
Abstract
Carbohydrate structures have been identified in eukaryotic and prokaryotic cells as glycoconjugates with communication skills. Their recently discussed role in various diseases has attracted high attention in the development of simple and convenient methods for oligosaccharide synthesis. In this review, recent approaches combining nature’s power for the design of tailor made biocatalysts by enzyme engineering and substrate engineering will be presented. These strategies lead to highly efficient and selective glycosylation reactions. The introduced concept shall be a first step in the direction to a glycosylation toolbox which paves the way for the tailor-made synthesis of designed carbohydrate structures.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oligosaccharides with biological background have attracted high interest both scientifically and industrially over the past years. They are involved in various processes like inflammation, viral and bacterial infections, cell–cell recognition and immune response (Varki 1993; Yun 1996; Wong 2005). Moreover, many oligosaccharides are currently produced for commercial markets (Nakakuki 2002), e.g. isomaltooligosaccharides (IMO; Buchholz and Seibel 2003), leucrose (O-α-d-glucopyranosyl-(1,5)-d-fructopyranoside; Buchholz et al. 1998) and palatinose (O-α-d-glucopyranosyl-(1,6)-d-fructopyranoside; Lina et al. 2002). Oligosaccharides are of interest in the fields of food, pharmacy and cosmetics due to their ability to prevent and treat diseases from various biological origins. Isomaltose for instance enhances cytokine interleukin (IL)-12 production by macrophages stimulated by Lactobacillus gasseri in vitro. Dietary IMO significantly increase the number of lactobacilli in the intestinal microflora (Mizubuchi et al. 2005). However, the access to defined tailor-made oligosaccharides in reasonable amounts is still in demand. In this review, we will present chemo-enzymatic approaches enhancing the accessibility of tailor-made oligosaccharides. We illustrate how combined chemical and enzymatic approaches are able to expand the library of accessible carbohydrate structures.
Tailor-made oligosaccharides by enzyme engineering
Altering the stereoselectivity of glycosyltransferases
In general, glycosyltransferases exhibit high stereo- and regiocontrol in their glycosylation reactions. But two longstanding questions about glycosyltransfer enzymes persist: How do glycosyltransferases control oligosaccharide versus polysaccharide synthesis and how do they achieve and direct their glycosidic linkage specificity?
These questions were recently addressed and studied on the basis of different classes of glycosyltransferases. The characterisation of a glucosyltransferase from Streptococcus oralis (Hellmuth et al. 2008) and two fructosyltransferases (FTFs) from Bacillus subtilis and Bacillus megaterium (Homann et al. 2007; Beine et al. 2008) gave further insights into fundamental aspects of glycosyl transfer mechanisms.
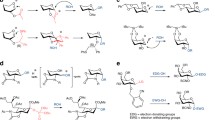
The glucosyltransferase R (GTFR) from S. oralis (E.C. 2.4.1.5) belongs to family 70 of glucosidhydrolases (GH) according to the carbohydrate active enzyme (CAZy) database (Cantarel et al. 2009). The wild-type enzyme utilises sucrose to transfer the glucose moiety to a growing polymer chain. The synthesised polysaccharide is a dextran with α-(1,6) glucosidic linkages (Demuth et al. 2002; Dols et al. 1998). A variety of approaches, involving both rational and random mutagenesis, have been used successfully to alter protein function (Bloom et al. 2005; Shao and Arnold 1996). However, examples of the expansion of the substrate specificity and the control of the regioselectivity of glycosyltransferases are rare (Shaikh and Withers 2008; Lairson et al. 2006). Three catalytic residues crucial for the transfer of glucose have been identified previously in family GH70 enzymes (van Hijum et al. 2006). Using sequence alignments, the corresponding catalytic amino acids in the GTFR were identified as D516 (putative catalytic nucleophile), E554 (putative acid/base catalyst) and D627 (putative transition state stabiliser; Seibel et al. 2006b). The putative nucleophile D516 is involved in formation of the covalent glucosyl–enzyme complexes (Mooser et al. 1991; MacGregor et al. 1996). The crucial role of this catalytic triad has been verified by site-directed mutagenesis experiments (Kato et al. 1992; Monchois et al. 1997; Kralj