
Abstract
A novel β-glucosidase (BGL)-producing strain was isolated and identified as Penicillium purpurogenum KJS506 based on its morphology and internal transcribed spacer (ITS) rDNA gene sequence. When rice straw and corn steep powder were used as carbon and nitrogen sources, respectively, the maximal BGL activity of 12.3 U ml−1, one of the highest levels among BGL-producing microorganisms was observed. The optimum temperature and pH for BGL production were 32 °C and 4, respectively. An extracellular BGL was purified to homogeneity by sequential chromatography of P. purpurogenum culture supernatants, and the purified BGL showed higher activity (V max = 934 U mg protein–1) than most BGLs from other sources. The complete ORF of bgl3 was cloned from P. purpurogenum by a modified thermal asymmetric interlaced polymerase chain reaction. The bgl3 gene consists of a 2,571-bp ORF and encodes a putative protein containing 856 amino acids with a calculated molecular mass of 89,624 Da. The putative gene product was identified as a member of glycoside hydrolase family 3. The present results should contribute to improved industrial production of BGL by P. purpurogenum KJS506.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cellulose, a linear polymer of d-glucose units linked by 1,4-ß-d-glucosidic bonds, is the main constituent of wood tissue and the most abundant renewable biomass available on earth. Microbial cellulases catalyzing the hydrolysis of plant polysaccharides are industrially important enzymes used to saccharify industrial and agricultural cellulose-containing residues, treat cellulose pulp wastes in the paper industry, enhance the extraction of fermentable substances in the beer brewing and alcohol fermentation industries, etc. (Da-Silva et al. 1997). Cellulases bring about the hydrolysis of cellulose by synergistic action of its constituent enzymes: (a) β-1,4-endoglucanase (1,4-β-d-glucan 4-glucanohydrolase; EC 3.2.1.4), which cleaves internal β-1,4-glycosidic bonds, (b) cellobiohydrolase (1,4-β-d-glucan cellobiohydrolase; EC 3.2.1.91), an exo-acting enzyme which releases cellobiose from reducing and non-reducing ends of cellulose, and (c) β-glucosidase (BGL; EC 3.2.1.21) that hydrolyzes cellobiose to glucose (Beguin and Aubert 1994; de Palma-Fernandez et al. 2002). BGL catalyzes the hydrolysis of β-O-glucosidic linkages of alkyl- and aryl β-glucosides as well as cellobiose and oligosaccharides. BGL does not act on cellulose directly; nevertheless, its activity is important for cellulolysis because it can alleviate the inhibitory action exerted by cellobiose on endoglucanases and exoglucanases (Bhat and Bhat 1997).
The physiological roles postulated for BGLs are extremely diverse: glucoside ceramide catabolism in human tissue, cell wall, pigment and cyanoglucoside metabolism, defense against pathogens in plants, and utilization of oligosaccharide substrates by many fungi and bacteria (Leclerc et al. 1987). BGLs are widely used in various biotechnological processes, including the production of ethanol from cellulosic agricultural residues and the synthesis of useful β-glucosides (Shinoyama et al. 1991).
In the process of cellulose hydrolysis, enzyme production is still the most crucial and costly step. Many fungal strains secrete higher amounts of cellulases than bacterial ones, with Trichoderma as the leading one. Other fungal species were shown to be interesting cellulase producers, such as Phanerochaete chrysosporium (Eriksson et al. 1974). Although the cellulolytic enzymes of Trichoderma reesei have been investigated thoroughly (Arja et al., 2004; Saloheimo et al. 1997), the amount of BGL secreted by this fungus is insufficient for effective conversion of cellulose to glucose (Workman and Day 1982). In addition, there is an increasing demand for the production of BGL in the conversion of cellulose to glucose for the subsequent production of fuel ethanol (Saha et al. 1994). BGL has been the focus of many researches recently because of their important roles in a variety of fundamental biological processes.
In the present study, a potent BGL-producing fungus was isolated and identified as P. purpurogenum KJS506 (KACC 93053P). We optimized the environmental conditions to enhance BGL production by P. purpurogenum, purified BGL from P. purpurogenum culture, and cloned the bgl-encoding gene by employing degenerate PCR and thermal asymmetric interlaced (TAIL)-PCR. The properties of the enzyme, including its substrate specificity, molecular form, and amino acid sequence analysis, revealed that this enzyme is a BGL, a member of glycoside hydrolase family 3.
Materials and methods
Isolation of microorganism
The soil samples collected from Sorak Mountain (South Korea) by the capillary tube method were diluted in sterile dilution solution (0.9% saline), aliquots were spread on potato dextrose agar plate, and the plates were incubated for 3 days. Initial screening for BGL-producing fungi was carried out in agar plates containing 10 mM MUG as described previously (Daenen et al. 2008). Based on the fluorescence observed, 20 strains were inoculated into 3 ml of the growth medium containing (g l−1) peptone 8, yeast extract 2, KH2PO4 5, K2H PO4 5, MgSO4·7H2O 3, Thiamin·HCl 0.005, and cellulose 20 (Sigma, MO, USA) and cultivated at 28 °C with agitation at 200 rpm for 5 days. BGL activity of the culture broth was analyzed using p-nitrophenyl-β-d-glucopyranoside (pNPG, Sigma, MO, USA) as described previously. One unit of pNPG-hydrolyzing activity was defined as the amount of enzyme equivalent to release 1 μmol of p-nitrophenol per minute. After analyses, the strain with the highest BGL activity was selected.
