
Abstract
To increase the thermostability of Rhizomucor miehei lipase, the software Disulfide by Design was used to engineer a novel disulfide bond between residues 96 and 106, and the corresponding double cysteine mutants were constructed. The R. miehei lipase mutant could be expressed by Pichia pastoris in a free secreted form or could be displayed on the cell surface. The new disulfide bond spontaneously formed in the mutant R. miehei lipase. Thermostability was examined by measuring of hydrolysis activity using 4-nitrophenyl caprylate as a substrate. The engineered disulfide bond contributed to thermostability in the free form of the R. miehei lipase variant. The variant displayed on the yeast cell surface had significantly increased residual hydrolytic activity in aqueous solution after incubation at 60°C for 5 h and increased synthetic activity in organic solvent at 60°C. These results indicated that yeast surface display might improve the stability of R. miehei lipase, as well as amplifying the thermostability through the engineered disulfide bond.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lipase-catalyzed reactions have received much attention in recent years, as they are increasingly used in biotechnology applications in the chemical, food, and pharmaceutical industries (Noel and Combes 2003; Weber et al. 2004, 2006; Keng et al. 2008). Regardless of process conditions, thermostability is often a key factor in successful industrial bioprocesses, since reaction rates typically increase exponentially with temperature, until the point of enzyme denaturation (Peterson et al. 2007). Thus, the engineering of thermostable lipases from biological entities for use in industrial reactors is an important goal. The most commonly used approaches for obtaining thermostable lipases are various immobilization techniques (Gandhi et al. 1996; Guisan et al. 2001; Palomo et al. 2003; Mateo et al. 2007; Iyer and Ananthanarayan 2008) and enzyme mutagenesis (Svendsen 2000; Eijsink et al. 2005). The combination of both has also been used to improve lipase thermostability (Costa et al. 2009).
Previous mutagenesis strategies to obtain a lipase with a higher thermostability have included introducing extra disulfide bonds (Yamaguchi et al. 1996) and directed evolution (Niu et al. 2006). Disulfide bonds make considerable contributions to protein stability (Yamaguchi et al. 1996; Mansfeld et al. 1997; Turunen et al. 2001; Shimizu-Ibuka et al. 2006; Mansfeld and Ulbrich-Hofmann 2007), mainly because of decreases in the conformational chain entropy of the denatured protein (Thornton 1981; Pace et al. 1988; Zhang et al. 1994). Many attempts have been made to enhance the stability of proteins by the introduction of novel disulfide bonds (Wells and Powers 1986; Matsumura and Matthews 1991; Kanaya et al. 1991; Wakarchuk et al. 1994; Yamaguchi et al. 1996; Clarke et al. 2000; Turunen et al. 2001; Mimura et al. 2005; Mansfeld and Ulbrich-Hofmann 2007). While some led to the expected stabilization (Kanaya et al. 1991; Wakarchuk et al. 1994; Yamaguchi et al. 1996; Turunen et al. 2001; Mansfeld and Ulbrich-Hofmann 2007), many variants showed no effect, or even destabilization, when compared to the native enzymes (Wells and Powers 1986; Matsumura and Matthews 1991; Clarke et al. 2000; Mimura et al. 2005). Possible reasons for detrimental effects include strain caused by steric contacts or because the introduced disulfide bond precluded required stereochemistry, or caused loss of favorable interactions after substitution of a residue with cysteine (Katz and Kossiakoff 1986; Mitchinson and Wells 1989).
Disulfide by Design is a program for designing novel protein disulfide bonds that is based on an algorithm previously developed to assist in protein fold recognition (Dombkowski and Crippen 2000). This method has made considerable contributions to identifying residue pairs that are likely to form a disulfide bond if particular amino acids are mutated to cysteines, but it requires that a crystal structure of the given protein is known. Cases where disulfide bonds have been successfully introduced using this program to create a stabilized protein include (βα)8-barrel protein (Ivens et al. 2002), Trichoderma reesei endo-1,4-beta-xylanase II (Fenel et al. 2004), a cold-adapted alkaline phosphatase (Ásgeirsson et al. 2007), and xylanase from Bacillus stearothermophilus (Jeong et al. 2007).
Yeast cell-surface display systems are convenient tools for applied biotechnology. The cell-surface display of lipases on yeast has been effective for the preparation of whole-cell biocatalysts (Matsumoto et al. 2002; Sato et al. 2002; Tanino et al. 2006). The productivity of lipase-displaying yeast cells could be improved, however, to increase their usefulness in large-scale manufacturing processes.
In this work, Rhizomucor miehei lipase with enhanced thermostability was generated for improving potential applications. Because the 3-dimensional (3-D) structure of R. miehei lipase had been determined by X-ray crystallography (Brady et al. 1990), an R. miehei lipase variant with a new disulfide bond could be engineered by Disulfide by Design and the indicated cysteine residues substituted before functionally expressing the R. miehei lipase variant in P. pastoris. Both a free form and a cell-surface-displayed form were generated, as a fusion with the N-terminal 874 residues of the Flo1p flocculation domain (FS). The two forms of the R. miehei lipase variant were subjected to thermostability assays using the commercial Lipozyme IM RM as a benchmark.