et al. 2004; Devulapalle et al. 1997). Based on structural modelling, a new “NNS” motif was identified in a reuteransucrase from Lactobacillus reuteri (GTFA). It putatively forms the enzyme active centre (Swistowska et al. 2007; Seibel et al. 2005). By changing this “NNS”-motif into the “SEV” motif, the stereoselectivity of a dextransucrase was directed from reuteran (α-1,4) to dextran (α-1,6) synthesis (Kralj et al. 2005). By a combination of rational and random mutagenesis, the stereoselectivity of the GTFR was altered drastically. The enzyme activity remained high (Hellmuth et al. 2008). For different glucosyltransferases, changes in the transglycosylation activity have been reported for mutations in a highly conserved motif (“RAHDSEV”) around the transition state stabiliser (Monchois et al. 1999; Kralj et al. 2005, 2006; Remaud-Simeon et al. 2000; Shimamura et al. 1994). This region was randomised by polymerase chain reaction mutagenesis in order to generate variants with novel acceptor substrate properties. The screen yielded a double (R624G/V630I) and a triple (R624G/V630I/D717A) variant forming a polymer with different chemical and physical properties. The wild-type polymer is water soluble. In contrast, the polymer of the variants is not soluble in water and has a different texture. Methylation analysis showed a shift from mainly α-(1,6)-linkages (62%, wild-type polymer) up to 46% of α-(1,3) glucosidic bonds (triple mutant polymer; Fig. 1). The corresponding single amino acid variants do not show any difference in polymer`s linkage types compared to the wild-type (D717A) or just up to 7% more α-(1,3) linkages (R624G and V630I). These combined rational and random mutagenesis results show the potential of enzyme engineering for the synthesis of tailor-made oligosaccharides.

GTFR variants forming oligosaccharides instead of polysaccharides (S628D) and α-(1,3) instead of α-(1,6) linkages (R624G/V630I/D717A)
Altering oligosaccharide versus polysaccharide synthesis
Hellmuth et al. performed a mutagenesis screen yielding a GTFR variant with drastically altered polymer formation properties (Hellmuth et al. 2008). The variant S628D did not form polymer anymore whereas the wild-type enzyme synthesises up to 80% dextran (Figs. 1 and 2). When glucose or fructose is added, the GTFR variant S628D synthesises isomaltose (O-α-d-glucopyranosyl-(1,6)-α,β-d-glucopyranose) or leucrose (d-glucopyranosyl-α-(1,5)-d-fructopyranose). The yield was 50% (25-fold increase compared to the wild-type enzyme) or 60%, respectively (Fig. 2). In contrast to the wild-type GTFR, the polymer formation is negligible (Fig. 2).

GTFR variant S628D synthesises the oligosaccharides isomaltose, palatinose and leucrose with glucose or fructose as acceptors
In a parallel approach, two FTFs from B. subtilis NCIMB 11871 and B. megaterium were engineered. The aim was to get control over the polysaccharide-forming activity. Natively, these FTFs catalyse the synthesis of the high molecular weight polysaccharide levan utilising sucrose as substrate. In the presence of acceptor molecules like galactose, xylose, fucose etc., low molecular weight fructo-oligosaccharides were synthesised (Avigad et al. 1957; Cheetham et al. 1989; Homann et al. 2007; Fig. 3). The FTFs belong to family GH 68 of the clan GH-J according to CAZy database (Cantarel et al. 2009). They are retaining glycosidases and have a typical five-bladed beta-propeller core structure forming a covalent intermediate with the fructosyl subunit in the active site (Meng and Fütterer 2003). In order to get insights into the enzymatic mechanism, random mutagenesis on the FTF from B. subtilis was performed (Beine et al. 2008). A screen for an altered polymer formation yielded a variant of asparagine in position 242. This amino acid is located in the +2 subsite of the enzyme’s active centre. For the B. subtilis FTF variant N242H as well as for the corresponding variants from B. megaterium (N252H, N252A), it was shown that the asparagine is crucial for polysaccharide formation (Beine et al. 2008; Homann et al. 2007). The crystal structure of SacB from B. subtilis with raffinose in the active site showed a potential influence on the third carbohydrate unit positioned in the +2 site (Meng and Futterer 2008). In former studies, arginine in position 360, which is also crucial for polymer synthesis, was identified in the +1 region of the FTF from B. subtilis (Chambert and Petit-Glatron 1991).

