
Abstract
A bacterial artificial chromosomal library of Nonomuraea sp. ATCC39727 was constructed using Escherichia coli–Streptomyces artificial chromosome (ESAC) and screened for the presence of dbv genes known to be involved in the biosynthesis of the glycopeptide A40926. dbv genes were cloned as two large, partially overlapping, fragments and transferred into the host Streptomyces lividans, thus generating strains S. lividans∷NmESAC50 and S. lividans∷NmESAC57. The heterologous expression of Nonomuraea genes in S. lividans was successfully demonstrated by using combined RT–PCR and proteomic approaches. MALDI-TOF analysis revealed that a Nonomuraea ABC transporter is expressed as two isoforms in S. lividans. Moreover, its expression may not require a Nonomuraea positive regulator at all, as it is present at similar levels in both clones even though S. lividans∷NmESAC57 lacks regulatory genes. Considered together, these results show that S. lividans expresses Nonomuraea genes from their own promoters and support the idea that S. lividans can be a good host for genetic analysis of Nonomuraea.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The expression of secondary metabolic gene clusters of actinomycetes in genetically and physiologically characterized hosts is emerging as a viable alternative to both classic strain and fermentation process development and molecular biological manipulation of the native producer strain (Doekel et al. 2002). The utility of Escherichia coli as a host for heterologous gene expression as well as metabolic engineering is unquestioned, but several limitations have been encountered, as E. coli is significantly different from the main natural producers of secondary metabolites. Thus, alternative organisms have been selected as heterologous hosts. Examples of heterologous expression of non-ribosomal peptides in Streptomyces have been reported (Zirkle et al. 2004). Streptomyces is more amenable to strain improvement, grows more rapidly than natural producers and presents a series of advantages over E. coli—actinomycetes have a complex array of promoters, can efficiently transcribe from heterologous promoters, have post-translational capabilities otherwise absent in E. coli and are suitable hosts for the expression of high G+C-content DNAs (Wilkinson et al. 2002). Furthermore, they might well have the biosynthetic apparatus and necessary primary precursors to support natural product synthesis from exogenous pathways.
Uncommon genera of actinomycetes have been investigated by microbial technologies for the discovery of novel bioactive metabolites (Donadio et al. 2002). As an example, Nonomuraea sp. was studied for the production of the industrially important glycopeptide antibiotic A40926, precursor of the semi-synthetic glycopeptide dalbavancin, currently under clinical development (Candiani et al. 1999; Steiert and Schmitz 2002). The A40926 biosynthetic gene cluster was demonstrated to be approximately 71 kb and contain 37 dbv ORFs (Sosio et al. 2003). To date, only a gene transfer system has been described for Nonomuraea sp. (Stinchi et al. 2003); however, this approach suffers from a low efficiency in terms of yield and is highly time consuming. To overcome these difficulties, we set out to search for new hosts for the heterologous expression of Nonomuraea genes. In this paper, we demonstrate that dbv genes can be expressed as part of large DNA segments introduced into Streptomyces lividans chromosome as a single copy. We report the construction of a genomic library of Nonomuraea in the shuttle pPAC-S2 (E. coli–Streptomyces artificial chromosome; ESAC) vector (Sosio et al. 2000), the identification of E. coli clones containing dbv genes and the expression of selected dbv genes in S. lividans, as monitored by genomic and proteomic methodologies.
Materials and methods
Bacterial strains, plasmids and culture conditions
Nonomuraea sp. ATCC 39727 and Streptomyces lividans ZX7 (John Innes Centre, Norwich, UK) were used for these studies. E. coli DH10B cells were obtained from Life Technologies, Gibco. The ESAC vector pPAC-S2 has been already described (Sosio et al. 2000).