Materials and methods
Strains, vectors, and media
P. pastoris GS115 and plasmid pPIC9k were purchased from Invitrogen (USA). The ProRML gene was amplified from plasmid pPIC9K-RML (Xu et al. 2008). Plasmid pouBis containing Flo1 gene was a gift from Prof. Blondin (France). Surface-display plasmid pKFS was constructed from plasmid pPIC9k and the N-terminal 874 residues of Flo1 (FS). P. pastoris GS115 was grown in complete YPD (1% yeast extract, 2% peptone, 2% glucose) or selective MD (1.34% yeast nitrogen base (YNB), 400 μg l−1 biotin, 2% dextrose, 2% agar) media. BMGY (1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1.34% YNB, 400 μg l−1 biotin, 1% glycerol) and BMMY (1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1.34% YNB, 400 μg l−1 biotin, 0.5% methanol) were used to culture the recombinant P. pastoris. Escherchia coli DH5α (supE44 ∆lacU169 hsdR17 recA1 endA1 gyrA96 thi-1 relA1; TaKaRa, Dalian) was used for the transformation of recombinant plasmids and was grown in Luria–Bertani medium (1% tryptone, 0.5% yeast extract, 1% NaCl, pH 7.0) with ampicillin (50 μg ml−1) added when necessary.
Disulfide bond design and plasmid construction
A model of R. miehei lipase (3TGL; http://www.rcsb.org/pdb) was analyzed using Disulfide by Design, which recognizes cysteine pairs that are in the proper orientation to form a disulfide bond (Dombkowski 2003). Swiss-PdbViewer (http://www.expasy.ch/spdbv/; Guex and Peitsch 1997) was used for viewing and manipulating the R. miehei lipase structures and models.
To facilitate purification of the lipases, the C-terminal hexahistidine (His6)-tagged ProRML was amplified using the primers 5′-ccggaattcgttccaattaagagacaatctaac-3′ with an EcoRI site and 5′-tattat gcggccgcttagtgatgatgatgatgatgagtacacaaaccagtgtt-3′ with a NotI site, using plasmid pPIC9K-RML as a template. The ProRML gene was ligated to the plasmid pPIC9k after digestion with EcoRI and NotI, and the resulting plasmid was designated pPICR. A plasmid containing His6-tagged ProRML with the Pro96Cys and Lys106Cys mutations was created by QuikChange mutagenesis (Stratagene) using pPICR as a template. The Pro96Cys mutation was created using the primer 5′-gacttttgtttgtgtttcttacccac-3′, and the Lys106Cys mutation was created using the primer 5′-gtttctggtacttgtgttcataaggg-3′. The mutagenesis oligonucleotides were synthesized by Sangon (Shanghai, China) and the mismatched sites for site-directed mutagenesis are underlined. After sequencing to verify the mutations, the resulting plasmid was designated pPICR66 (Fig. 1a) and the mutated RML named mRML66.

a To construct plasmid pPICR, the C-terminal His6-tagged ProRML was link to pPIC9K after enzymatic digestion with EcoRI and NotI; the mutated plasmid pPICR66 was created through QuikChange mutagenesis (Stratagene) using plasmid pPICR as a template. b To construct plasmids for cell-surface display, ProRML and PromRML66 were linked to the plasmid pKFS to form a complete open reading frame with FS
To display R. miehei lipase and its variant mRML66 on the P. pastoris cell surface, both RML and mRML66 were amplified using the primer 5′-ccggaattcgttccaattaagagacaatctaac-3′ with an EcoRI site and 5′-cgccgc gcggccgc ttaagtacacaaaccagtgttaatac-3′ with a NotI site using pPICR and pPICR66 as templates. Subsequently, the two fragments were ligated to the surface-display plasmid pKFS after digestion with EcoRI and NotI, and the resultant plasmids were designated as pKFSR and pKFSR66 (Fig. 1b). All plasmids were verified by sequencing by Sangon (Shanghai, China).
Transformation of P. pastoris and expression of lipases
Lithium chloride-competent GS115 was obtained as described by the Pichia Expression Kit Instruction Manual (Invitrogen 1997). P. pastoris GS115 harboring recombinant plasmids was spread on BMGY plates for 1 day at 30°C and then transferred onto BMMY plates supplemented with 0.5% tributyrin before adding 100 μl methanol every 24 h to induce expression of R. miehei lipase and the variant mRML66. P. pastoris GS115 harboring pPIC9K and pKFS were spread on plates and cultured as controls. Lipase activity was determined by clear zones around the colonies.
Expression and purification of lipases
Fresh P. pastoris transformants were inoculated into 50 ml BMGY medium in a 500-ml flask. After 24 h of cultivation, the culture was centrifuged at 6,000×g for 10 min and resuspended in BMMY medium containing 0.5% methanol. In order to maintain the induction of R. miehei lipase and mRML66, 100% methanol was added every 24 h to the culture to a final concentration of 0.5%. Cells were incubated for 6 days at 30°C at 250 rpm shaking.
After centrifugation of the culture medium at 10,000×g for 10 min at 4°C to remove the yeast cells, the supernatant was ultrafiltered with a Millipore Amicon ultra-15 10K centrifugal device. The concentrate was collected and diluted in 5 ml of buffer A (20 mM sodium phosphate containing 500 mM NaCl and 20 mM imidazole, pH 8.0) before purification with an ÄKTA purifier system using a Ni2+-charged 1-ml HisTrap® FF Crude column (GE Healthcare) equilibrated with buffer A. Lipases were eluted with buffer B (buffer A plus 500 mM imidazole). Purified lipases were stored at −20°C in 15% glycerol until use. Samples were boiled in 120 mM Tris-Cl buffer (pH 6.8) containing 2% sodium dodecyl sulfate (SDS), 3% glycerol, and 1% β-mercaptoethanol (β-ME) for 10 min. Samples were centrifuged at 10,000×g for 5 min, and supernatants subjected to a 12% SDS-polyacrylamide gel electrophoresis (PAGE) with 0.1% SDS (Laemmli 1970) in a vertical slab gel apparatus (Bio-Rad, USA).