Identification of microorganism
Fatty acid composition was analyzed by a gas chromatography (Agilent 6890 N, CA, USA), and the identification of the isolated strain was determined using the MIDI database. For the sequence analysis, the ITS1-5.8S–ITS2 rDNA region of the fungus was amplified by PCR using primer set pITS1 (5′-TCCGTAGGTGAACCTGCCG-3′) and pITS4 (5′-TCCTCCGCTTATTGATATGC-3′). The 700-bp amplicon thus obtained was cloned and sequenced. The sequences were proofread, edited, and merged into composite sequences using the PHYDIT program (version 3.1). The identified strain P. purpurogenum KJS506 was deposited at the Korean Agricultural Culture Collection (KACC) and was given the KACC accession number 93053P.
Culture conditions
For flask culture, the mycelia of P. purpurogenum KJS506 were inoculated into 100 ml of potato dextrose broth. Pre-cultures (5 ml) were inoculated into 200 ml of cellulolytic medium in a fermenter. The effect of carbon or nitrogen source on BGL production was investigated after 6 days of cultivation in flasks containing medium composed of 50 g l−1 of carbon sources and various nitrogen sources. The concentration of nitrogen source was adjusted to the same content of nitrogen using Kjeldahl method. The effect of temperature and pH on BGL production was analyzed in a 7-l fermenter with varying the growth temperatures (24 to 36 °C) and pHs (3.0 to 7.0). For fermenter culture, the mycelia of P. purpurogenum were inoculated into 100 ml of potato dextrose broth. Pre-cultures (50 ml) were inoculated into 3 l of cellulolytic medium in a 7-l fermenter. This culture media contained (g l−1) peptone 8, yeast extract 2, KH2PO4 5, K2HPO4 5, MgSO4·7H2O 3, Thiamin·HCl 0.005, and microcrystalline cellulose 20. Other conditions were the same as described in fermenter culture. All the fermentation trials were carried out in triplicate. Fungal biomass was determined by dry cell weight method (Coulibaly et al. 2002). The culture broth was filtered through a filter paper (No. 3, Whatman, Kent, UK), followed by oven drying at 105 °C.
Enzyme assay
BGL activity was assayed using pNPG as substrate. The enzymatic reaction mixtures (1 ml) containing 100 µl of enzyme solution and 10 mM pNPG (final concentration) in 100 mM sodium acetate buffer (pH 5.0) were incubated for 15 min at 50 °C. The amount of p-nitrophenol released was measured at A415 (ε 415 = 17.0 mM−1 cm−1) after addition of 2 M Na2CO3 to the reaction mixtures. One unit of pNPG-hydrolyzing activity was defined as the amount of enzyme equivalent to release 1 µmol of p-nitrophenol per minute. The activity was also examined with cellobiose or cello-oligosaccharides as substrate by measuring the amount of produced glucose using GOD–POD method (Lin et al. 1999).
Purification of β-glucosidase
Cells from the culture broth were harvested by centrifugation at 10,000×g for 30 min. The supernatants were combined, concentrated, and desalted by ultrafiltration through a polyether sulfone membrane (10 kDa cutoff) in a stirred cell (Amicon Inc. Beverly, MA). The dialyzed enzyme solution was subjected to chromotography. All chromatographic separations were performed using a BioLogic FPLC system (Bio-Rad, CA) as described previously (Leite et al. 2007). DEAE SepharoseTM (1.6 × 10 cm, Amersham Biosciences), MonoQ ion exchange column Fast Flow column 5/50 GL (1.0 × 10 cm, Amersham Biosciences), and hydroxyapatite column (1.0 × 10 cm, Amersham Biosciences) were used sequentially for the purification of BGL. All procedures were performed at 4 °C, and 50 mM potassium phosphate buffer (pH 7.0) containing 1 mM dithiothreitol (DTT). Protein was measured by the the Bradford method, using bovine serum albumin as a standard. Protein in the column effluents was monitored by measuring the absorbance at 280 nm. Glycoprotein nature of protein in polyacrylamide gel were detected using GelCode glycoprotein staining kit (Pierce Biotechnology, USA) as described in the manufacturer’s instructions.
N-terminal and internal amino acid sequencing of bgl3
The purified enzyme (1.5 μg) was separated by 10% SDS-PAGE and blotted onto a PVDF membrane. Automated Edman degradation of the enzyme protein was performed. The N-terminal amino acid sequence was determined by Edman degradation with an automatic protein sequencer (model 491A; Applied Biosystems, Division of PerkinElmer) at The National Instrumentation Center for Environmental Management (Seoul, South Korea). The sliced protein spot was also analyzed by nano-LC-MS/MS for internal amino acid sequencing (Shevchenko et al. 1996). To identify protein sequence, homology search method was employed with a MS data analysis program, SEQUEST (ThermoFinnigan, San Jose, CA) against fungi protein database obtained from National Center for Biotechnology Information protein sequence database. The partial amino acid sequence was used to identify analogous proteins through a BLAST search of the nonredundant protein database.