Preparation of high molecular weight DNA
Nonomuraea mycelium was grown for 5 days at 30°C in RARE3 medium (Sosio et al. 2003) and plugs were prepared following the protocols reported by Evans and Dyson (1993). DNA–agarose plugs were dialysed 3 times against 10 ml of HE buffer (10 mM HEPES–NaOH, 1 mM EDTA pH 8), for 1 h. Each plug was incubated in 160 μl M buffer (100 mM TRIS–HCl, 0.5 M NaCl, 10 mM dithioerythritol, pH 7.5) containing 0.5 U Sau3AI (Roche), at 4°C, for 4 h. Samples were added to 2.6 μl of 1 M MgCl2 and incubated at 37°C, for 4 min. Then, samples were added to 29 μl of 0.5 M EDTA, and 15 μl of proteinase K (20 mg/ml) and warmed at 37°C, for 30 min. After repeated washing with HE buffer, plugs were melted at 65°C for 10 min. DNA contained in melted agarose solution was quantified by loading 10-μl aliquots on a 1% (w/v) agarose gel; the remaining solutions were transferred to 45°C. For all further pipetting steps, tips with the ends cut off were used to reduce DNA shearing. GELase (Epicentre Technologies) was added (1.5 U for 100 μl of DNA solution) and mixtures were incubated at 45°C, for 1 h. After a brief spin, tubes were put on ice.
pPAC-S2 library preparation
About 100 ng of Sau3AI-digested Nonomuraea DNA was ligated to 20 ng of BamHI-digested and dephosphorylated pPAC-S2 vector, prepared as described in Alduina et al. 2003 (approximately 2:1 molar ratio considering an average insert size of 60 kb) at 16°C, overnight, using 400 U T4 DNA ligase (Biolabs). The ligation mixture was desalted using Millipore filters (type VS, 0.025 mm) and then used to transform E. coli DH10B electrocompetent cells with a BioRad Gene Pulser instrument (2.5 kV, 100 Ω, 25 μFa). Recombinant clones were designated as NmESACs. They were stored at −80°C in LB containing 20% (v/v) glycerol in individual wells of microtiter plates.
NmESAC DNAs were prepared by the alkaline lysis method (Sambrook et al. 1989), digested with DraI and fractionated on a 0.8% (w/v) agarose gel by pulsed field gel electrophoresis (PFGE) (4 s for 4 h; 20 s for 14 h; at 160 V in 0.5X TBE, 7°C). DNA sequencing of selected NmESACs, purified with the Plasmid Maxi kit (Qiagen) was performed by the SEQLAB Sequence Laboratories (Göttingen, Germany).
DNA manipulation and Southern blot analysis
DNA manipulations and Southern hybridisations were performed according to standard protocols (Sambrook et al. 1989). Colony hybridisations were carried out according to the protocol for the Hybond-N+ membrane kit (Amersham Pharmacia Biotech). The hybridisation probes, used for the identification of NmESAC clones containing dbv genes, were randomly primed labelled (Rediprime II, Amersham Pharmacia Biotech) internal fragments of 951 and 1,249 bp, respectively, of the Nonomuraea dbv ORF1 and ORF37. The quality of the library was checked using randomly primed labelled internal fragments Nonomuraea hrdB and Streptomyces coelicolor dnaK (558 and 700 bp, respectively). HrdB and dnaK encode the vegetative sigma factor and the dnaK heat shock protein in S. coelicolor (Kang et al. 1997; Puglia et al. 1995). The fragments were obtained by PCR using the primers listed in Table 1. The hrdB primers were kind gifts of Oliver Puk, Tübingen University. The partial hrdB sequence has been submitted to the dbGSS of GenBank (accession no. AY838272).
Transformation of S. lividans
Protoplast formation, transformation and regeneration from S. lividans were carried out by standard procedures (Kieser et al. 2000). S. lividans strains were cultured according to Puglia et al. (1995), using thiostrepton (50 μg/ml). Samples (1 and 10 ml for RNA and protein extraction, respectively) were taken from each culture grown for 42 h and collected by centrifugation for 15 min at 6,000 g. The mycelium was used to extract total RNA and proteins.
RNA isolation and RT–PCR analysis
Collected mycelium was immediately frozen at −80°C and, after at least 2 h, broken by using P-buffer containing lysozyme (1 mg/ml). RNA isolation was performed using the RNeasy Midi kit (QIAGEN), according to the manufacturer’s instructions. Residual genomic DNA was removed by DNase digestion (Roche). RT–PCR was performed using the Superscript One-Step RT–PCR kit (Invitrogen) with 0.1 μg total RNA as template. The primers are listed in Table 1. For each reaction, a negative control containing only Taq polymerase was included. The S. lividans hrdB gene was used as internal control (Kang et al. 1997). The identities of the RT–PCR products were confirmed by sequencing.