Synthesis of novel fructo-oligosaccharides
Tailor-made oligosaccharides by substrate engineering
Synthesis of sucrose analogues
Chemically modified substrates together with certain promiscuity of glycosyltransferases to accept substrate analogues expand the diversity of tailor-made oligosaccharides (Seibel et al. 2006a, b). Bacterial glycosyltransferases of the non-Leloir type are able to use easily accessible substrates like sucrose as monosaccharide donor. It was shown that both GTFs and FTFs are able to glycosylate various kinds of acceptors, e.g. alcohols, amino acids, mono- and disaccharides (Seibel et al. 2005, 2006b). The enzymatic synthesis of sucrose analogues has recently been established for a variety of structures. The FTF from B. subtilis transfers the fructosyl residue of the substrate sucrose to different monosaccharide acceptors (d-mannose, d-galactose, 2-deoxy-d-glucose, d-fucose, d-xylose) to yield the β-d-fructofuranosyl-α-d-glycopyranosides (d-Man-Fru, d-Gal-Fru, d-2-deoxy-Glc-Fru, d-Fuc-Fru, d-Xyl-Fru; Cheetham et al. 1989; Seibel et al. 2006c). A range of these structures have been formed in high yields (up to 300 g/L), with respect to the quasi-equilibrium, under kinetic control and optimum conditions (Beine et al. 2008). Even l-glycopyranosides as acceptors led to the formation of β-d-fructofuranosyl-β-l-glycopyranoside (l-Glc-Fru, l-Gal-Fru, l-Fuc-Fru l-Xyl-Fru, Rha-Fru). These sucrose analogues have a β-(1,2)-glycosidic linkage (Seibel et al. 2006a). Additionally, a range of new sucrose derivatives can be obtained in aqueous medium from 3-ketosucrose by chemo-enzymatic approaches. Agrobacterium tumefaciens elaborates a dehydrogenase, which oxidises sucrose specific at the 3-position. The product synthesised is α-d-ribo-hex-3-ulopyranosyl-β-d-fructofuranoside (3-ketosucrose; Fig. 3; Stoppok et al. 1992). Subsequent chemical steps provide access to a range of products, including α-d-allopyranosyl-β-d-fructofuranoside by selective hydration (Pietsch et al. 1994; Timme et al. 1998).
From sucrose analogues to novel functional oligosaccharides
Short-chain fructo-oligosaccharides like kestose and nystose are well known for their health benefits in human nutrition (Yun 1996). Until today, there has been no convenient and effective method to produce tailor-made fructo-oligosaccharides. Recently, a strategy for a two-step enzymatic synthesis of novel 1-kestose and 1-nystose analogues has been investigated by Zuccaro et al. A genetically optimised strain of Aspergillus niger was used to overexpress the β-fructofuranosidase Suc1. This enzyme transforms sucrose and sucrose analogues with high yield to the 1-kestose and 1-nystose analogues. They are headed with different monosaccharides (galactose, mannose, xylose, fucose; Fig. 3) of potential interest (Zuccaro et al. 2008). The concerted action of the two enzymes led to the highly efficient synthesis of defined fructo-oligosaccharides. These novel structures may provide beneficial prebiotic and immunostimulating effects.