Characterization of purified BGL
The optimal pH of BGL activity was determined by incubating the purified enzyme at 70 °C for 15 min in different buffers: citrate (100 mM, pH 3–4), sodium acetate (100 mM, pH 4–6), and phosphate (100 mM, pH 6–8). To determine the optimal temperature, the enzyme was incubated in sodium acetate buffer (100 mM, pH 5) for 30 min at different temperatures from 30 to 80 °C. To determine the thermostability of BGL activity, the purified enzyme was incubated at different temperatures (40, 50, 60, 70, and 80 °C) in the absence of substrate. After incubating them for certain periods of time (0–72 h), the residual BGL activity was determined as described above. The values of the Michaelis constant (K m) and the maximum velocity (V max) were determined for BGL by incubating in 100 mM sodium acetate buffer pH 5.0 at 65 °C with pNPG and cellobiose at concentrations ranging from 0.5 to 50 mM. Values for K m and V max were determined using non linear regression. Inhibition of BGL by glucose was determined in the presence of pNPG as the substrate. The specificity of purified enzyme towards different substrates was tested under standard assay conditions.
PCR cloning and DNA sequencing of P. purpurogenum β-glucosidase gene (bgl3)
Genomic DNA was isolated using Wizard nucleic acid purification kit (Promega). Degenerate primers, BGLDF: TGGCCNTT(C/T)GCNGA(T/C)GCNGT and BGLDR: CT(A/G/C)GTNACNCCNGA(A/G)GAC, were designed based on the partial peptide sequences (LYLWPFAD and PYLVTPEQ ) obtained from nano-LC-MS/MS sequencing. PCR was performed as follows: All PCRs contained 1× buffer, 4 µL of 10 mM dNTPs, 10 pmol/µL of each primer, and 5 U of Taq DNA polymerase in 100 µL. Amplification was performed in a TC-512 programmable gradient thermal controller (Techne, Staffordshire, UK) with 1 cycle of 94 °C for 5 min followed by 35 cycles of denaturation (60 s at 94 °C), annealing (60 s at 45–55 °C), and extension (180 s at 72 °C), with a final extension of 72 °C for 10 min. For analysis, 10 μL of reaction mixture was electrophoresed on a 1% agarose gel and stained with ethidium bromide solution (5 μg ml−1). The purified PCR products were ligated to T&A cloning vector (RBC Bioscience, Taipei, Taiwan) and sequenced with M13 primers from both strands at Macrogen (Seoul, South Korea). DNA from agarose gel was extracted using Dyne Power Gel extraction column (Dynebio Ltd, Seoul, South Korea) according to manufacturer’s instruction. Cloning of the full-length bgl3 gene by TAIL-PCR
Genomic DNA of P. purpurogenum was used as the template. To isolate the 5′-end of the bgl3 gene fragment, TAIL-PCR was performed according to the protocol developed by Liu and Whittier (1995). Three random degenerate primes (RP) such as RP1 (5′-NTGCANTNT-GCNGT-3′), RP2 (5′-NGTCAGNNNGANANGA-3′), and RP3 (5′-NGTGNGANANCANCA-3′), in which N represents A/G/C/T, were designed. Three interlaced specific reverse primers complementary to the known nucleotide sequence (863-bp PCR product) were synthesized (SPR1: CAAGTTGCACGTCCACACCTTTGCCACGG, SPR2: CTCTCACACCGAGTGG-TGAATCCTG, SPR3: GGGAAGGCAGTATTGTAATCAGCT). The tertiary PCR products were separated by electrophoresis on 1.0% agarose gels. The PCR product was purified and cloned into the T&A vector, and then its nucleotide sequence was determined. Similarly, three interlaced specific sense primers according to the known nucleotide sequence were designed (SPF1: GATGTACGTGGTGACCACGVVGSSGTCG-TCCGT, SPF2: GGCTG-CTCTGACCGCGGCTGTGACAACGGTAC, SPF3: TCCCTTACCTGGTCACCCCTGAA-GATCT) to isolate the 3′-end of bgl3 fragment. Three random degenerate primers were also used.
To amplify the bgl3 cDNA, total RNA was isolated (TRI reagent, Sigma, MO, USA) from P. purpurogenum culture grown with cellulose as carbon source. cDNA-encoding GH3 BGL was cloned using Maxime RT-PCR PreMix Kit (iNtRON, Korea) with primers BGLF: TGCGGAACAGCTTATTGATTT and BGLR: CTACTTTCCGATGTTGAGAGC following the manufacturer’s instructions.
DNA manipulations and sequence analysis
Database similarity searches were performed using the National Centre for Biotechnological Information (NCBI) online program BLAST (Altschul et al. 1990) against protein (BlastX) and nucleotide (BlastN) sequences stored in GenBank. Multiple sequence alignments were done by Clustal W program. The protein sequence was analyzed by ExPASy server (Gasteiger et al. 2003).