Determination of protein concentration
The Bradford method was used to measure protein concentration in solutions. A 20% (v/v) solution of dye reagent (HOU-BIO, Hong Kong) was prepared with distilled water. A sample of 0.01 ml was added into 0.99 ml of Bradford solution. The test tubes were incubated at ambient temperature for 10 min. The quantity of protein in solution was determined by spectrophotometer (595 nm) using bovine serum albumin as the standard protein.
Confirmation of the de novo disulfide bond in the variant
SDS-PAGE was used to discriminate between the reduced and nonreduced lipases by difference in Stokes’ radius (Anthony et al. 2002). Samples were incubated for 1 h at room temperature in 60 mM Tris-HCl buffer (pH 8.0) containing 1% SDS and 3% glycerol in the presence (reducing conditions) or absence (nonreducing conditions) of 20 mM β-ME. Then samples were adjusted to pH 6.8 and centrifuged at 10,000×g for 5 min; supernatants were subjected to 12% SDS-PAGE.
Analysis of lipase activity
The commonly used procedure to investigate lipase activity uses 4-nitrophenyl caprylate (pNPC; Sigma) and the release of p-nitrophenol, measured spectrophotometrically at 405 nm. Free lipases and cell-surface-displayed lipases were dissolved in lipase assay buffer (50 mM Tris-HCl, pH 8.0), mixed with substrate solution (0.0625 mM pNPC, 0.1% Triton X-100 in lipase assay buffer), and reacted for 5 min before measuring OD405 with a kinetic microplate reader. Reactions were terminated by the addition of 20 μl of 20% trichloroacetic acid (Sigma) solution. After centrifugation at 10,000×g for 1 min, a 200-μl aliquot of the resultant supernatant was placed in a 96-well plate and measured. Product yields were determined from a standard curve prepared using p-nitrophenol as a standard. One unit of activity was defined as the amount of enzyme that released l μmol of pNP per minute from pNPC under the assay conditions. Each sample was assayed in triplicate and the average value was determined.
Enzymatic properties and kinetic parameters of free R. miehei lipase and R. miehei lipase variant
The optimum pH for the free R. miehei lipase and mRML66 was determined using pH buffer solutions ranging from 7.0 to 9.0. The optimum temperature of R. miehei lipase and mRML66 was determined by measuring the activity at pH 8.0 in a water bath at temperatures ranging from 30°C to 50°C. All the reactions were carried out for 5 min before the activity was calculated.
The kinetic parameters K m and V max were determined for lipase activity. Enzyme assays with 50 μl of free lipases were performed in Tris-HCl buffer, pH 8.0, with pNPC concentrations from 6.25 to 62.5 μM. The data obtained were plotted according to the method of Lineweaver–Burk to determine K m and V max graphically (Sharma et al. 2002).
Thermostability of lipases in aqueous solution
To determine the thermostability of free R. miehei lipase and mRML66 and further confirm the new disulfide bond in mRML66, the purified lipases of free R. miehei lipase and mRML66 were incubated in 50 mM Tris-HCl buffer (pH 8.0) in the presence or absence of 20 mM β-ME at room temperature for 1 h and maintaining the solution at 60°C in a water bath with periodic shaking. Samples were taken every 2 min for the measurement of residual lipase activity under standard assay conditions (pH 8.0, 40°C); 50 μl aliquots were diluted into 1 ml of the activity assay solution.
For displayed lipases, yeast cells harvested from culture medium were washed twice with 50 mM Tris-HCl (pH 8.0) and resuspended in 50 mM Tris-HCl buffer (pH 8.0). The thermostability of the displayed lipases and the commercial Lipozyme IM RM standard were determined by keeping a suspension in a 60°C water bath with periodic shaking and removing samples every hour for the measurement of residual lipase activity.
Thermostability of lipases used for synthesis of ethyl caproate in organic solvent
The esterification reaction was performed by fully dehydrating all reagents in advance using zeolite. As substrate, 0.25 M ethanol and 0.2 M caproic acid were dissolved in heptane. A total of 200 mg (dry weight) of yeast displaying R. miehei lipase or commercial Lipozyme IM RM was suspended in 10 ml of the substrate mixture in a 50-ml conical flask, stoppered to prevent evaporation of solvents and reactants. The reaction was carried out at 60°C at 200 rpm. To obtain the initial synthetic activity of lipases, the samples were collected after 10 min of reaction and centrifuged at 10,000×g for 10 min. The supernatant or a final concentration of 0.04% butyl acetate as an internal standard were mixed and dissolved in 1 ml of n-hexane. A 2.0-μl aliquot of the treated sample was subjected to gas chromatography (Agilent 7890 A; Agilent chromatography workstation, hydrogen flame ionization detector; DB-FFAP capillary column; USA) to quantify the amount of ethyl caproate produced by the esterification reaction. The temperature for gasification and detection was 260°C, and the capillary column was maintained at 50°C for 1 min, then warmed to 60°C at a rate of 10°C min−1, then to 68°C at a rate of 40°C min−1, then to 95°C at a rate of 20°C min−1, followed by 200°C at a rate of 40°C min−1, maintained for 2 min. High-purity N2 was used as a carrier gas, the pressure of the precolumn was 58,800 Pa, the flow rate of H2 was 40 ml min−1, and the air was 400 ml min−1. One unit of synthetic activity was defined as the synthesis of l μmol of ethyl caproate per minute.