Sucrose analogues have also been evaluated as donor substrates for many bacterial FTFs (Biedendieck et al. 2007; Seibel et al. 2006c). For example, the wild-type FTF from B. subtilis converted 52% of the sucrose analogue galactosylfructosid into transglycosylation products. These are almost exclusively levan-type polysaccharides (Beine et al. 2008). Also, Xyl-Fru as a substrate yielded new xylosyl-fructo-oligosaccharides (Xyl-(Fru)n-Fru (n = 1–26) mixtures). As previoulsy discussed, the substitution of the glucose moiety to a different glycopyranosyl unit like xylose influences structural and biochemical properties of the new molecule. In contrast to short-chain fructo-oligosaccharides, the glycopyranosyl residue in polysaccharides may not influence its structure and properties significantly. Consequently, in a recent approach, substrate and enzyme engineering were combined. A random mutagenesis approach of the FTF from B. subtilis yielded the variant N242H (Fig. 4; Beine et al. 2008). In contrast to the wild-type enzyme, the variant N242H with Xyl-Fru as substrate synthesised one main product. It was identified as the 6-kestose analogue Xyl-Fru2 (β-d-fructofuranosyl-(2,6)-β-d-fructofuranosyl-(2,1)-α-d-xylopyranoside; Fig. 3; Beine et al. 2008). As 6-kestose and xylo-oligosaccharides have strong prebiotic activity, this novel product has most likely a beneficial impact on the mammalian gut system’s probiotics.

Active centre of the levansucrase from B. subtilis with docked substrate analogue xylosyl-fructosid (Xyl-Fru; previously unpublished data). The position of Asn242 is shown in subsite +2. Docking procedure was performed with Autodock 4 (Scripps Institute, La Jolla, CA, USA), visualisation by pymol (DeLano 2002). With Xyl-Fru as substrate, the wild-type levansucrase forms dominantly a polyfructoside (left) while variant N242H (right) synthesises the 6-kestose analogue
Substrate-directed stereoselectivity of glycosyltransferases
In a new strategy using substrate engineering, the stereoselectivity of the glucosyltransferases GTFR (from S. oralis) and GTFA (from L. reuteri) was changed without engineering the enzyme itself. For this approach, acceptor substrates were designed which allow the control of linkage specificity by the enzyme and further chemical reactions. The blockage of the preferred transglycosylation site by 6-O-p-toluenesulfonyl switched the glycosylation activity of the GTFR from the major α-(1,6)- to the minor α-(1,3)-activity (Fig. 5; Hellmuth et al. 2007). This principal was extended to a two-step chemo-enzymatic synthesis. The chemoselectivity of the GTFR is guided from α-1,6 to α-1,2, α-1,3 or α-1,4 linkage. Furthermore, the p-toluenesulfonyl group can be substituted via a SN2 mechanism by diverse nucleophilic thio-sugars (Blanchard et al. 2007; Hellmuth et al. 2007). The two-step reaction cascade enables the successful construction of various complex glycoconjugates containing thio-glycosidic linkages. The substituents are glycopyranosides of choice (e.g. galactose, glucose, neuraminic acid; Fig. 5; Hellmuth et al. 2007). The concerted application of thio-sugars helps to expand the structural diversity of conventionally synthesised oligosaccharides (Pachamuthu and Schmidt 2006; Hellmuth et al. 2007). Because of the stability of thio-glycosidic bonds against enzymatic and acidic cleavage, thio-glycosides have been considered very promising candidates for the preparation of carbohydrate-based therapeutics (Pachamuthu and Schmidt 2006). Thio-oligosaccharides have already been described as cytostatic agents with tumour-inactivating capabilities (Witczak et al. 2003). Further expansion offers the allyl group which can be transformed into an aldehyde by ozonolysis. This gives the opportunity of coupling the oligosaccharide to an aglycon-like proteins, drugs or solid carriers or to other functionalised carbohydrates.

Acceptor substrate-directed synthesis followed by nucleophile substitution
Concluding remarks and future prospects
The novel strategies in this review for the synthesis of glycoconjugates shall open new opportunities for answering tantalising questions like how carbohydrates influence the immune system, infection and inflammation processes or cell signalling. For investigations of carbohydrate interactions with proteins and even living systems, one has to synthesise target structures. For this purpose, an expansion of today’s existing library of accessible oligosaccharide structures is necessary. A combination of enzyme and substrate engineering offers the generation of diverse structures which can be investigated (Table 1). Enzyme engineering alone is already a powerful tool for the synthesis of tailor-made oligosaccharides. This has been demonstrated with the glucosyltransferase GTFR and the FTFs from B. subtilis and B. megaterium. These enzymes were switched from polymer to oligosaccharide synthesis by the substitution of only one amino acid. Mutagenesis studies also yielded a GTFR variant that shifts its linkage specificity from native α-(1,4)- to mainly α-(1,3)-bonds. The stereoselectivity of the GTFA from L. reuteri was engineered to form α-(1,6)-bonds instead of the reuteran α-(1,4)-linkages.