Homology modeling
The three-dimensional homology model of P. purpurogenum BGL was generated using Build Homology Models (MODELER) module in Discovery Studio 2.5 (DS 2.5, Accelrys Software Inc., San Diego, CA). Crystal structure of barley β-d-glucan exohydrolase (1IEQ) was used as template. The comparative modeling was to generate the most probable structure of the query protein by the alignment with template sequences, simultaneously satisfying spatial restraints and local molecular geometry. Sequence identity between target and template was found to be 22% according to BLAST parameters. Fitness of the models sequence in their current 3D environment was evaluated by Profiles-3D Score/Verify Protein (MODELER) as implemented in DS 2.5. Discrete optimized protein energy score in MODELER was also calculated to determine the quality of protein structures. Profile-3D score was 178.23 against 197.15 maximum expected score. The root mean square deviation (RMSD) between the models and template was calculated by superimposing on template crystal structure for the reliability of the models, and RMSD was 1.7 Å based on C-alpha atoms. The generated structure was improved by subsequent refinement of the loop conformations by assessing the compatibility of an amino acid sequence to known PDB structures using the Protein Health module in DS 2.5. The geometry of loop regions was corrected using Refine Loop/MODELER. Finally, the best quality model was chosen.
Results
Identification of the isolated strain
Among 340 strains screened for BGL activity, 20 strains were selected based on the fluorescence observed when the agar plates containing 10 mM of MUG were exposed under UV. Out of 20 isolates, an efficient BGL-producing microorganism was selected for further study. Sequencing of the ITS rDNA region of the isolated fungus was performed, and the ITS sequence was submitted to GenBank with the accession no. GQ292537. The strain showed the highest identity (99%) with P. purpurogenum. Phylogenetic relationships were inferred using alignment and cladistic analyses of homologous nucleotide sequences of known microorganisms, and the approximate phylogenetic position of the strain is shown in Fig. 1. The probability that the isolated fungus and P. purpurogenum belong to the same branch is 99%. In addition, the composition of the cellular fatty acids of the isolated strain was similar to those of the Penicillium species (Table 1). The colonies are flat, filamentous, and velvety or cottony in texture. The conidiophores (3–3.5 μm in diameter) are simple or branched and are terminated by clusters of flask-shaped phialides. Based on its morphology, the composition of its cellular fatty acids, and a comparison of the ITS rDNA gene sequences, the isolated strain was identified as a strain of P. purpurogenum and was named as P. purpurogenum KJS506.

The phylogenetic dendrogram for P. purpurogenum KJS506 and related strains based on the ITS rDNA sequence. Numbers following the names of the strains are accession numbers of published sequences
Optimization of carbon and nitrogen sources for BGL production in flask culture
Various carbon (cellulose, rice straw, xylan, Avicel, CMC, cellobiose, glucose, maltose, lactose, and sucrose) and nitrogen (peptone, tryptone, corn steep powder (CSP), yeast extract, urea, potassium nitrate, and sodium nitrate) sources were tested for BGL production by P. purpurogenum KJS506. Rice straw and CSP were found to be optimum C and N sources for BGL production. When nitrogen sources were combined, an inhibitory effect on BGL activity was observed. To evaluate the effect of the initial concentrations of carbon and nitrogen sources on cell growth and BGL production in culture, various concentrations of rice straw and CSP were tested (data not shown). BGL production was maximal (12.3 U ml−1) at 50 g l−1 of rice straw and 10 g l−1 of CSP in the production medium.
Effect of pH and temperature on BGL production in a fermenter
The influence of culture medium pH on BGL production was studied for pH values in the range from 3.0 to 6.5 at 30 °C. As the pH of the culture medium was increased from 3.0 to 5.0, BGL activity increased with the maximal activity of 17.8 U ml−1 at pH 4.0. After pH 4.0, BGL production was significantly decreased (Fig. 2a). In addition, there were distinct differences in cell growth between the cultures grown at pH 4.0 (26 g l−1) and those grown at pH 6.0 (15 g l−1).

a Effect of pH on the production of BGL in P. purpurogenum culture. Cultivation of P. purpurogenum KJS506 was performed for 6 days in a 7-l fermenter. Cultivation temperature and rpm were controlled as 30 °C and 200 rpm, respectively. b Effect of temperature on the production of BGL in P. purpurogenum culture. Cultivation pH and rpm were controlled as pH 4.0 and 200 rpm, respectively. BGL activity (filled circles), DCW (unfilled circles)
To investigate the effect of temperature on BGL production, P. purpurogenum KJS506 was grown at various temperatures (24–34 °C) in a fermenter that contained rice straw (50 g l−1) medium at pH 4.0 (Fig. 2b). P. purpurogenum showed significantly higher BGL activity (26.4 U ml−1) at 32 °C than at 28 °C (13.6 U ml−1), which suggests that temperature is a critical factor for BGL production in this strain.