Results
Generation and confirmation of the introduced disulfide bond
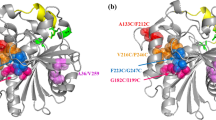
To improve the thermophilic profile of R. miehei lipase, the protein structure file in PDB format (3TGL) was analyzed. The output showed 39 residue pairs satisfying the geometric constraints for disulfide bonds, including the three original disulfide bonds (residues 29–268, 40–43, 235–244) that stabilize the molecule (Brady et al. 1990; Table 1). The results showed that the software was effective in predicting the residue pairs to form disulfide bonds. Next, to select the most promising candidates for new disulfide bonds, the locations of residues that would be changed to cysteine were investigated. Two amino acids, Pro96 and Lys106, were on the β-sheet (Fig. 2), could form a loop to stabilize the active site lid region, and had a strong potential for disulfide bond formation when substituted with cysteine (estimated χ 3 torsion angle of 129.87°). Furthermore, the active site lid region containing Pro96 and Lys106 is reported to be related to the activation of R. miehei lipase (Peters and Bywater 2002), and the positions of the amino acids are far from the catalytic center of R. miehei lipase by tertiary structure analysis (Fig. 2a). Cysteines were modeled in the two positions and the resulting 3TGL model containing the Cys96–Cys106 disulfide was viewed and manipulated using Swiss-PdbViewer. The 3-D structure predicted that Cys96 and Cys106 would be exposed and the disulfide bond would be generated successfully (Fig. 2b). Consequently, the mutations were quantified with the GROMOS96 (van Gunsteren et al. 1996) using Swiss-PdbViewer with the GROMOS96 43B1 parameters set, and the resulting models with mutations were subjected to energy minimization analysis. This resulted in significant predicted reduction of calculated total free energy, from −12,604.277 to −14,235.750 kJ mol−1, meaning that the variant was predicted to be more stable than the native lipase.

a Molecular graphic cartoon showing the main chain of R. miehei lipase. The catalytic triad (Ser144, Asp203, and His257) and the two mutated sites, Pro96 and Lys106, were labeled in yellow. b Stereo view of the disulfide bond and its surrounding. The disulfide bond was shown in yellow. The green dashed lines indicate hydrogen bonding network around disulfide bond
Based on these observations, we introduced a disulfide bond between positions Pro96 and Lys106 by substituting the two residues with cysteine by site-directed mutagenesis. The wild-type lipase and the resulting variant were designated R. miehei lipase and mRML66, respectively. The selected variants were constructed and successfully expressed in P. pastoris. The C-terminal His6-tagged R. miehei lipase and mRML66 were expressed in GS115 and purified with a Ni2+-chelating affinity column. SDS-PAGE analysis revealed that the molecular mass of purified R. miehei lipase and mRML66 was about 32 kDa (Fig. 3), consistent with the reported molecular mass (Wu et al. 1996).

SDS-PAGE analysis of R. miehei lipase and mRML66 purified by the C-terminal His6-tag. Samples were boiled with 120 mM Tris-Cl buffer, pH 6.8, containing 2% SDS, 3% glycerol, and 1% β-ME for 10 min. The sample buffers were centrifuged at 10,000×g for 5 min, resolved on 12% polyacrylamide gel, and then stained with Coomassie blue R-250. Lane M protein molecular mass markers (TaKaRa). Sizes in kilodaltons are indicated on the left; lane 1 purified R. miehei lipase, lane 2 purified mRML66
P. pastoris is known to be capable of producing disulfide bonds in proteins. To verify that a disulfide had formed, samples of the purified proteins were subjected to SDS-PAGE under reducing and nonreducing conditions. Reduced and nonreduced R. miehei lipase showed almost no difference in mobility, probably because the three original disulfide bonds are buried in the core of the molecule and solvent inaccessible and the mild treatment of the samples could not open the interior disulfide bonds. This would result in identical conformations for reduced and nonreduced R. miehei lipase. mRML66 ran at a lower apparent molecular mass than R. miehei lipase (Fig. 4), probably due to the addition of the two cysteine residues, because both the Pro96 and Lys106 in R. miehei lipase are exposed to the surface and have longer side chains than cysteine residues. The substitution of proline with cysteine would cause some conformation changes, because proline has an exceptional conformational rigidity compared to other amino acids, so mRML66 would be expected to have a smaller Stokes’ radius than R. miehei lipase. The treatment of R. miehei lipase and mRML66 for SDS-PAGE did not result in full denaturation. SDS is not expected to fully bind to the lipases to form protein–SDS complexes (Fig. 3). It is expected to bind only to the exposed side chains and not to the exposed disulfide bond, because of the lack of side chains. Therefore, in addition to a smaller Stokes’ radius, lower predicted SDS binding to mRML66 would cause mRML66 to have a greater SDS-PAGE mobility than R. miehei lipase. An approximately 1-kDa difference in mobility was observed between reduced and nonreduced mRML66. The new disulfide bond was exposed to the surface and could be easily reduced in the presence of β-ME, thus causing a conformational change between reduced and nonreduced mRML66. The difference in mobility may be due to nonreduced mRML66 having a more regular Stokes’ radius that caused slightly accelerated migration in SDS-PAGE (Hartig et al. 2005; Truscott et al. 2007). Another explanation is that the restricted conformation of the nonreduced form might be resistant to degradation compared to the reduced form, or less SDS binding might result in slightly accelerated migration, causing the nonreduced form to appear smaller than the reduced form.

