
Abstract
Despite the availability of genome data and recent advances in methionine regulation in Corynebacterium glutamicum, sulfur metabolism and its underlying molecular mechanisms are still poorly characterized in this organism. Here, we describe the identification of an ORF coding for a putative regulatory protein that controls the expression of genes involved in sulfur reduction dependent on extracellular methionine levels. C. glutamicum was randomly mutagenized by transposon mutagenesis and 7,000 mutants were screened for rapid growth on agar plates containing the methionine antimetabolite d,l-ethionine. In all obtained mutants, the site of insertion was located in the ORF NCgl2640 of unknown function that has several homologues in other bacteria. All mutants exhibited similar ethionine resistance and this phenotype could be transferred to another strain by the defined deletion of the NCgl2640 gene. Moreover, inactivation of NCgl2640 resulted in significantly increased methionine production. Using promoter lacZ-fusions of genes involved in sulfur metabolism, we demonstrated the relief of l-methionine repression in the NCgl2640 mutant for cysteine synthase, o-acetylhomoserine sulfhydrolase (metY) and sulfite reductase. Complementation of the mutant strain with plasmid-borne NCgl2640 restored the wild-type phenotype for metY and sulfite reductase.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The two sulfur-containing amino acids methionine and cysteine are key elements in animal feed. While methionine is an essential amino acid, animals are able to convert supplemented methionine into cysteine. Both amino acids are not only essential for protein biosynthesis, but are also precursors of various metabolites such as glutathione, S-adenosylmethionine, polyamines and biotin, and are involved as the methyl group donor in numerous cellular processes. Since concentrations of methionine and cysteine are often low in edible plant sources (Nikiforova et al. 2002), methionine is an important feed additive, with an annual production in the range of 300,000 t, third to glutamate and lysine (Leuchtenberger 1996). Unlike other bulk amino acids, methionine is produced by chemical synthesis and thus is the last of the major commercial amino acids that is not produced by fermentation.
Corynebacterium glutamicum strains have proven to be effective producers of l-lysine and l-threonine (Sahm et al. 1996; Hermann 2003) which, together with l-methionine, belong to the aspartate family of amino acids. Although lysine titers of 120 g/l or more are routinely obtained in industrial fermentations with C. glutamicum, no such process exists for methionine. The key step that distinguishes the biosynthesis of lysine and methionine is the incorporation of sulfur into the carbon skeleton. The common source of sulfur is sulfate, that has to be taken up, activated and reduced by the consumption of 7 mol ATP and 8 mol NADPH per mole of methionine (Neidhardt et al. 1990). In terms of cellular energy demands, this makes methionine the most expensive amino acid.
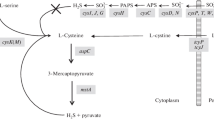
Genes involved in methionine biosynthesis are scattered on the genome of C. glutamicum (Ruckert et al. 2003), while genes involved in assimilatory sulfate reduction are clustered and at least partially organized in the cys-operon (Fig. 1). Sulfite reductase (NCgl2718) generates sulfide and is one of the genes under the control of the putative cys-operon promoter. Sulfide is then incorporated into o-acetylhomoserine via direct sulfhydrylation (MetY), or into o-acetylserine to give cysteine via the transsulfhydrylation pathway (cysteine synthase CysK, NCgl2473; Fig. 1). C. glutamicum and related organisms use both pathways for methionine biosynthesis (Hwang et al. 2002; Lee and Hwang 2003). A diversity of regulatory mechanisms controlling the metabolic flux through these pathways has been described (Lee and Hwang 2003). Tight regulation avoids uneconomic depletion of cellular energy, but deregulation is indispensable for the overproduction of methionine. A detailed understanding of the regulatory mechanisms involved in sulfhydrylation is thus essential for the future rational design of methionine-producing strains.