Characterization of BGL purified from P. purpurogenum culture
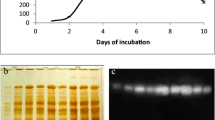
The enzyme from the culture supernatant of P. purpurogenum grown on rice straw was purified 16-fold to homogeneity with an overall enzyme yield of 17.3% and a specific activity of 875 U mg protein−1. The molecular weight of purified BGL as determined by SDS-PAGE is about 110 kDa (Fig. 3). The pH and temperature optimum for purified BGL was found to be 5.0 and 65 °C, respectively (Fig. 4). The purified BGL showed t 1/2 value of 10 h at 65 °C. The purified BGL showed wide substrate specificity and was active on different cellulosic substrates (Table 2). The enzyme readily hydrolyzed cellooligosaccharides including cellotriose, cellotetraose, and cellopentaose, and glucose was the major product. The substrate specificity of P. purpurogenum BGL was in agreement with those recorded for BGLs from other microorganisms (Table 3). Therefore, the purified P. purpurogenum BGL is a β-glucosidase, rather than an endo-type cellulase or cellobiohydrolase, which hydrolyzes cellooligosaccharides to cellobiose. The hydrolyzing activity of the enzyme was much higher with substrates containing β-1,4 linkages, such as pNPG and pNPgal, than with substrates containing β-1,2 linkages, such as oNPG and oNPgal. It showed maximum specific activity towards pNPG (875 U mg protein−1).

SDS-PAGE of BGL purified from the P. purpurogenum culture. Lane 1 shows glycoprotein specific staining of the purified BGL from P. purpurogenum; lane 2 shows SDS-PAGE of the purified BGL; and lane M is the marker

a Effect of pH on the activity of purified BGL from P. purpurogenum. The enzyme activity was assayed by the standard assay method by changing the buffer to obtain the desired pH. The buffers used were citrate (pH 3.0 to 4.0), sodium acetate (pH 4.0 to 6.0), and phosphate (pH 6.0 to 7.0). b Effect of temperature on the activity of purified BGL from P. purpurogenum. The enzyme was assayed at various temperatures by the standard assay method. Each value represents the mean of triplicate measurements and varied from the mean by not more than 15%
The initial velocities were determined in the standard assay mixture at pH 5.0. The initial velocities were determined in the standard assay mixture at pH 5.0. The substrates tested had hyperbolic saturation curves, and the kinetic parameters were determined using nonlinear regression fitting of the Michaelis–Menten equation (Prism 5, Graphpad Software, CA, USA). The K m and V max values obtained for pNPG by P. purpurogenum BGL under standard assay conditions were 5.1 mM and 934 U mg protein−1, respectively. The product inhibition studies, under nonsaturating conditions, showed that glucose inhibited BGL competitively with Ki value of 21.5 mM. Table 3 compares the properties of P. purpurogenum BGL with those of reported BGLs. P. purpurogenum BGL has higher specific activity and lower K m values than those of most other BGLs.
Cloning of the full-length bgl3 gene by TAIL-PCR
The purified enzyme (1.5 μg) was separated by 10% SDS-PAGE and blotted onto a PVDF membrane, and the N-terminal amino acid sequence was obtained by automated protein sequencer. Automated Edman degradation of the purified P. purpurogenum BGL gave the N-terminal amino acid sequence YSPPAYPTPWASGAGEWAQ, which showed a significant similarity (78%) to T. aurantiacus BGL enzyme in GH3. A partial 850-bp amplicon was obtained by PCR using a degenerate primer pair, BGLDF and BGLDR, which were designed based on the peptide sequences identified by nano-LC-MS/MS sequencing. The partial gene sequence (bgl3) of P. purpurogenum contained the BGL-like domain (GFVMTD) that is found in the GH3 family.
To amplify the DNA fragments upstream and downstream of bgl3 gene, the specific primers (SPF1, 2, and 3; SPR1, 2, and 3) were designed based on the sequence of the partial bgl3. The strategy followed for the amplification of flanking regions by TAIL-PCR is shown in Fig. 5. Tertiary PCR of 3′ TAIL-PCR with SPF3 primer and RP1 random primer resulted in amplification of a 700-bp fragment. Similarly, tertiary PCR of 5′ TAIL-PCR with SPR3 primer and RP2 random primer resulted in a 500-bp fragment. The PCR fragments were gel eluted and then sequenced on both strands. NCBI nucleotide BLAST result confirmed that the sequence belongs to GH3 BGL. The N-terminal and C-terminal sequences were partially overlapped with already sequenced internal fragment. Similarly, another round of TAIL-PCR was performed to amplify the full-length region of bgl3 gene. Finally, PCR with GH3ORFF and GH3ORFR resulted in 2,845 bp amplicon, which was cloned and confirmed. The nucleotide sequence of bgl3 was submitted to GenBank (Accession no: GQ475527).