The engineering of the substrate further expanded the library of tailor-made oligosaccharide structures. Enzymatically derived sucrose analogues acted as novel substrates for FTFs yielding products with potential benefit for the gut and immune system. By blocking the C6-position in a glucose moiety with p-toluenesulfonyl group, the specificity of the glucosyltransferases GTFR and GTFA were altered. They were switched from the wild-type α-(1,6)-specificity to α-(1, 3)- or α-(1,4)-linkages, respectively. Chemical substitution of the p-toluenesulfonyl group by monosaccharides of choice further expanded the library of accessible tailor-made oligosaccharides.
Enzyme and substrate engineering together is a powerful combination for the creation of various tailor-made oligosaccharide structures. The synthesis of naturally occurring oligosaccharides and their analogues envisage the possibility to explore the growing field of glycobiology. New powerful methods such as glycoarrays or metabolic engineering support this research field a lot.
References
Avigad G, Feingold DS, Hestrin S (1957) Enzymatic synthesis and reactions of a sucrose isomer α-D-galactopyranosyl-β-D-fructofuranoside. J Biol Chem 224:295–307
Beine R, Moraru R, Nimtz M, Na'amnieh S, Pawlowski A, Buchholz K, Seibel J (2008) Synthesis of novel fructooligosaccharides by substrate and enzyme engineering. J Biotechnol 138:33–41
Biedendieck R, Beine R, Gamer M, Jordan E, Buchholz K, Seibel J, Dijkhuizen L, Malten M, Jahn D (2007) Export, purification, and activities of affinity tagged Lactobacillus reuteri levansucrase produced by Bacillus megaterium. Appl Microbiol Biotechnol 74:1062–1073
Blanchard S, Armand S, Couthino P, Patkar S, Vind J, Samain E, Driguez H, Cottaz S (2007) Unexpected regioselectivity of Humicola insolens Cel7B glycosynthase mutants. Carbohydr Res 342:710–716
Bloom JD, Meyer MM, Meinhold P, Otey CR, MacMillan D, Arnold FH (2005) Evolving strategies for enzyme engineering. Curr Opin Struct Biol 15:447–452
Buchholz K, Seibel J (2003) Isomaltooligosaccharides. In: Eggleston G, Cote GL (eds) Oligosaccharides in food and agriculture. Oxford University Press, Washington, DC, pp 63–75
Buchholz K, Noll-Borchers M, Schwengers D (1998) Production of leucrose by dextransucrase. Starch 50:162–164
Cantarel B, Coutinho P, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:233–238
Chambert R, Petit-Glatron MF (1991) Polymerase and hydrolase activities of Bacillus subtilis levansucrase can be separately modulated by site-directed mutagenesis. Biochem J 279:35–41
Cheetham PSJ, Hacking AJ, Vlitos M (1989) Synthesis of novel disaccharides by a newly isolated fructosyltransferase from Bacillus subtilis Enz. Microb Technol 11:212–219
DeLano WL (2002) The PyMOL molecular graphics system. DeLano Scientific LLC, San Carlos, CA
Demuth K, Jördening HJ, Buchholz K (2002) Oligosaccharide synthesis by dextransucrase: new unconventional acceptors. Carbohydr Res 337:1811–1820
Devulapalle KS, Goodman SD, Gao Q, Hemsley A, Mooser G (1997) Knowledge-based model of a glucosyltransferase from the oral bacterial group of mutans streptococci. Protein Sci 6:2489–2493
Dols M, Remaud-Simeon M, Willemot RM, Vignon M, Monsan P (1998) Characterization of the different dextransucrase activities excreted in glucose, fructose, or sucrose medium by Leuconostoc mesenteroides NRRL B-1299. Appl Environ Microbiol 64:1298–1302