The split pathway of sulfur incorporation in methionine biosynthesis. Sulfide is incorporated either via direct sulfhydrolation that is catalyzed by MetY (a), or by the transsulfhydrolation pathway (b). lacZ fusions were generated for promoters of the boxed gene products. The promoter of NCgl2718 governs the expression of genes involved in assimilatory sulfate reduction organized in a gene cluster (cys-operon, NCgl2715–NCgl2720). Cysteine synthase (NCgl2473) is not part of this cluster. Ask Aspartate kinase, AsDH aspartate semialdehyde dehydrogenase, CysK cysteine synthase, Hom homoserine dehydrogenase, MetA homoserine acetyltransferase, MetB cystathionine γ-synthase, MetC cystathionine β-lyase, MetH methionine synthase, MetY O-acetylhomoserine sulfhydrolase, MetK S-adenosylmethionine synthase, NCgl2715 sulfate-adenosyltransferase subunit 1, NCgl2716 sulfate-adenosyltransferase subunit 2, NCgl2717 PAPS-reductase, NCgl2718 sulfite reductase annotated as putative nitrite reductase
There are only scattered reports on methionine-producing variants of C. glutamicum. Since 1975, when Kase and Nakayama (1975) reported on a multistep procedure using random mutagenesis and antimetabolite selection, no further improvement has been reported. Recently, a putative transcriptional repressor (McbR) involved in the regulation of the metabolic network directing the synthesis of sulfur containing amino acids was identified and a mcbR knockout strain was constructed (Rey et al. 2003). Still, methionine levels obtained in both examples fall short of commercial relevance.
Here, we report the identification of a novel regulator of methionine biosynthesis by screening a transposon library of C. glutamicum for ethionine-resistant strains. As a structural analogue of methionine, d,l-ethionine cannot be metabolized and thus mimics high concentrations of methionine. Consequently, methionine biosynthesis is downregulated and the organism finally starves from methionine depletion. One of several mechanisms to circumvent antimetabolite toxicity is to outcompete the toxic agent by overproduction of the natural metabolite (Mondal and Chatterjee 1994). Overproducing strains are typically based on mutations that relieve the feed-back inhibition of biosynthetic proteins (Kase and Nakayama 1975). Here, we screened for overproduction upon Tn5531-inactivation of genes involved in regulatory cascades.
Materials and methods
Bacterial strains, media and plasmids
C. glutamicum ATCC14752 or ATCC13032 were routinely cultivated in CGXII minimal medium (Keilhauer et al. 1993). Escherichia coli DH5α was used for standard cloning and E. coli ET12567 for plasmid amplification when plasmids were destined to be transformed in C. glutamicum. Strains and plasmids used in this study are listed in Table 1. Luria broth (LB) supplemented with appropriate antibiotics (50 μg/ml kanamycin, 50 μg/ml chloramphenicol, 100 μg/ml ampicillin) was the standard medium for E. coli strains. LB medium supplemented with 4 mM MgSO4 and 10 mM KCl (Psi-broth) was the recovery medium for chemically transformed E. coli. The recovery medium for electroporated C. glutamicum strains was LB with brain heart infusion and sorbitol (LBHIS; Liebl et al. 1989). Plasmid pCGL0040 (GenBank accession no. U53587) was used as the donor for Tn5531 (IS1207 Kmr) and was amplified in E. coli ET12567.
Recombinant DNA technologies
The transformation of E. coli cells with plasmid DNA was performed with chemically competent E. coli DH5α or ET12567. Cells were prepared according to the rubidium chloride method (http://micro.nwfsc.noaa.gov/protocols/) and transformed as described by Sambrook et al. (1989). Preparation of competent C. glutamicum cells and electrotransformation was done as described elsewhere (Liebl et al. 1989; Ankri et al. 1996a).
Genomic DNA was isolated using the Wizard genomic DNA purification kit (Promega). Plasmid preparation from E. coli cells was routinely done with the QIAprep miniprep kit (Qiagen). Restriction endonucleases were from Roche Diagnostics. Digested DNA fragments were recovered from agarose gels with the QIAEX II gel extraction kit (Qiagen). Standard DNA techniques were performed as described by Sambrook et al. (1989). DNA sequencing was performed using the Global edition IR2 system (LI-COR, Lincoln, Neb.).