Schematic representation of steps followed in TAIL-PCR. To amplify the N-terminal sequence of partial bgl3, primers SPR1, 2, and 3 with random primer RP2 were used in the primary, secondary and tertiary PCR, respectively. Similarly, to amplify the 3′ flanking region of partial bgl3, primers SPF1, 2, and 3 with random primer RP1 were used in the primary, secondary, and tertiary PCR, respectively
Nucleotide sequence analysis of bgl3
The bgl3 cDNA (2,571 bp) encodes a protein composed of 856 amino acid residues with a predicted pI of 4.6. The bgl3 gene has four introns with an average length of 68 bp. At the N terminus of the deduced sequence (Fig. 6), a putative signal sequence (MRNSLLISLAVAALAEGKA) was identified by the SignalP 3.0 server system (http://www.cbs.dtu.dk/services/), with cleavage predicted to occur after 19th amino acid (Ala) of the pre-protein. Conserved domain search (RPSBLAST) analysis confirmed the presence of N-terminal catalytic domain (pfam00933) and C-terminal domain (PF01915) of GH3 BGL. The deduced amino acid sequence of P. purpurogenum BGL3 contains several possible N-glycosylation sites (Asn 253, 254, and 554). The Mr of the BGL3 protein calculated from the deduced amino acid sequence was 89,624 Da, compared with 110 kDa as determined by SDS-PAGE. The difference can be ascribed to the fact that BGL3 is produced in P. purpurogenum as a glycoprotein, which is confirmed by glycoprotein staining of the purified BGL (Fig. 3). A homology search revealed that the deduced BGL3 had 88%, 72%, 69%, 65%, and 64% amino acid identity with the BGLs of P. marnefii (GenBank accession No. XP_002144089), Talaromyces emersonii (AAL69548), Thermoascus aurantiacus (AAZ95587), A. aculeatus (P48825), and Penicillium decumbens (ACD86466), respectively. All of the homologous sequences belong to GH3, which suggests that P. purpurogenum BGL3 is also a member of GH3.


Sequence alignment of P. purpurogenum BGL with other cellulases using ClustalW. The accession numbers are: XP_002144089 (P. marneffei), AAL69548.3 (T. emersonii), XP_0012695 (A. clavatus), ABX79552 (T. aurantiacus), and ABP88968 (P. brasillianum). GH3 active site sequence is marked by an arrow; the predicted signal peptide sequence is shown in box. The putative nucleophile in the catalytic center is marked by a star
Homology modeling and superimposition
Using crystal structure of barley β-d-glucan exohydrolase (1IEQ) as template, the homology model of P. purpurogenum BGL (accession no. GQ475527) was created with DS2.5 software. Model validation was carried out using different validation tool. Ramachandran’s plot calculation was computed with PROCHECK program. Calculated Ramachandran’s plot suggested 83.5%, 12.6%, and 2.4% of residues in derived model are in most favored, additional allowed, generously allowed, and disallowed region, respectively. Altogether, 98.5% of residues are placed in the combined favored and allowed categories. A model structure with a high percentage of residues in the combine favored, and allowed categories are likely to represent a good protein fold. Thus, PROCHECK validates the folding integrity of our model and indicates that the model structure derived from the template (1IEQ) is of higher quality in terms of protein fold. Profile-3D score of model was 178.23 against 197.15 maximum expected score. Built model was also evaluated by superimposing on template crystal structure and RMSD was 1.7 Å based on C-alpha atoms. Active site residues were compared and superimposed on the template structure. When catalytic domain was superimposed on the crystal structure of barley β-d-glucan exohydrolase (Fig. 7), the conserved catalytic residues (Arg152, Lys185, His186, Glu201, Met238, Asp273, Trp274, and Trp442) in the P. purpurogenum BGL model have similar orientations and locations to the residues in barley β-d-glucan exohydrolase.

Molecular modeling of the active site pocket of P. purpurogenum BGL. Active site residues (green color) were superimposed on template 1IEQ (yellow color). Based on superimposition, the active site residues of P. purpurogenum BGL (Arg152, Lys185, His186, Glu201, Met238, Asp273, Trp274, and Trp442) and those of barley β-d-glucan exohydrolase 1IEQ (Arg158, Lys206, His207, Glu220, Met250, Asp285, Trp286, and Trp430) have similar orientation and position
Discussion
In the present study, we report the characterization of BGL from a new strain of P. purpurogenum, which we have designated as KJS506 strain. The strain produced a high level of BGL (12.3 U ml−1) when rice straw and CSP were used as the carbon and nitrogen sources, respectively. The higher enzymatic activity noted in the presence of rice straw may be attributed to the deregulation of global carbon metabolism regulator proteins derived from the carbon catabolite-derepressing medium (Mach and Zeilinger 2003). When environmental conditions such as pH and temperature were additionally optimized, the maximal BGL activity of 26.4 U ml−1, one of the highest levels among BGL-producing microorganisms, was observed.
Table 3 shows a comparison of the properties of BGLs from various sources. Based on substrate specificity, β-glucosidases have been classified as (1) aryl β-glucosidases, which act on aryl-glucosides, (2) true cellobiases, which hydrolyze cellobiose to release glucose, and (3) broad substrate specificity β-glucosidases, which act on a spectrum of substrates. P. purpurogenum BGL showed significant activity for both aryl-glucosides and cellooligo-saccharides, indicating that it belongs to broad substrate specificity β-glucosidases. P. purpurogenum BGL had a comparable K m value of 5.1 mM for pNPG. In comparison, pNPG K m values of purified BGLs from other fungi range 0.2 to 21.7 (Table 3). An extracellular BGL purified from P. purpurogenum was a monomer with a molecular mass of 110 kDa. The high molecular mass of P. purpurogenum BGL was also in agreement with those of many extracellular BGLs characterized from other fungal sources (Himmel et al. 1993; Kempton and Withers 1992; Kuriyama et al. 1995).