2D–gel electrophoresis and mass spectrometry analysis
For protein extraction, mycelium was sonicated according to Puglia et al. (1995). After dialysis against distilled water at 4°C and acetone precipitation at −20°C, proteins were redissolved in isoelectric focusing (IEF) buffer containing 8 M urea, 4% (w/v) CHAPS and 1% (w/v) 1,4-dithioerythritol and stored at −80°C until used again.
2D–gel electrophoresis was carried out as described by the manufacturer (Amersham Pharmacia Biotech). In the first dimension, we used 18-cm IPG strips, pH 4–7, and the Ettan IPGphor system. The IPG strips were rehydrated for 1 h in IEF buffer containing 500 μg of sample proteins, 0.5% (w/v) ampholytes and 1% (w/v) bromophenol blue. For protein separation, a 30-V pre-step was performed for 10 h, followed by IEF carried out for 74,850 V-h with a maximum voltage of 8,000 V. All the steps were performed at 20°C using 50 μA per strip. After IEF, the IPG strips were saturated with an equilibration buffer containing 6 M urea, 30% (v/v) glycerol, 2% (w/v) SDS, 0.05 M TRIS–HCl pH 6.8 and 2% (w/v) DTE for 12 min, in order to resolubilize proteins. The –SH groups were then blocked by substituting DTE with 2.5% (w/v) iodoacetamide in the equilibrating buffer. The focused proteins were then separated on 12.5% polyacrylamide gels (SDS-PAGE) at 10°C in a Hoefer Dalt vertical system, using a maximum setting of 40 μA and 110 V per gel. The spots were detected using silver staining according to Shevchenko et al. (1996), except that 0.08% (w/v) sodium thiosulphate for 5 min and 0.4% (w/v) silver solution were used.
Image analysis was performed using ImageMaster 2D platinum version 5.0 (Amersham Biosciences). Spot detection was optimised automatically using an auto-detection device and then edited manually. Images were matched using reproducible landmarks and normalised to total spot volume for quantification.
Spots from 2D gels were excised, triturated and washed with water. Proteins were reduced, S-alkylated and digested with trypsin as previously reported (Talamo et al. 2003). Digested aliquots were removed and subjected to a desalting/concentration step on a μZipTipC18 cartridge (Millipore, Bedford, Mass.), using acetonitrile as eluent. Peptide mixtures were loaded on the MALDI target using the dried droplet technique and α-cyano-4-hydroxycinnamic acid as matrix, and analysed with a Voyager-DE PRO mass spectrometer (Applied Biosystems, Framingham, Mass.). Internal mass calibration was performed with peptides derived from trypsin autoproteolysis. PROWL and ProteinProspector software packages (Zhang and Chair 2000; Clauser et al. 1999) were used to identify spots unambiguously from NCBI and SWISS-PROT non-redundant sequence databases. Candidates from peptide matches were further evaluated by comparison with their experimental masses and pIs obtained from 2D gels.
Results
Construction of a Nonomuraea ESAC library
Similarly to that of other actinomycetes, DNA from Nonomuraea sp. ATCC 39727 undergoes degradation during PFGE (Beyazova et al. 1995; R. Alduina et al., unpublished results). Thus, we modified the method already reported for the preparation of large insert libraries of S. coelicolor and Planobispora rosea (Alduina et al. 2003). Using unfractionated Nonomuraea DNA, we were able to obtain 2,051 recombinant clones (NmESACs), resulting from different independent ligations. All the clones were individually analysed by PFGE. Seven hundred and thirty-six recombinant clones carried inserts larger than 30 up to 155 kb, with an average insert size of 57 kb. In total, these clones encompass 42 Mb of Nonomuraea DNA. Assuming a Nonomuraea chromosome of about 8 Mb, as in many other actinomycetes (Redenbach et al. 2000), this library represents about a five-fold coverage of its genome.