SDS-PAGE analysis of R. miehei lipase and mRML66 under reducing and nonreducing conditions. Samples were digested with 60 mM Tris-Cl buffer, pH 8.0, containing 1% SDS and 3% glycerol in the presence (reducing conditions) or absence of 20 mM β-ME (nonreducing conditions) for 1 h at room temperature. Lane 1 reduced R. miehei lipase, lane 2 nonreduced R. miehei lipase, lane 3 reduced mRML66, lane 4 nonreduced mRML66. Lane M is protein molecular mass markers (TaKaRa). Sizes in kilodaltons are indicated on the right
Enzymatic properties and kinetic parameters of free R. miehei lipase and mRML66
To compare the enzymatic properties and kinetic parameters of R. miehei lipase and mRML66, the two His6-tagged lipases were assayed as described in “Materials and methods” section. As shown in Table 2, the optimum pH of R. miehei lipase and mRML66 was estimated to be 8.0, the same as the lipase without the His6-tag (Xu et al. 2008), and the optimum temperatures were 40°C for R. miehei lipase and 43°C for mRML66. The specific activity was 194.5 ± 2.8 U mg−1 for R. miehei lipase and 239.4 ± 5.0 U mg−1 for mRML66. mRML66 showed altered kinetic parameters compared to R. miehei lipase. The K m values for R. miehei lipase and mRML66 were 22.0 ± 1.7 and 25.9 ± 1.3 μM, respectively. The catalytic constant (k cat) was calculated to be 7,721.5 ± 188.4 min−1for R. miehei lipase and 9,595.4 ± 290.9 min−1 for mRML66. These results indicated that the presence of the introduced novel disulfide bond changed the reaction velocity and substrate affinity of mRML66. Furthermore, the specific activity of mRML66 was slightly higher than R. miehei lipase, while displaying, in some cases, identical or more reactive characteristics, indicating that the conformation was stabilized by the introduced disulfide bond.
Thermostability of lipases in aqueous solution
To determine the effect of the introduced disulfide bond on the thermostability of R. miehei lipase, purified R. miehei lipase and mRML66 were incubated at 60°C and hydrolysis activity examined at various times. The substrate hydrolyzing ability of both lipases decreased with increase in incubation time. However, the thermostability of free mRML66 was improved, with half-times approximately fivefold that of native R. miehei lipase at 60°C, as determined by pNPC hydrolysis. The thermostability of R. miehei lipase before and after treatment with the reducing agent β-ME was almost the same, which indicated that the conformation was not changed by β-ME and the three original disulfides were not reduced. While the β-ME-treated mRML66 showed a lower thermostability than the nonreduced mRML66, the result suggested that the newly designed disulfide in this study was reduced by β-ME and caused the loss of its thermostability (Fig. 5a). This result further confirmed the formation of the new disulfide bond in mRML66. In this manner, it was possible to rapidly screen for the presence of new disulfide bond.

a Time courses of residual activity of free R. miehei lipase (open diamond) and mRML66 (open square) in the absence of β-ME, or free R. miehei lipase (closed diamond) and mRML66 (closed square) in the presence of β-ME. One milliliter of enzyme solution, 50 μg of purified lipases were incubated in 50 mM Tris-HCl buffer (pH 8.0) in the presence or absence of 20 mM β-ME at room temperature for 1 h and then incubated at 60°C over time. After incubation, 50 aliquots were diluted into 1 ml of the activity assay solution. b Time courses of residual activity of displayed R. miehei lipase (open diamond), displayed mRML66 (open square), and commercial Lipozyme IM RM (open triangle) in aqueous solution at 60°C. The relative activity indicates residual lipase activity which was calculated relative to the initial lipase activity, which was defined as 100%. The data points represent the average of three independent experiments
The thermostability of surface-displayed lipases in aqueous solution was also analyzed. The maximum activities of displayed R. miehei lipase and mRML66 for hydrolysis of pNPC were 105.3 and 112.3 U g−1 dry cell weight, respectively. Irreversible thermal inactivation of displayed R. miehei lipase and displayed mRML66 was determined by incubation in aqueous buffer at 60°C. Interestingly, R. miehei lipase and mRML66 displayed on the yeast cell surface showed not only a high thermostability but also a remarkable increase in activity (Fig. 5b), reaching a value approximately 1.6 times the initial value after 1 h incubation at 60°C. Similarly, Tanino et al. (2006) reported that the activity of Rhizopus oryzae lipase displayed on yeast using the same anchor system increased to about 6.5-fold of the initial value after 4 h at 60°C. The thermostability of displayed mRML66 against irreversible inactivation was significantly higher than displayed R. miehei lipase, with residual activities of 47% and 88% of the initial activity, respectively, after incubation at 60°C for 5 h. This result suggested that the thermostability of displayed lipases was superior to their free form. In this investigation, the initial activity of commercial Lipozyme IM RM was about 10 U/g. When its thermostability was tested in the same conditions, the result indicated that both the surface-displayed R. miehei lipase and mRML66 showed higher activity and tolerance than Lipozyme IM RM in aqueous solution.
Thermostability of lipases in the synthesis of ethyl caproate in organic solvent
Esterification of ethanol and caproic acid to ethyl caproate by displayed lipases was analyzed. Ethanol and caproic acid could easily access the surface-displayed lipases. Cells displaying lipases were therefore used for ethyl caproate production without any permeabilizing treatment. To compare the catalytic ability of lipases, the initial synthetic activities of displayed R. miehei lipase, displayed mRML66, and Lipozyme IM RM were determined to be 27.7, 78.8, and 243.2 U g−1 dry cell weight, respectively. This indicated that the disulfide bond increased the synthetic ability of displayed mRML66, but its activity was lower than commercial Lipozyme IM RM. The conversion rate increased with reaction time at 60°C, and the caproic acid molar conversion rate for displayed mRML66 and Lipozyme IM RM was about three- or fourfold higher than displayed R. miehei lipase, after a 0.5-h reaction (Fig. 6). However, the caproic acid molar conversion of displayed mRML66 and Lipozyme IM RM reached 96% after 6 h at 60°C, while the conversion rate of displayed R. miehei lipase increased slowly after 3 h. This indicated that displayed R. miehei lipase was almost inactivated after 3 h at 60°C, while displayed mRML66 remained active. It also indicated that the introduced disulfide bond stabilized the conformation of R. miehei lipase in organic solvent. Although Lipozyme RM IM showed a higher initial synthetic activity than displayed mRML66 in organic solvent, displayed mRML66 showed a higher final conversion rate for caproic acid than Lipozyme RM IM after 9 h of reaction, indicating a better thermostability.