Based on amino acid sequence similarities, glycosidases have been classified into several families, with most BGLs belonging to either family 1 or family 3 (Henrissat et al. 1995; Henrissat 1998). Cellulases are generally composed of two or more discrete domains: a core which contains the catalytic domain, a highly conserved cellulose-binding domain which is involved in binding to crystalline cellulose, and connecting these two domains, a hinge which tends to be glycosylated. When P. purpurogenum BGL was compared with other related enzymes, the catalytic domain showed the highest identity with A. aculeatus BGL (66%), and both BGLs belong to GH3. P. purpurogenum BGL contains GFVMTD residues, and the peptide is highly conserved motif in GH3 (Makoto et al. 1988). In the fragment GFVMTD, an aspartic acid residue has been shown to be highly conserved and confirmed to be the active-site residue of BGLs in GH3 (Wong et al. 1998). Comparative modeling of the three-dimensional structure indicated that the catalytic nucleophile (Asp-285 of barley enzyme) is conserved across the GH3 family. In addition, the conserved active site residues in the P. purpurogenum BGL model have similar orientations and locations in the template GH3 enzyme 1IEQ (Fig. 7). The evidence from enzymology, bioinformatics, and homology modeling studies strongly suggest that P. purpurogenum BGL should be classified as a member of GH3.
In conclusion, a potent BGL-producing P. purpurogenum KJS506 was identified. Under the optimal culture condition of P. purpurogenum in a 7-l fermenter, a BGL-specific activity of 54 U mg protein−1, one of the highest among BGL-producing microorganisms, was obtained. The purified BGL from P. purpurogenum showed higher specific activity and lower K m values than those of most other BGLs. P. purpurogenum bgl3 encoding gene was cloned by TAIL-PCR method in combination with degenerate PCR. The TAIL-PCR approach is a simple and efficient technique for genome walking in plant molecular biology (Liu and Huang 1998; Liu and Whittier 1995) and does not require any restriction or ligation steps. Our results indicate that TAIL-PCR would facilitate the full-length gene cloning in fungal strains. Based on the enzymology and bioinformatics, P. purpurogenum BGL was classified as a member of GH3. This now sets the stage for more detailed investigations of this novel BGL, such as expression of bgl3 gene in heterologous host system followed by protein engineering studies. The present results should contribute to better industrial production of BGL by P. purpurogenum KJS506.
Reference
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Arja MJL, Vesa J, Raija L (2004) Three cellulases from Melanocarpus albomyces for textile treatment at neutral pH. Enzyme Microb Technol 34:332–341
Bahia A, Ali G (2006) Characterization of novel beta-glucosidase from a stachybotrys strain. Appl Microbiol Biotechnol 32:191–197
Beguin P, Aubert JP (1994) The biological degradation of cellulose. FEMS Microbiol Rev 13:25–58
Bhat MK, Bhat S (1997) Cellulose degrading enzymes and their potential industrial applications. Biotechnol Adv 15:583–620
Coulibaly L, Naveau H, Agathos SN (2002) A tanks-in-series bioreactor to simulate macromolecule-laden wastewater pretreatment under sewer conditions by Aspergillus niger. Water Res 36:3941–3948
Daenen L, Saison D, Sterckx F, Delvaux FR, Verachtert H, Derdelinckx G (2008) Screening and evaluation of the glucoside hydrolase activity in Saccharomyces and Brettanomyces brewing yeasts. J Appl Microbiol 104:478–88
Da-Silva R, Gomes E, Franco CML (1997) Pectinases, hemicelulase e cellulases substrate, production application no processamento de alimentos. Bol SBCTA 31:249–250
de Palma-Fernandez ER, Gomes E, da Silva R (2002) Purification and characterization of two beta-glucosidases from the thermophilic fungus Thermoascus aurantiacus. Folia Microbiol (Praha) 47:685–690
Eriksson KE, Pettersson B, Westermark U (1974) Oxidation: an important enzyme reaction in fungal degradation of cellulose. FEBS Lett 49:282–285
Galas E, Romanowska I (1996) Production purification and characterization of a highly glucose-tolerant novel ß-glucosidase from Candida peltata. Appl Environ Microbiol 62:3165–3170
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31:3784–3788
Henrissat B (1998) Glycosidase families. Biochem Soc Trans 26:153–156
Henrissat B, Callebaut I, Fabrega S, Lehn P, Mornon JP, Davies G (1995) Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc Natl Acad Sci U.S.A. 92:7090–7094
Himmel ME, Adney WS, Fox JW, Mitchell DJ, Baker JO (1993) Isolation and characterization of two forms of beta-D-glucosidase from Aspergillus niger. Appl Biochem Biotechnol 39–40:213–225