The quality of the library was evaluated by colony hybridisation on 288 randomly chosen clones (corresponding to a two-fold coverage of the Nonomuraea genome) using S. coelicolor dnaK and Nonomuraea hrdB genes, each expected as a single copy in the genome (Ahmad et al. 2000; R. Alduina unpublished results). These hybridisations led to the identification of two NmESACs hybridising to hrdB and two additional ones to dnaK, consistent with a random representation of Nonomuraea DNA in the library.
Introduction of portions of the dbv cluster into S. lividans
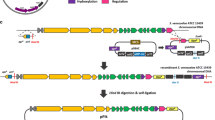
To identify NmESACs containing dbv genes, the library was screened by colony and Southern hybridisations using probes derived from the first and the last gene of the dbv cluster, ORF1 and ORF37, respectively (asterisks, Fig. 1a). Out of seven positive clones (carrying inserts from 50 to 60 kb), two clones, namely NmESAC50 and NmESAC57, were chosen for end-sequencing. NmESAC50 (Fig. 1b) carried a 52,802-bp insert ranging from 198 bp upstream to dbv ORF1 up to 4,069 bp of ORF25. NmESAC57 (Fig. 1c) contained a 44,998-bp DNA fragment extending from 10,734 bp of ORF17 to approximately 10 kb past ORF37, falling outside of the region sequenced (Sosio et al. 2003). Thus, NmESAC50 and NmESAC57 are expected to contain 24 and 20 complete dbv ORFs, respectively, and share a 26-kb overlapping fragment extending from dbv ORF18 to ORF24.

Schematic representation of Nm-Escherichia coli–Streptomyces artificial chromosome (ESAC) clones. a Organization of the 37 ORFs of A40926 biosynthetic gene cluster. Asterisks indicate ORFs used as probes for colony hybridisation. b, c Regions from the dbv cluster cloned in NmESAC50 and NmESAC57, respectively. The squares delimit the ends of the clones and the triangles indicate ORFs targeted by RT–PCR
NmESAC50, NmESAC57 and pPAC-S2 vector were transferred into S. lividans ZX7, where the vector is expected to integrate site-specifically into the chromosome (Sosio et al. 2000). One clone from each transformation, named S. lividans∷NmESAC50, S. lividans∷NmESAC57 and S. lividans∷ESAC, was randomly selected for further characterisation. The correct site-specific integration in the S. lividans chromosome and the integrity of the dbv inserts were verified by Southern hybridisation of BamHI-digested genomic DNA with pPAC-S2, NmESAC50 and NmESAC57 DNAs as probes. These hybridisations (data not shown) demonstrated that the vector was successfully integrated in the attB site of S. lividans chromosome and that the dbv inserts were integrally transferred from E. coli to Streptomyces.
RT–PCR analysis
Heterologous expression of dbv genes was analysed by RT–PCR analysis. Specific primers were designed to detect the transcripts of dbv ORFs 4, 6, 14, 19, 24 and 33 (triangles, Fig. 1b, c). RT–PCR analysis showed that ORFs 4, 6 and 14 were transcribed in S. lividans∷NmESAC50 (Fig. 2a–c), ORF33 was transcribed in S. lividans∷NmESAC57 (Fig. 2f), and ORFs 19 and 24 in both clones (Fig. 2d, e), as expected according to the dbv regions cloned. No signals were detected in S. lividans and S. lividans∷ESAC (lanes 1 and 2, Fig. 2a–g). It should be noted that, because of their orientation and/or distance from NmESAC ends, all dbv ORFs are expected to be transcribed from their own promoters. Interestingly, we can surmise that expression of dbv ORFs 19, 24 and 33 is quantitatively independent from a Nonomuraea positive regulator, which is present in NmESAC50 but not in NmESAC57. Moreover, expression of dbv genes was also detected at 18 h (data not shown), when S. lividans is in the first rapid growth phase (Puglia et al. 1995).