Time course of esterification reaction using displayed R. miehei lipase (open diamond), displayed mRML66 (open square), and commercial Lipozyme IM RM (open triangle) and GS115/pKFS (multiplication symbol) in organic solvent. The reactions were carried out in a 50-ml conical flask with stopper containing 10 ml of n-heptane, 0.2 M caproic acid, 0.25 M ethanol, and 0.2 g whole-cell biocatalyst or commercial Lipozyme IM RM at 200 rpm and 60°C. Molecular sieves (3 Å) were added to the reaction system to remove the water produced in the reaction. Percentages of caproic acid molar conversion were plotted against reaction time. The data points represent the average of three independent experiments
Discussion
R. miehei lipase has been frequently used for hydrolysis and transesterification and is a model for the molecular modeling of other homologous lipases, because of the extensive knowledge of its 3-D structure (Brady et al. 1990). In this study, we determined the most effective cysteine residues for replacement in R. miehei lipase, using a disulfide bond design program and the 3-D structure model. We investigated the effect of an introduced disulfide bond on the enzymatic properties of free lipases and on lipases anchored to the yeast cell surface with FS. Cell-surface display is an in vivo immobilization that cannot only be prepared easily by simple cultivation and separation of cells but also stabilizes and immobilizes the enzyme (Jung et al. 2006). Thermostability generally results from molecular rigidity introduced by attachment to a rigid support and creation of a protected microenvironment. Previous studies on displaying R. miehei lipase on the Saccharomyces cerevisiae cell surface using α-agglutinin as an anchor protein showed little improvement in thermostability (Zhang et al. 2007). P. pastoris is used for the expression of heterogeneous proteins because it has a high growth rate and is easily cultivated on simple inexpensive media. Its major advantage is that it is appropriate for protein folding, with the capability to produce disulfide bonds and glycosylation. The results of this study also indicated that R. miehei lipase displayed on the P. pastoris cell surface with FS improved its thermostability. Furthermore, stabilization improved with the introduced engineered disulfide bond. This could be due to addition to the molecular rigidity by the disulfide bond and the result of immobilization. Hydrophobic effects and hydrogen bonds may also play a role in the stabilization (Fig. 2b). The formation of intramolecular disulfide bond may result in structural changes that alter the short-range order of mRML66.
The thermostability is of particular interest since it has a marked effect on the lipase activity. While the disulfide bond has a strong effect on the thermostability of mRML66, excessive rigidity in this region may influence the catalytic activity. In this investigation, hydrolytic and synthetic tests allowed the comparison of whole cell lipases and immobilized commercial Lipozyme IM RM, with very promising results for the yeast cell-surface-displayed mRML66. Although the synthetic activity of displayed mRML66 was lower than Lipozyme IM RM, mRML66 had a significantly improved activity and thermostability over displayed R. miehei lipase and was feasible for high-temperature industrial reactions.
Lipozyme RM IM had better synthesis activity in organic solvent but a poorer hydrolysis activity in aqueous solution, than yeast cell-surface-displayed lipases. The difference was probably attributable to the anion-exchange resin, which was a hydrophobic support whose surface would improve the contact of lipases and substrate in organic solvent, while the yeast cell surface on which R. miehei lipase and its variant were displayed was highly glycosylated and hydrophilic (Tanino et al. 2006).
In summary, a strategically placed disulfide bond made the R. miehei lipase more thermostable and able to resist thermal inactivation, suggesting it would be more effective in resisting the extremely harsh conditions of industrial processes.
References
Anthony LC, Dombkowski AA, Burgess RR (2002) Using disulfide engineering to study conformational changes in the β′260–309 coiled-coil region of E. coli RNA polymerase during σ70 binding. J Bacteriol 184:2634–2641. doi:https://doi.org/10.1128/JB.184.10.2634-2641.2002
Ásgeirsson B, Adalbjörnsson BV, Gylfason GA (2007) Engineered disulfide bonds increase active-site local stability and reduce catalytic activity of a cold-adapted alkaline phosphatase. Biochim Biophys Acta 1774:679–687. doi:https://doi.org/10.1016/j.bbapap.2007.03.016
Brady L, Brzozowski AM, Derewenda ZS (1990) A serine protease triad forms the catalytic centre of a triacylglycerol lipase. Nature 343:767–770
Clarke J, Hounslow AM, Bond CJ, Fersht AR, Daggett V (2000) The effects of disulfide bonds on the denatured state of barnase. Protein Sci 9:2394–2404
Costa L, Brissos V, Lemos F, Ramôa Ribeiro F, Cabral JM (2009) Enhancing the thermal stability of lipases through mutagenesis and immobilization on zeolites. Bioprocess Biosyst Eng 32:53–61. doi:https://doi.org/10.1007/s00449-008-0220-x