Hellmuth H, Hillringhaus L, Höbbel S, Kralj S, Dijkhuizen L, Seibel J (2007) Highly efficient chemoenzymatic synthesis of novel branched thiooligosaccharides by substrate direction with glucansucrases. Chembiochem 8:273–276
Hellmuth H, Wittrock S, Kralj S, Dijkhuizen L, Hofer B, Seibel J (2008) Engineering the glucansucrase GTFR enzyme reaction and glycosidic bond specificity: toward tailor-made polymer and oligosaccharide products. Biochemistry 47:6678–6684
Homann A, Biedendieck R, Gotze S, Jahn D, Seibel J (2007) Insights into polymer versus oligosaccharide synthesis: mutagenesis and mechanistic studies of a novel levansucrase from Bacillus megaterium. Biochem J 407:189–198
Kato C, Nakano Y, Lis M, Kuramitsu HK (1992) Molecular genetic analysis of the catalytic site of Streptococcus mutans glucosyltransferases. Biochem Biophys Res Commun 189:1184–1188
Kralj S, van Geel-Schutten GH, van der Maarel MJ, Dijkhuizen L (2004) Biochemical and molecular characterization of Lactobacillus reuteri 121 reuteransucrase. Microbiology 150:2099–2112
Kralj S, van Geel-Schutten IG, Faber EJ, van der Maarel MJ, Dijkhuizen L (2005) Rational transformation of Lactobacillus reuteri 121 reuteransucrase into a dextransucrase. Biochemistry 44:9206–9216
Kralj S, Eeuwema W, Eckhardt TH, Dijkhuizen L (2006) Role of asparagine 1134 in glucosidic bond and transglycosylation specificity of reuteransucrase from Lactobacillus reuteri 121. FEBS J 273:3735
Lairson LL, Watts AG, Wakarchuk WW, Withers SG (2006) Using substrate engineering to harness enzymatic promiscuity and expand biological catalysis. Nat Chem Biol 2:724–728
Lina BA, Jonker D, Kozianowski G (2002) Isomaltulose (Palatinose): a review of biological and toxicological studies. Food Chem Toxicol 40:1375–1381
MacGregor EA, Jespersen HM, Svensson B (1996) A circularly permuted alpha-amylase-type alpha/beta-barrel structure in glucan-synthesizing glucosyltransferases. FEBS Lett 378:263–266
Meng G, Fütterer K (2008) Donor substrate recognition in the raffinose-bound E342A mutant of fructosyltransferase Bacillus subtilis levansucrase. BMC Struct Biol 8:16
Meng G, Fütterer K (2003) Structural framework of fructosyl transfer in Bacillus subtilis levansucrase. Nat Struct Biol 10:935–941
Mizubuchi H, Yajima T, Aoi N, Tomita T, Yoshikai Y (2005) Isomalto-oligosaccharides polarize Th1-like responses in intestinal and systemic immunity in mice. J Nutr 135:2857–2861
Monchois V, Remaud-Simeon M, Russell RR, Monsan P, Willemot RM (1997) Characterization of Leuconostoc mesenteroides NRRL B-512F dextransucrase (DSRS) and identification of amino-acid residues playing a key role in enzyme activity. Appl Microbiol Biotechnol 48:465–472
Monchois V, Willemot RM, Monsan P (1999) Glucansucrases: mechanism of action and structure-function relationships. FEMS Microbiol Rev 23:131–151
Mooser G, Hefta SA, Paxton RJ, Shively JE, Lee TD (1991) Isolation and sequence of an active-site peptide containing a catalytic aspartic acid from two Streptococcus sobrinus alpha-glucosyltransferases. J Biol Chem 266:8916–8922
Nakakuki T (2002) Present status and future of functional oligosaccharide development in Japan. Pure Appl Chem 74:1245–1251
Pachamuthu K, Schmidt RR (2006) Synthetic routes to thiooligosaccharides and thioglycopeptides. Chem Rev 106:160–187
Pietsch M, Walter W, Buchholz K (1994) Regioselective synthesis of new sucrose derivatives via 3-ketosucrose. Carbohydr Res 254:183–194