RT–PCR analysis. a–g Results of RT–PCR on RNAs from Streptomyces lividans (lane 1), S. lividans∷ESAC (lane 2), S. lividans∷NmESAC50 (lane 3), S. lividans∷NmESAC57 (lane 4), and on Nonomuraea DNA (lane 5). RT–PCR conditions for: dbv ORF4 (a), dbv ORF6 (b), dbv ORF14 (c), dbv ORF19 (d), dbv ORF24 (e), dbv ORF33 (f) and hrdB (g). g Asterisks indicate negative controls for each sample
2D-PAGE and mass spectrometry analysis
To investigate the expression of dbv genes also at a protein level, the S. lividans∷NmESAC50, S. lividans∷NmESAC57 and S. lividans∷ESAC proteomes were analysed by 2D-PAGE. An experimental range of pI and molecular weight was selected for good resolution of the S. lividans proteins (Fig. 3a) and, at the same time, the detection of the expected 15 and 18 Nonomuraea proteins from NmESAC50 and NmESAC57 transformants, respectively. Gels were compared and matched using as landmarks seven spots (arrows 1–7, Fig. 3a) present in all clones that were identified by peptide mass fingerprint and tandem mass spectrometry analysis as S. lividans isocitrate dehydrogenase (spot 1), putative large secreted protein (spot 2), trigger factor (spot 3), succinyl CoA synthetase α-chain (spot 4), hypothetical protein SCO4636 (spot 5), putative condensing enzyme (spot 6) and 50S ribosomal protein L7/L12 (spot 7). All protein landmarks, apart from putative condensing enzyme and putative large secreted protein, showed values of pI and molecular weight very similar to those of their homologous S. coelicolor counterparts (Novotna et al. 2003; Hesketh et al. 2002).

2D-PAGE analysis. a 2D gel image of proteins extracted from S. lividans∷ESAC. b–d Representative 2D gel region comprising statistically significant changes in the proteome of S. lividans∷ESAC, S. lividans∷NmESAC50 and S. lividans∷NmESAC57, respectively. Arrows 1–7 Protein landmarks, arrows 8–10 TipA proteins, arrows 11–12 dbv ORF19 isoforms
Comparative analysis with respect to S. lividans∷ESAC allowed the detection of 13 and 18 spots associated with protein components differentially expressed in S. lividans∷NmESAC50 and S. lividans∷NmESAC57 proteomes, respectively. Interestingly, in the best resolved area, five spots (arrows 8–12, Fig. 3c–d) were common to both clones and were subjected to mass spectrometric characterization. Among these, two spots (arrows 11 and 12, Fig. 3c–d) with the same molecular weight (25 kDa) but different pI values (5.4 and 5.55) were identified as isoforms of the Nonomuraea ABC transporter dbv ORF19 (sequence coverage 76% and 38%, respectively). The dbv ORF19 protein together with dbv ORFs 21 and 24 was expected to be present in the proteome of both clones. The more abundant spot (arrow 11, Fig. 3c–d) showed an experimental pI value (5.4) very similar to that predicted from the protein sequence; the less abundant one (arrow 12, Fig. 3c–d) migrated at a more basic pI value, probably as a result of a still uncharacterised post-translational modification.
The remaining three spots were identified as isoforms of TipAS (arrows 9 and 10, Fig. 3c–d) and TipAL (arrow 8, Fig. 3c–d) S. lividans proteins.
TipAL and TipAS, two alternative in-frame translational products of tipA, have been reported to be induced by diverse cyclic thiopeptide antibiotics, such as thiostrepton, in different Streptomyces strains (Chiu et al. 1999). Although thiostrepton was added to all cultures at the same concentration, we found significant quantitative increased levels of TipAS and TipAL only in the two strains carrying ESACs with large inserts.
Discussion
In this paper we report the construction of a bacterial artificial chromosomal (BAC) library of Nonomuraea sp. ATCC39727 using ESAC and, after integration of the vector into the chromosome of S. lividans, the expression of Nonomuraea genes in this genetically accessible host. Nonomuraea sp. produces the industrially important glycopeptide antibiotic A40926, precursor of dalbavancin, currently under clinical development (Candiani et al. 1999; Steiert and Schmitz 2002). A high-quality, high molecular weight genomic library from Nonomuraea was constructed. The procedure used overcame DNA degradation during PFGE, a phenomenon that frequently occurs in actinomycetes (Beyazova et al. 1995). To our knowledge, the NmESAC library is the first BAC library of an actinomycete, whose DNA undergoes degradation during PFGE.
Two NmESAC clones containing part of the 71-kb biosynthetic dbv gene cluster for the glycopeptide antibiotic A40926 were identified and transferred into S. lividans. By using combined genomic and proteomic approaches, we demonstrated gene expression of Nonomuraea DNA cloned into S. lividans.