The genome database used to identify ORFs and promoters was ERGO (Integrated Genomics, Chicago, Ill.). The NCgl numbers refer to the C. glutamicum ATCC13032 genome sequence in the GenBank database at the National Center for Biotechnology Information (NCBI; GenBank accession no. NC_003450). The promoters deduced from the ERGO database for cysteine synthase cysK (NCgl2473; promoter position 2721625–2721822), sulfite reductase (NCgl2718; promoter position 3005188–3005389) and o-acetylhomoserine-sulfhydrolase (metY; NCgl0625; promoter position 667771–668107) were PCR-amplified and fused by XhoI and BamHI linkers to promoterless lacZ on plasmid pClik (Cmr), opened by the same restriction enzymes. The resulting plasmids were termed pClik-PcysK, pClik-Pcys and pClik-PmetY, respectively (Table 1). A unique BglII site on these plasmids was used to introduce PCR-amplified NCgl2640, using primer pair 2640-fwd-BglII (5′-CCGCTGCTGCTGGTGGCGCTAGATCTGCTAACGGC-3′) and 2640-rev-BglII (5′-ATGTGTTGGGAGATCTCTTAAGTTATTTAGTCCAG-3′). The amplified DNA fragment comprised putative regulatory elements located up to 370 bp upstream of the NCgl2640 gene.
Transposon mutagenesis, screening and localization of transposon insertion sites
Plasmid pCGL0040 was isolated from E. coli ET12567 and C. glutamicum ATCC14752 was transformed with the plasmid by electroporation. Transposon insertion mutants were selected by plating on LBHIS containing 20 μg/ml kanamycin. All mutants were pooled, washed twice with sterile 0.9% NaCl and plated on CGXII containing kanamycin (20 μg/ml) and ethionine (7.5 g/l) in 100-μl aliquots of a 106 dilution of the pool. The most rapidly growing colonies were selected for further analysis.
For the localization of the transposon insertion sites, genomic DNA of the mutants was isolated and digested with EcoRI, as described by Simic et al. (2001). Insertion sites were determined by cloning transposon–chromosome junction sites into EcoRI-digested pUC18. Plasmids from kanamycin-resistant clones were isolated and subsequently sequenced with oligonucleotide Tn5531-Eco (5′-CGGGTCTACACCGCTAGCCCAGG-3′; Simic et al. 2001). The sequences thus obtained were analyzed using the BLASTn program applied to the NCBI GenBank sequence and the ERGO database. Different sequence analysis tools (http://www.expasy.org/, http://npsa-pbil.ibcp.fr or the protein family database, PFAM at http://pfam.wustl.edu/) were used for pattern and profile searches with the NCgl2640 sequence.
Chromosomal deletion of NCgl2640 in C. glutamicum ATCC13032
Using the two primer pairs 2640-SacB1 (5′-GAGAGGGCCCATCAGCAGAACCTGGAACC-3′) with 2640-SacB2 (5′-GATCCAGAGGTCCACAACC-3′) and 2640-SacB3 (5′-GATGGTTCAAGACGAACTCC-3′) with 2640-SacB4 (5′-GAGAGTCGACCAGAATCAATTCCAGCCTTC-3′), the upstream and downstream region of NCgl2640 was PCR-amplified from chromosomal DNA of C. glutamicum ATCC13032. The resulting fragments were digested with ApaI/XbaI and SpeI/SalI, respectively, and cloned together into the nonreplicative vector pClik-SacB, that was digested with ApaI/SalI, yielding plasmid pSdel-NCgl2640. C. glutamicum ATCC13032 was transformed by electroporation with the nonreplicative plasmid pSdel-NCgl2640. Kanamycin-resistant clones contained chromosomally integrated plasmids. Subsequently, we selected for loss of the plasmid by screening for sucrose-resistant mutants according to Schaefer et al. (1994). The deletion was verified by PCR analysis and Southern blotting.
LacZ activity measurements
Selected mutant strains of C. glutamicum were transformed with the plasmids pClik-PcysK, pClik-PmetY and pClik-Pcys or the NCgl2640-complemented derivatives thereof (Table 1). Transformants were grown in CGXII minimal medium containing chloramphenicol (15 μg/ml) in the presence or absence of 10 mM l-methionine. Cells were grown to an optical density at 600 nm (OD600) of 1–3 and analyzed for β-Gal activity, as described by Sambrook et al. (1989). Assays were done in triplicates in four independent test series.
