621–624
Koyama Y, Ichikawa T, Nakano E (1996) Cloning, sequence analysis, and expression in Escherichia coli of the gene encoding the Candida utilis urate oxidase (uricase). J Biochem 120:969–973
Laskowski R, MacArthur M, Moss D, Thornton J (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291
Legoux R, Delpech B, Dumont X, Guillemot JC, Ramond P, Shire D, Caput D, Ferrara P, Loison G (1992) Cloning and expression in Escherichia coli of the gene encoding Aspergillus flavus urate oxidase. J Biol Chem 267:8565–8570
Li J, Chen Z, Hou L, Fan H, Weng S, Xu C, Ren J, Li B, Chen W (2006) High-level expression, purification, and characterization of non-tagged Aspergillus flavus urate oxidase in Escherichia coli. Protein Expr Purif 49:55–59
McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674
Nishimura H, Yoshida K, Yokota Y, Matsushima A, Inada Y (1982) Physicochemical properties and states of sulfhydryl groups of uricase from Candida utilis. J Biochem 91:41–48
Oda M, Satta Y, Takenaka O, Takahata N (2002) Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol 19:640–653
Osman AM, Del Corso A, Ipata PL, Mura U (1989) Liver uricase in Camelus dromedarius: purification and properties. Comp Biochem Physiol B 94:469–474
Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM (eds) Macromolecular crystallography, part A. Academic, New York, pp 307–326
Pfrimer P, de Moraes LM, Galdino AS, Salles LP, Reis VC, De Marco JL, Prates MV, Bloch C Jr, Torres FA (2010) Cloning, purification, and partial characterization of Bacillus subtilis urate oxidase expressed in Escherichia coli. J Biomed Biotechnol. doi:10.1155/2010/674908
Suzuki H, Verma DP (1991) Soybean nodule-specific uricase (Nodulin-35) is expressed and assembled into a functional tetrameric holoenzyme in Escherichia coli. Plant Physiol 95:384–389
Wu XW, Lee CC, Muzny DM, Caskey CT (1989) Urate oxidase: primary structure and evolutionary implications. Proc Natl Acad Sci USA 86:9412–9416
Wu X, Wakamiya M, Vaishnav S, Geske R, Montgomery C Jr, Jones P, Bradley A, Caskey CT (1994) Hyperuricemia and urate nephropathy in urate oxidase-deficient mice. Proc Natl Acad Sci USA 91:742–746
Zhang L, Xiang H, Gao J, Hu J, Miao S, Wang L, Deng X, Li S (2010) Purification, characterization, and crystallization of the adhesive domain of SdrD from Staphylococcus aureus. Protein Expr Purif 69:204–208
Acknowledgments
The diffraction data sets were collected at Beamline BL17U of Shanghai Synchrotron Radiation Facility. This research was supported by Scientific Research Common Program of Beijing Municipal Commission of Education (grant no. KM200710025029) and National Natural Science Foundation of China, National Laboratory Special fund (no. 2060204). We are grateful to Prof. Maojun Yang of Tsinghua University for his technical assistance with data collection, processing, valuable comments, and critical discussion. We thank Prof. Mark Barlam of Nankai University and Prof. Wei Ding of Capital Medical University for their revising the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Xiaojuan Liu, Mingjie Wen, and Jing Li contributed equally to this work.
Rights and permissions
About this article
Cite this article
Liu, X., Wen, M., Li, J. et al. High-yield expression, purification, characterization, and structure determination of tag-free Candida utilis uricase. Appl Microbiol Biotechnol 92, 529–537 (2011). https://doi.org/10.1007/s00253-011-3244-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3244-0