The expected transcripts from ESAC inserts were detected for all the ORFs analysed. The translational product of one of these ORFs was also detected as demonstrated by the proteomic approach. This ORF is the Nonomuraea ABC transporter encoded by dbv ORF19 and is present in S. lividans as two isoforms. Different isoforms of ABC transporter genes have been recently described by proteomic studies in Streptomyces coelicolor (Hesketh et al. 2002). The presence of isoforms have been ascribed to post-translational modifications. In contrast, no mass spectrometry information on the structural differences between Nonomuraea dbv ORF19 isoforms was obtained.
Interestingly, from our transcriptional and proteomic data we can surmise that the expression of dbv ORF19 in S. lividans is quantitatively independent from a Nonomuraea positive regulator, since this gene was expressed at similar levels in both clones, even in the clone that does not contain any dbv regulators. Thus, the expression of dbv ORF19 may not require a positive regulator at all, or an S. lividans regulator may drive its expression.
Comparative proteomic analysis allowed us to define altered gene expression profiles for the host. The up-regulated proteins were identified as TipAS and TipAL isoforms. TipAL and TipAS are two alternative in-frame translational products of tipA. These proteins are induced in different Streptomyces strains by diverse cyclic thiopeptide antibiotics (Chiu et al. 1999). An altered expression level of TipA does not seem to influence either dbv ORF19 expression or, presumably, the expression of other dbv ORFs. Further investigations are necessary to understand tipA induction.
To our knowledge, this is the first report on the expression of Nonomuraea genes in the model host S. lividans. This library is a valuable tool for genetic analysis of Nonomuraea in a different background such as Streptomyces. Moreover, the S. lividans NmESAC clones can be considered useful biotechnological tools for further manipulations to generate novel peptides with improved properties, through combinatorial biosynthesis (Keller and Schauwecker 2003).
References
Ahmad S, Selvapandiyan A, Bhatnagar RK (2000) Phylogenetic analysis of Gram-positive bacteria based on grpE, encoded by the dnaK operon. Int J Syst Evol Microbiol 50:1761–1766
Alduina R, De Grazia S, Dolce L, Salerno P, Sosio M, Donadio S, Puglia AM (2003) Artificial chromosome libraries of Streptomyces coelicolor A3(2) and Planobispora rosea. FEMS Microbiol Lett 218:181–186 DOI 10.1016/S0378-1097(02)01125-4
Beyazova ML, Brodsky BC, Shearer MC, Horan AC (1995) Preparation of actinomycete DNA for pulse-field gel electrophoresis. Int J Syst Bacteriol 45:852–854
Candiani G, Abbondi M, Borgonovi M, Romano G, Parenti F (1999) In-vitro and in-vivo antibacterial activity of BI 397, a new semi-synthetic glycopeptide antibiotic. J Antimicrob Chemother 44:179–192
Chiu ML, Folcher M, Katoh T, Puglia AM, Vohradsky J, Yun BS, Seto H, Thompson CJ (1999) Broad spectrum thiopeptide recognition specificity of the Streptomyces lividans TipAL protein and its role in regulating gene expression. J Biol Chem 274:20578–20586
Clauser KR, Baker PR, Burlingame AL (1999) Role of accurate mass measurement (+/−10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem 71:2871–2882
Doekel S, Eppelmann K, Marahiel MA (2002) Heterologous expression of nonribosomal peptide synthetases in B. subtilis: construction of a bi-functional B. subtilis/E. coli shuttle vector system. FEMS Microbiol Lett 216:185–191 DOI 10.1016/S0378-1097(02)00993-X
Donadio S, Monciardini P, Alduina R, Mazza P, Chiocchini C, Cavaletti L, Sosio M, Puglia AM (2002) Microbial technologies for the discovery of novel bioactive metabolites. J Biotechnol 99:187–198 DOI 10.1016/S0168-1656(02)00209-2