Isolation of DNA-binding proteins
The principle of isolating DNA-binding proteins by DNA-affinity chromatography using magnetic beads is essentially described by Gabrielsen and Huet (1993) and a detailed protocol for C. glutamicum is available (Rey et al. 2003). We basically followed the latter protocol with a few exceptions. All buffers except the elution buffers were supplemented with 2.5 mM l-methionine. Crude extracts of cells grown in the presence or absence of 10 mM l-methionine were prepared separately and combined thereafter. Immediately after cell disruption, crude extract was protected against proteolysis by a protease inhibitor cocktail (phenylmethylsulfonyl fluoride, aprotinin, leupeptin; Rosenberg 1996). After ultracentrifugation of the crude extract (200,000 g, 40 min, 4°C), the protein solution was desalted by gel filtration (Sephadex G25). Biotinylated PCR-amplified promoter-DNA was immobilized on streptavidin-coated Dynabeads (M270; Dynal Biotech). As a negative control fragment, we amplified a 460-bp fragment from the upstream region of the groES gene of C. glutamicum. The washing buffer contained high amounts of unspecific competitor DNA (0.4 mg/ml salmon testes DNA; Sigma). 1D-SDS-PAGE was done with a 4% stacking gel and a 12% separative gel (Schägger and von Jagow 1987) and proteins were stained with colloidal Coomassie brilliant blue G-250 (Neuhoff et al. 1988). The protocol for tryptic digestion and matrix assisted laser desorption/ionization–time of flight (MALDI-TOF) analysis was essentially that described by Hermann et al. (2001).
Determination of extra- and intracellular methionine concentrations
Methionine was quantified as its o-phthaldialdehyde derivative by high-pressure liquid chromatography (HPLC; Molnar-Perl 2001). C. glutamicum was grown to the stationary phase in 500-ml shake-flasks with culture volumes of 50–100 ml at 30°C and 225 rpm. Cells were removed by centrifugation (10,000 g, 10 min, 4°C) and methionine concentrations were determined by HPLC analysis.
For the determination of intracellular methionine levels, cells were separated from the bulk liquid and inactivated by silica oil centrifugation, as described by Ebbighausen et al. (1989), and subsequently disrupted by sonification or by a blue-capped Ribolyser (FastPrep; Q-Biogene). Soluble protein in the supernatant was measured by a Bradford-type assay (Bradford 1976). The intracellular content of soluble protein in C. glutamicum was empirically determined as 250 mg/ml. Based on this value, we calculated the total internal cell volume of a sample in order to determine methionine concentrations upon HPLC analysis.
Results
Selection and identification of ethionine-tolerant transposon mutants
We hypothesized that ethionine tolerance can be acquired by overproduction of methionine, which in turn can be achieved by inactivation of a repressor involved in the regulation of methionine biosynthesis. Therefore, we transformed C. glutamicum ATCC14752 with pCGL0040 as the donor of Tn5531 by electroporation. About 7,000 mutants were obtained on LBHIS plates containing kanamycin. All mutants were scraped off the plates, pooled, washed and plated onto CGXII-agar plates containing 7.5 g/l d,l-ethionine plus kanamycin. We plated about 100,000 colony-forming units (CFU) to ensure multiple recoveries of putatively ethionine-resistant mutants. Growth of the wild type was inhibited by 6 g/l ethionine for at least 4 days. After 2 days, 11 kanamycin- and ethionine-resistant mutants were isolated. All mutants contained the identical Tn5531 insertion in ORF NCgl2640. This mutation was termed 14752∷2640; and one clone was selected for further experimentation (Table 1).
Sequence analysis of transposon insertion loci
The site of transposon insertion in strain 14752∷2640 was located in the C-terminal half of the putative protein at position 2,918,026/2,918,027 (GenBank accession no. NC_003450). NCgl2640 was separated from NCgl2639 by 7 bp, so both genes are presumably organized as an operon (Fig. 2). NCgl2639 is annotated as a putative hydrolase or acetyltransferase (GenBank). NCgl2640 encodes a protein of 42 kDa and homology searches identified more than 25 putative bacterial proteins of significant homology (e-value <2e−20), none of which were functionally assigned. A conserved domain search identified a consensus pattern for proteins of unknown function (COG2170) with high significance and for motif 04107 of the PFAM database that is characteristic for the glutamate-cysteine ligase family. No further consensus patterns were detected, in particular none for DNA-binding proteins.