Remaud-Simeon M, Willemot R, Sarcabal P, Potocki de Montalk G, Monsan P (2000) Glucansucrases: molecular engineering and oligosaccharide synthesis. J Mol Catal B Enzymatic 10:117–128
Seibel J, Moraru R, Götze S (2005) Biocatalytic and chemical investigations in the synthesis of sucrose analogues. Tetrahedron 61:7081–7086
Seibel J, Beine R, Moraru R, Behringer C, Buchholz K (2006a) A new pathway for the synthesis of oligosaccharides by the use of non-Leloir glycosyltransferases. Biocat Biotrans 24:157–165
Seibel J, Hellmuth H, Hofer B, Kicinska AM, Schmalbruch B (2006b) Identification of new acceptor specificities of glycosyltransferase R with the aid of substrate microarrays. Chembiochem 7:310–320
Seibel J, Moraru R, Götze S, Buchholz K, Na'amnieh S, Pawlowski A, Hecht HJ (2006c) Synthesis of sucrose analogues and the mechanism of action of Bacillus subtilis fructosyltransferase (levansucrase). Carbohydr Res 341:2335–2349
Shaikh FA, Withers SG (2008) Teaching old enzymes new tricks: engineering and evolution of glycosidases and glycosyl transferases for improved glycoside synthesis. Biochem Cell Biol 86:169–177
Shao Z, Arnold FH (1996) Engineering new functions and altering existing functions. Curr Opin Struct Biol 6:513–518
Shimamura A, Nakano YJ, Mukasa H, Kuramitsu HK (1994) Identification of amino acid residues in Streptococcus mutans glucosyltransferases influencing the structure of the glucan product. J Bacteriol 176:4845–4850
Stoppok E, Matalla K, Buchholz K (1992) Microbial modification of sugars as building blocks for chemicals. Appl Microb Biotechnol 36:604–610
Swistowska AM, Gronert S, Wittrock S, Collisi W, Hecht HJ, Hofer B (2007) Identification of structural determinants for substrate binding and turnover by glucosyltransferase R supports the permutation hypothesis. FEBS Lett 581:4036–4042
Timme V, Buczys R, Buchholz K (1998) Kinetic investigations on the hydrogenation of 3-ketosucrose. Starch/Stärke 50:29–32
van Hijum SA, Kralj S, Ozimek LK, Dijkhuizen L, van Geel-Schutten IG (2006) Structure-function relationships of glucansucrase and fructansucrase enzymes from lactic acid bacteria. Microbiol Mol Biol Rev 70:157–176
Varki A (1993) Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 3:97
Witczak ZJ, Kaplon P, Dey PM (2003) Thio-sugars VII. Effect of 3-deoxy-4-S-(β-D-gluco- and β-D-galactopyranosyl)-4-thiodisaccharides and their sulfoxides and sulfones on the viability and growth of selected murine and human tumor cell lines. Carbohydr Res 338:11–18
Wong CH (2005) Protein glycosylation: new challenges and opportunities. J Org Chem 70:4219–4225
Yun JW (1996) Fructooligosaccharides—occurrence, preparation and application. Enzyme Microb Technol 19:107–117
Zuccaro A, Götze S, Kneip S, Dersch P, Seibel J (2008) Tailor-made fructooligosaccharides by a combination of substrate and genetic engineering. Chembiochem 9:143–149
Acknowledgement
The authors gratefully acknowledge the funding of this work by the Arbeitsgemeinschaft industrieller Forschungsvereinigungen (AiF) and the Deutsche Forschungsgemeinschaft (DFG).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Homann, A., Seibel, J. Towards tailor-made oligosaccharides—chemo-enzymatic approaches by enzyme and substrate engineering. Appl Microbiol Biotechnol 83, 209–216 (2009). https://doi.org/10.1007/s00253-009-1989-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-1989-5