Evans M, Dyson P (1993) Pulsed-field gel electrophoresis of Streptomyces lividans DNA. Trends Genet 9:72
Hesketh AR, Chandra G, Shaw AD, Rowland JJ, Kell DB, Bibb MJ, Chater KF (2002) Primary and secondary metabolism, and post-translational protein modifications, as portrayed by proteomic analysis of Streptomyces coelicolor. Mol Microbiol 46:917–932 DOI 10.1046/j.1365-2958.2002.03219.x
Kang JG, Hahn MY, Ishihama A, Roe JH (1997) Identification of sigma factors for growth phase-related promoter selectivity of RNA polymerases from Streptomyces coelicolor A3(2). Nucleic Acids Res 25:2566–2573
Keller U, Schauwecker F (2003) Combinatorial biosynthesis of non-ribosomal peptides. Comb Chem High Throughput Screen 6:527–540
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. John Innes Centre, Norwich
Novotna J, Vohradsky J, Berndt P, Gramajo H, Langen H, Li XM, Minas W, Orsaria L, Roeder D, Thompson CJ (2003) Proteomic studies of diauxic lag in the differentiating prokaryote Streptomyces coelicolor reveal a regulatory network of stress-induced proteins and central metabolic enzymes. Mol Microbiol 48:1289–1303 DOI 10.1046/j.1365-2958.2003.03529.x
Puglia AM, Vohradsky J, Thompson CJ (1995) Developmental control of the heat-shock stress regulon in Streptomyces coelicolor. Mol Microbiol 17:737–746
Redenbach M, Scheel J, Schmidt U (2000) Chromosome topology and genome size of selected actinomycetes species. Antonie van Leeuwenhoek 78:227–235
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M (1996) Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc Natl Acad Sci USA 93:14440–14445
Sosio M, Giusino F, Cappellano C, Bossi E, Puglia AM, Donadio S (2000) Artificial chromosomes for antibiotic-producing actinomycetes. Nat Biotechnol 18:343–345 DOI 10.1038/73810
Sosio M, Stinchi S, Beltrametti F, Lazzarini A, Donadio S (2003) The gene cluster for the biosynthesis of the glycopeptide antibiotic A40926 by Nonomuraea species. Chem Biol 10:541–549 DOI 10.1016/S1074-5521(03)00120-0
Steiert M, Schmitz FJ (2002) Dalbavancin (Biosearch Italia/Versicor). Curr Opin Invest Drugs 3:229–233
Stinchi S, Azimonti S, Donadio S, Sosio M (2003) A gene transfer system for the glycopeptide producer Nonomuraea sp ATCC39727. FEMS Microbiol Lett 225:53–57 DOI 10.1016/S0378-1097(03)00490-7
Talamo F, D’Ambrosio C, Arena S, Del Vecchio P, Ledda L, Zehender G, Ferrara L, Scaloni A (2003) Proteins from bovine tissues and biological fluids: defining a reference electrophoresis map for liver, kidney, muscle, plasma and red blood cells. Proteomics 3:440–460 DOI 10.1002/pmic.200390059
Wilkinson CJ, Hughes-Thomas ZA, Martin CJ, Bohm I, Mironenko T, Deacon M, Wheatcroft M, Wirtz G, Staunton J, Leadlay PF (2002) Increasing the efficiency of heterologous promoters in actinomycetes. J Mol Microbiol Biotechnol 4:417–426
Zhang WZ, Chair BT (2000) Profound. An expert system for protein identification using mass spectrometric peptide mapping information. Anal Chem 72:2482–2489
Zirkle R, Ligon JM, Molnar I (2004) Heterologous production of the antifungal polyketide antibiotic soraphen A of Sorangium cellulosum So ce26 in Streptomyces lividans. Microbiology 150:2761–2774 DOI 10.1099/mic.0.27138-0
Acknowledgements
We thank Margherita Sosio for helpful suggestions and Maurizio Noto for technical assistance. This work was supported by the European Union via the MEGA-TOP project QLK3-1999-00650 and the COMBIG-TOP project LSHB-CT-2003-503491. We acknowledge our colleagues participating in these projects for stimulating discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Alduina, R., Giardina, A., Gallo, G. et al. Expression in Streptomyces lividans of Nonomuraea genes cloned in an artificial chromosome. Appl Microbiol Biotechnol 68, 656–662 (2005). https://doi.org/10.1007/s00253-005-1929-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-005-1929-y