Dombkowski AA (2003) Disulfide by design: a computational method for the rational design of disulfide bonds in proteins. Bioinformatics 19:1852–1853
Dombkowski AA, Crippen GM (2000) Disulfide recognition in an optimized threading potential. Protein Eng 13:679–689
Eijsink VGH, Gaseidnes S, Borchert TV, van den Burg B (2005) Directed evolution of enzyme stability. Biomol Eng 22:21–30. doi:https://doi.org/10.1016/j.bioeng.2004.12.003
Fenel F, Leisola M, Janis J, Turunen O (2004) A de novo designed N-terminal disulphide bridge stabilizes the Trichoderma reesei endo-1, 4-beta-xylanase II. J Biotechnol 108:137–143. doi:https://doi.org/10.1016/j.jbiotec.2003.11.002
Gandhi NN, Vijayalakshmi VV, Sawant SB, Joshi JB (1996) Immobilization of Mucor miehei lipase on ion exchange resin. Chem Eng J 61:149–156
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723
Guisan JM, Sabuquillo P, Fernandez-Lafuente R, Fernandez-Lorente G, Mateo C, Halling PJ, Kennedy D, Miyata E, Re D (2001) Preparation of new lipases derivatives with high activity–stability in an hydrous media: adsorption on hydrophobic supports plus hydrophilization with polyethylenimine. J Mol Catal B-Enzym 11:817–824. doi:https://doi.org/10.1016/S1381-1177(00)00011-4
Hartig GRS, Tran TT, Smythe ML (2005) Intramolecular disulphide bond arrangements in nonhomologous proteins. Protein Sci 14:474–482. doi:https://doi.org/10.1110/ps.04923305
Invitrogen (1997) A manual of methods for expression of recombinant proteins in Pichia pastoris, Catalog No. K1710-01. Invitrogen, San Diego
Ivens A, Mayans O, Szadkowski H, Jurgens C, Wilmanns M, Kirschner K (2002) Stabilization of a (βα) 8-barrel protein by an engineered disulfide bridge. Eur J Biochem 269:1145–1153
Iyer PV, Ananthanarayan L (2008) Enzyme stability and stabilization—aqueous and non-aqueous environment. Proc Biochem 43:1019–1032. doi:https://doi.org/10.1016/j.procbio.2008.06.004
Jeong MY, Kim S, Yun CW, Choi YJ, Cho SG (2007) Engineering a de novo internal disulfide bridge to improve the thermal stability of xylanase from Bacillus stearothermophilus No. 236. J Biotechnol 127:300–309. doi:https://doi.org/10.1016/j.jbiotec.2006.07.005
Jung HC, Kwon SJ, Pan JG (2006) Display of a thermostable lipase on the surface of a solvent-resistant bacterium, Pseudomonas putida GM730, and its applications in whole-cell biocatalysis. BMC Biotechnol 6:23–31. doi:https://doi.org/10.1186/1472-6750-6-23
Kanaya S, Katsuda C, Kimura S, Nakai T, Kitakuni E, Nakamura H, Katayanagi K, Morikawa K, Ikehara M (1991) Stabilization of Escherichia coli ribonuclease H by introduction of an artificial disulfide bond. J Biol Chem 266:6038–6044
Katz BA, Kossiakoff A (1986) The crystallographically determined structures of atypical strained disulfides engineered into subtilisin. J Biol Chem 261:15480–15485
Keng PS, Basri M, Ariff AB, Abdul Rahman MB, Abdul Rahman RN, Salleh AB (2008) Scale-up synthesis of lipase-catalyzed palm esters in stirred-tank reactor. Bioresour Technol 99:6097–6104. doi:https://doi.org/10.1016/j.biortech.2007.12.049
Laemmli UK (1970) Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 227:680–685
Mansfeld J, Ulbrich-Hofmann R (2007) The stability of engineered thermostable neutral proteases from Bacillus stearothermophilus in organic solvents and detergents. Biotechnol Bioeng 97:672–679. doi:https://doi.org/10.1002/bit.21292
Mansfeld J, Vriend G, Dijkstra BW, Veltman OR, Van den Burg B, Venema G, Ulbrich-Hofmann R, Eijsinki VGH (1997) Extreme stabilization of athermolysin-like protease by an engineered disulfide Bond. J Biol Chem 272:11152–11156
Mateo C, Palomo JM, Fernandez-Lorente G, Guisan JM, Fernandez-Lafuente R (2007) Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzyme Microb Technol 40:1451–1463. doi:https://doi.org/10.1016/j.enzmictec.2007.01.018
Matsumoto T, Fukuda H, Ueda M (2002) Construction of yeast strains with high cell surface lipase activity by using novel display systems based on the Flo1p flocculation functional domain. Appl Environ Microbiol 68:4517–4522. doi:https://doi.org/10.1128/AEM.68.9.4517-4522.2002
Matsumura M, Matthews BW (1991) Stabilization of functional proteins by introduction of multiple disulfide bonds. Methods Enzymol 202:336–356
Mimura H, Nakanishi Y, Maeshima M (2005) Disulfide-bond formation in the H(+)-pyrophosphatase of Streptomyces coelicolor and its implications for redox control and enzyme structure. FEBS Lett 579:3625–3631. doi:https://doi.org/10.1016/j.febslet.2005.05.045
Mitchinson C, Wells JA (1989) Protein engineering of disulfide bonds in subtilisin BPN'. Biochemistry 28:4807–4815
Niu WN, Li ZP, Zhang DW, Yu MR, Tan TW (2006) Improved thermostability and the optimum temperature of Rhizopus arrhizus lipase by directed evolution. J Mol Catal B-Enzym 43:33–39. doi:https://doi.org/10.1016/j.molcatb.2006.04.013
Noel M, Combes D (2003) Rhizomucor miehei lipase: differential scanning calorimetry and pressure/temperature stability studies in presence of soluble additives. Enzyme Microb Technol 33:299–308. doi:https://doi.org/10.1016/S0141-0229(03)00123-6