Genomic context of NCgl2640. GenBank annotations available for the open reading frames: NCgl2638 similarities to multisubunit Na+/H+ antiporter, NCgl2639 similar to predicted hydrolases or acyltransferases, alpha/beta hydrolase superfamily, NCgl2640 hypothetical protein/uncharacterized BCR, NCgl2641 hypothetical protein, no annotation. Tn5531 Transposon 5531 (IS1207). Filled triangle Site of insertion of Tn5531 in NCgl2640
Verification of the ethionine-resistance phenotype
C. glutamicum ATCC14752 and the mutant strain were cultivated in shake-flasks in CGXII medium containing 3 g/l glucose in the presence or absence of 7.5 g/l d,l-ethionine. Growth of the mutant in the presence of ethionine was indistinguishable from growth without ethionine or growth of the wild type without ethionine (Fig. 3).

Growth of C. glutamicum wild type and mutant grown in CGXII medium in the presence or absence of ethionine. The glucose concentration was 3 g/l, the d,l-ethionine concentrations was 7.5 g/l. Filled symbols Ethionine present during cultivation, open symbols cultivation without ethionine. Filled triangles Wild type, filled circles mutant strain
To exclude second-site mutations that may have caused the ethionine-resistance phenotype, we tested the effect of NCgl2640 inactivation in C. glutamicum ATCC13032. NCgl2640 was excised by homologous recombination and selection of sucrose-tolerant mutant strains. This mutant, designated 13032∷2640, was readily resistant to d,l-ethionine at 7.5 g/l, while no growth of wild-type ATCC13032 was detected on these plates. Thus, we demonstrated that the ethionine-resistance phenotype depended on inactivation of NCgl2640.
Increased l-methionine levels in the NCgl2640 mutant
The resistance of C. glutamicum 14752∷2640 to high ethionine concentrations may be due to upregulated biosynthesis of l-methionine. To test this hypothesis, we analyzed the production of l-methionine in the wild type and mutant in CGXII minimal medium batch cultures. Generally, the mutant accumulated about twice the amount of methionine compared with the wild type, both intra- and extracellularly (Fig. 4).

Extra- and intracellular l-methionine levels in C. glutamicum wild type and the NCgl2640 knockout mutant in CGXII medium. Dark bars C. glutamicum wild type, white bars NCgl2640 knockout mutant
Altered expression levels of methionine biosynthesis genes in the NCgl2640 mutant
We assumed that tight regulation of sulfur incorporation would be a key element in methionine biosynthesis. Hence, inactivation of a repressor of sulfur gene expression would cause elevated methionine levels that confer ethionine resistance. Therefore, we decided to elucidate the impact of the NCgl2640 knockout on the expression levels of metY, cysK and cys-operon genes. For this purpose, strain 14752∷2640 and the wild type were transformed with the lacZ-reporter plasmids pClik-PcysK, pClik-PmetY and pClik-Pcys (Table 1) and grown to an OD600 of 3.0 in CGXII medium with or without 10 mM l-methionine. Gene expression was then detected by monitoring LacZ activity.
In the wild type, the presence of methionine reduced the expression levels of all examined genes, but expression of the cys-operon was completely abolished. In the mutant strain, significant derepression was observed for the cys-operon (Fig. 5). A complementation assay was used to exclude the polar effects of the inactivation of NCgl2640 on the adjacent NCgl2639 (hydrolase or acetyltransferase; Fig. 2). Expression of NCgl2640 from medium-copy-number plasmids under the control of its endogenous promoter (370 bp of the upstream sequence were included) completely restored the wild-type phenotype in the mutant, i.e. methionine-induced repression of the cys-operon (Fig. 5).