Joo AR et al (2009) Purification and characterization of a beta-1,4-glucosidase from a newly isolated strain of Fomitopsis pinicola. Appl Microbiol Biotechnol 83:285–294
Karnchanatat A et al (2007) Purification and biochemical characterization of an extracellular beta-glucosidase from the wood-decaying fungus Daldinia eschscholzii (Ehrenb.:Fr.) Rehm. FEMS Microbiol Lett 270:162–170
Kaur J, Bhupinder SC, Badhan AK, Ghatora KS (2007) Purification and characterization of β-glucosidase from Melanocarpus sp MTCC 3922. Electron J Biotechnol 10:261–270
Kempton JB, Withers SG (1992) Mechanism of Agrobacterium beta-glucosidase: kinetic studies. Biochemistry 31:9961–9969
Kuriyama K, Tsuchiya K, Murui T (1995) Some properties of transglycosylation activity of sesame β-glucosidase. Biosci Biotechnol Biochem 59:1142–1143
Leclerc MAA, Ratomahenina R, Galzy P (1987) Yeast β-glucosidases. Biotechnol Genet Eng Rev 5:269–295
Leite RSR, Gomes E, Da-Silva R (2007) Characterization and comparison of thermostability of purified β-glucosidases from a mesophilic Aureobasidium pullulans and thermophilic Thermoascus aurantiacus. Process Biochem 42:1101–1106
Lin J, Balakrishna P, Suren S (1999) Purification and biochemical characterization of β-glucosidase from a thermophilic fungus, Thermomyces lanuginosus - SSBP. Biotechnol Appl Biochem 30:81–87
Liu YG, Huang N (1998) Efficient amplification of insert end sequences from bacterial artificial chromosome clones by thermal asymmetric interlaced PCR. Plant Mol Bio Rep 16:175–181
Liu YG, Whittier RF (1995) Thermal asymmetric interlaced PCR: Automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 25:674–681
Lymar ES, Li B, Renganathan V (1995) Purification and characterization of a cellulose-binding (beta)-glucosidase from cellulose-degrading cultures of Phanerochaete chrysosporium. Appl Environ Microbiol 61:2976–2980
Mach R, Zeilinger S (2003) Regulation of gene expression in industrial fungi Trichoderma. Appl Microbiol biotechnol 60:515–522
Magalhaes PO, Ferraz A, Milagres AF (2006) Enzymatic properties of two β-glucosidases from Ceriporiopsis subvermispora produced in biopulping conditions. J Appl Microbiol 101:480–486
Makoto M, Isao O, Sakuzo F, Ichiro Y (1988) Nucleotide sequences of Saccharomycopsis fibuligera genes for extracellular β-glucosidases as expressed in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 54:3147–3155
Rashid MH, Siddiqui KS (1997) Purification and characterization of a beta-glucosidase from Aspergillus niger. Folia Microbiol (Praha) 42:544–550
Saha BC, Freer SN, Bothast RJ (1994) Production, purification, and properties of a thermostable beta-glucosidase from a color variant strain of Aureobasidium pullulans. Appl Environ Microbiol 60:3774–3780
Saloheimo M, Nakari-Setala T, Tenkanen M, Penttila M (1997) cDNA cloning of a Trichoderma reesei cellulase and demonstration of endoglucanase activity by expression in yeast. Eur J Biochem 249:584–591
Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem 68:850–858
Shinoyama HTV, Ando A, Fujii T, Sasaki M, Doi Y, Yasui T (1991) Enzymatic synthesis of useful alkyl-β-glucosides. Agri Biol Chem 55:1679–1681
Stahl P, Klug M (1996) Characterization and differentiation of filamentous fungi based on fatty acid composition. Appl Environ Microbiol 62:4136–4146
Valaskova V, Baldrian P (2006) Degradation of cellulose and hemicelluloses by the brown rot fungus Piptoporus betulinus-production of extracellular enzymes and characterization of the major cellulases. Microbiology 152:3613–3622
Wei DL, Kirimura K, Usami S, Lin TH (1996) Purification and characterization of an extracellular beta-glucosidase from the wood-grown fungus Xylaria regalis. Curr Microbiol 33:297–301
Wong WK, Ali A, Chan WK, Ho V, Lee NT (1998) The cloning, expression and characterization of a cellobiase gene encoding a secretory enzyme from Cellulomonas biazotea. Gene 207:79–86
Workman WE, Day DF (1982) Purification and properties of beta-glucosidase from Aspergillus terreus. Appl Environ Microbiol 44:1289–1295
Yan TS, Lin CL (1996) Purification and characterization of a glucose-tolerant β-glucosidase from Aspergillus niger CCRC31494. Biosci Biotechnol Biochem 61:965–970
Acknowledgment
This study was supported by a grant (code 2008A0080126) from ARPC. This subject was also supported by Korea Ministry of Environment as “The GAIA Project”.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jeya, M., Joo, AR., Lee, KM. et al. Characterization of β-glucosidase from a strain of Penicillium purpurogenum KJS506. Appl Microbiol Biotechnol 86, 1473–1484 (2010). https://doi.org/10.1007/s00253-009-2395-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2395-8