Pace CN, Grimsley GR, Thomson JA, Barnett BJ (1988) Conformational stability and activity of ribonuclease T1 with zero, one, and two intact disulfide bonds. J Biol Chem 263:11820–11825
Palomo JM, Muñoz G, Fernandez-Lorente G, Mateo C, Fuentes M, Guisan JM, Fernandez-Lafuente R (2003) Modulation of Mucor miehei lipase properties via directed immobilization on different hetero-functional epoxy resins. Hydrolytic resolution of (R, S)-2-butyroyl-2-phenylacetic acid. J Mol Catal B Enzym 21:201–210. doi:https://doi.org/10.1016/S1381-1177(02)00224-2
Peters GH, Bywater RP (2002) Essential motions in a fungal lipase with bound substrate, covalently attached inhibitor and product. J Mol Recognit 15:393–404. doi:https://doi.org/10.1002/jmr.579
Peterson ME, Daniel RM, Danson MJ, Eisenthal R (2007) The dependence of enzyme activity with temperature: determination and validation of parameters. Biochem J 402:331–337. doi:https://doi.org/10.1042/BJ20061143
Sato N, Matsumoto T, Ueda M, Tanaka A, Fukuda H, Kondo A (2002) Long anchor using Flo1 protein enhances reactivity of cell surface-displayed glucoamylase to polymer substrates. Appl Microbiol Biotechnol 60:469–474. doi:https://doi.org/10.1007/s00253-002-1121-6
Sharma R, Soni SK, Vohra RM, Gupta LK Gupta JK (2002) Purification and characterization of a thermostable alkaline lipase from a new thermophilic Bacillus sp. RSJ-1. Proc Biochem 37:1075–1084. doi:https://doi.org/10.1016/S0032-9592(01)00316-8
Shimizu-Ibuka A, Matsuzawa H, Sakai H (2006) Effect of disulfide-bond introduction on the activity and stability of the extended-spectrum class A β-lactamase Toho-1. Biochim Biophys Acta 1764:1349–1355
Svendsen A (2000) Lipase protein engineering. Biochim Biophys Acta 1543:223–238
Tanino T, Fukuda H, Kondo A (2006) Construction of a Pichia pastoris cell-surface display system using Flo1p anchor system. Biotechnol Prog 22:989–993. doi:https://doi.org/10.1021/bp060133+
Thornton JM (1981) Disulphide bridges in globular proteins. J Mol Biol 151:261–287
Truscott SM, Lybarger L, Martinko JM, Mitaksov VE, Kranz DM, Connolly JM, Fremont DH, Hansen TH (2007) Disulfide bond engineering to trap peptides in the MHC class I binding groove. J Immunol 178:6280–6289
Turunen O, Etuaho K, Fenel F, Vehmaanpera J, Wu X, Rouvinen J, Leisola M (2001) A combination of weakly stabilizing mutations with a disulfide bridge in the α-helix region of Trichoderma reesei endo-1, 4-β-xylanase II increases the thermal stability through synergism. J Biotechnol 88:37–46. doi:https://doi.org/10.1016/S0168-1656(01)00253-X
van Gunsteren WF, Billeter SR, Eising AA, Hünenberger PH, Krüger P, Mark AE, Scott WRP, Tironi IG (1996) Biomolecular simulation: the GROMOS96 manual and user guide. vdf Hochschulverlag, ETH Zurich, Zurich
Wakarchuk WW, Sung WL, Campbell RL, Cunningham A, Watson DC, Yaguchi M (1994) Thermostabilization of Bacillus circulans xylanase by the introduction of disulfide bonds. Protein Eng 7:1379–1386
Weber N, Klein E, Vosmann K, Mukherjee KD (2004) Mono-thioesters and di-thioesters by lipase-catalyzed reactions of alpha, omega-alkanedithiols with palmitic acid or its methyl ester. Appl Microbiol Biotechnol 64:800–805. doi:https://doi.org/10.1007/s00253-004-1604-8
Weber N, Bergander K, Fehling E, Klein E, Vosmann K, Mukherjee KD (2006) Copolymeric polythioesters by lipase-catalyzed thioesterification and transthioesterification of alpha, omega-alkanedithiols. Appl Microbiol Biotechnol 70:290–297. doi:https://doi.org/10.1007/s00253-005-027-5
Wells JA, Powers DB (1986) In vivo formation and stability of engineered disulfide bonds in subtilisin. J Biol Chem 261:6564–6570
Wu XY, Jääskeläinen S, Linko Y-Y (1996) Purification and partial characterization of Rhizomucor miehei lipase for ester synthesis. Appl Biochem Biotech 59:145–158
Xu SW, Lin Y, Han ZL, Liang SL, Han SY (2008) High level expression of Rhizomucor miehei lipase gene in Pichia pastoris. Food and Ferment Ind 34:10–14 (In Chinese)
Yamaguchi S, Takeuchi K, Mase T, Oikawa K, McMullen T, Derewenda U, McElhaney RN, Kay CM, Derewenda ZS (1996) The consequences of engineering an extra disulfide bond in the Penicillium camembertii mono-and diglyceride specific lipase. Protein Eng 9:789–795
Zhang T, Bertelsen E, Alber T (1994) Entropic effects of disulphide bonds on protein stability. Nat Struct Biol 1:434–438
Zhang WG, Han SY, Wei DZ, Lin Y, Wang XN (2007) Functional display of Rhizomucor miehei lipase on surface of Saccharomyces cerevisiae with higher activity and its practical properties. J Chem Technol Biotechnol 3:329–335. doi:https://doi.org/10.1002/jctb.1814
Acknowledgments
The work was supported by the grant from National Natural Science Foundation of China (No. U0773001) and Ministry of Science and Technology of the People’s Republic of China (National “863” Project No. B1070230)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Han, Zl., Han, Sy., Zheng, Sp. et al. Enhancing thermostability of a Rhizomucor miehei lipase by engineering a disulfide bond and displaying on the yeast cell surface. Appl Microbiol Biotechnol 85, 117–126 (2009). https://doi.org/10.1007/s00253-009-2067-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2067-8