Influence of the NCgl2640 knockout on methionine-dependent expression of cysteine synthase (cysK), o-acetylhomoserine sulfhydrolase (metY) and the cys-operon. C. glutamicum ATCC14752 (wild type), mutant strain 14752∷2640 and mutant strains complemented with plasmid-borne NCgl2640 (∷2640-cpl), harboring reporter plasmids pClik-PcysK, pClik-PmetY or pClik-Pcys were grown in CGXII medium (3 g/l glucose) in the presence (white bars) or absence (dark bars) of l-methionine (10 mM). Promoter activity was determined by LacZ activity reporter assays and quantified in Miller units
NCgl2640 does not bind to the cysK, metY and cys-operon promoters
We clearly demonstrated that expression of the cys-operon was modulated by NCgl2640. Since classic DNA-binding motifs could not be identified, we employed DNA-affinity purification in a pull-down assay to investigate whether NCgl2640 could bind to the respective promoter regions. PCR-amplified and bead-immobilized promoters were incubated in the presence of 2.5 mM l-methionine with combined crude extracts of C. glutamicum cells cultivated in the presence or absence of 10 mM l-methionine. Proteins that were eluted from the promoters at high salt concentrations (>200 mM) were separated by 1D-SDS-PAGE and analyzed by MALDI-TOF. Employing a similar approach, Rey et al. (2003) isolated the McbR-repressor along with four additional proteins that appeared to bind specifically to the metY-promoter. We largely confirmed these results in our study. In addition, we found that one of the proteins, exopolyphosphatase, reported by Rey et al. (2003) to bind specifically to the metY-promoter, also binds to the control promoter of groES, indicating unspecific binding. Moreover, we could show that McbR also binds to promoters of cysK and the cys-operon; but binding to the cys-operon promoter was low compared to the metY-promoter (Fig. 6). Consistent in both studies, however, NCgl2640 was not detected, indicating that a direct DNA–protein interaction of NCgl2640 is not involved in the observed NCgl2640-mediated regulation.

SDS-PAGE of proteins binding to the putative control-promoter regions of groES (a) and to putative promoter regions of the C. glutamicum genes for cysteine synthase (cysK), o-acetylhomoserine sulfhydrolase (metY) and genes of the putative cys-operon (b). 1 Single-strand binding protein, 2 exopolyphosphatase, 3 diadenosine-tetraphosphate hydrolase, 4 DNA-polymerase III α-subunit, 5 DNA-polymerase I, 6 TetR-like regulator McbR that was shown to be involved in the regulation of methionine biosynthesis (Rey et al. 2003), 7 exopolyphosphatase (degradation product), 8 exopolyphosphatase, 9 ATP-phosphoribosyltransferase, 10 DNA-polymerase I. Except for exopolyphosphatase and DNA-polymerase I, none of the identified proteins bound to the groES control-promoter fragment (Fig. 6a). GAP-DH Glyceraldehyde-3-phosphate dehydrogenase used as additional marker-protein (37 kDa), M protein marker
Discussion
Using Tn5531 mutagenesis and antimetabolite selection, we demonstrated that a single gene inactivation event renders C. glutamicum tolerant to high concentrations of ethionine and increases methionine biosynthesis. Classic strain development encompasses several rounds of random mutagenesis and antimetabolite selection (Sauer 2001); and the structural analogue of methionine, ethionine, is the major antimetabolite used for the selection of methionine overproducers (Lawrence et al. 1968; Kase and Nakayama 1975; Tani et al. 1988). Ethionine inhibition of methionine biosynthesis can be overcome by mutations that relieve feedback inhibition, e.g. by altering allosteric binding sites, but the resulting resistance is typically based on combinations of unidentified mutations. Here, we demonstrate that inactivation of NCgl2640 is sufficient to confer high-level tolerance to ethionine. To the best of our knowledge, this is the first report on a single-gene knockout that confers ethionine resistance.
Doubling of the methionine pool apparently suffices to confer resistance to a roughly 10-fold excess of the antimetabolite. The observed increase in extracellular methionine levels equals that reported recently for the mcbR mutant of C. glutamicum (Rey et al. 2002). Thus, inactivation of the putative master regulator of methionine biosynthesis has a similar effect on methionine production as the inactivation of NCgl2640, which underlines the importance of additional regulatory mechanisms in methionine and sulfur metabolism.
Based on lacZ-promoter assays, NCgl2640 was shown to exhibit a regulatory function in methionine biosynthesis that is most pronounced for genes involved in sulfur metabolism (cys-operon). In contrast to the putative transcriptional repressor McbR, whose activity does not appear to be directly induced by methionine, NCgl2640-based regulation is clearly dependent on extracellular methionine. Two arguments render a direct transcriptional regulation by NCgl2640 unlikely. First, no DNA-binding motif was detected, and second, we isolated McbR but not NCgl2640 with the DNA-affinity purification assay. Thus, NCgl2640 is probably an indirectly acting regulatory element involved in the repression of sulfur metabolism genes.
Two hypotheses on the actual regulation mechanism of NCgl2640 can be formulated from its annotation in the genome sequence of C. glutamicum. In the first, NCgl2640 might modulate gene expression indirectly via the putative master regulator McbR that was shown to bind directly to the metY-promoter (Rey et al. 2003). Based on one annotation of NCgl2640 as a putative glutamyl-cysteine-ligase, it might be speculated that interference with glutathione biosynthesis might affect McbR directly or indirectly, resulting in altered expression levels of McbR-controlled genes. We observed a less pronounced effect of the inactivation of NCgl2640 on cysK and metY expression levels compared with cys-operon. By a proteomic analysis, Rey et al. (2003) showed that the levels of CysK, MetK and MetY were significantly altered in a mcbR knockout strain, although the levels of proteins encoded in the cys-operon were not affected. Thus, McbR seems to play a minor role in NCgl2640-mediated regulation.
The second hypothesis is based on the alternative annotation as a GTPase-activating protein published by Kyowa Hakko Kogyo Co. Ltd (GenBank accession no. BAC00130) and listed in ERGO, a commercial genome database that currently covers more than 600 genomes. GTPase-activating proteins are elements of GTPase-dependent regulation cascades that indirectly control gene expression (Voncken et al. 1995; Donovan et al. 2002; Litvak and Selinger 2003), mostly in eukaryotes (Caldon and March 2003) but also in bacteria (Lerner and Inouye 1991; Zhang and Inouye 2002). Thus, NCgl2640 may act as the methionine-sensing element that specifically enhances the GTP-hydrolyzing activity of its putative target GTPase that is involved in the regulation of methionine biosynthesis.
References
Ankri S, Reyes O, Leblon G (1996a) Electrotransformation of highly DNA-restrictive corynebacteria with synthetic DNA. Plasmid 35:62–66
Ankri S, Serebrijski I, Reyes O, Leblon G (1996b) Mutations in the Corynebacterium glutamicum proline biosynthetic pathway: a natural bypass of the proA step. J Bacteriol 178:4412–4419
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Caldon CE, March PE (2003) Function of the universally conserved bacterial GTPases. Curr Opin Microbiol 6:135–139
Donovan S, Shannon KM, Bollag G (2002) GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta 1602:23–45
Ebbighausen H, Weil B, Krämer R (1989) Isoleucine excretion in Corynebacterium glutamicum: evidence for a specific efflux carrier system. Appl Microbiol Biotechnol 31:184–190
Follettie MT, Peoples OP, Agoropoulou C, Sinskey AJ (1993) Gene structure and expression of the Corynebacterium flavum N13 ask-asd operon. J Bacteriol 175:4096–4103
Gabrielsen OS, Huet J (1993) Magnetic DNA affinity purification of yeast transcription factor. Methods Enzymol 218:508–525
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580
Hermann T (2003) Industrial production of amino acids by coryneform bacteria. J Biotechnol 104:155–172
Hermann T et al (2001) Proteome analysis of Corynebacterium glutamicum. Electrophoresis 22:1712–1723
Hwang BJ, Yeom HJ, Kim Y, Lee HS (2002) Corynebacterium glutamicum utilizes both transsulfuration and direct sulfhydrylation pathways for methionine biosynthesis. J Bacteriol 184:1277–1286
Kase H, Nakayama K (1975) l-Methionine production by methionine analog-resistant mutants of Corynebacterium glutamicum. Agric Biol Chem 39:153–160
Keilhauer C, Eggeling L, Sahm H (1993) Isoleucine synthesis in Corynebacterium glutamicum: molecular analysis of the ilvB-ilvN-ilvC operon. J Bacteriol 175:5595–5603
Lawrence DA, Smith DA, Rowbury RJ (1968) Regulation of methionine synthesis in Salmonella typhimurium: mutants resistant to inhibition by analogues of methionine. Genetics 58:473–492
Lee HS, Hwang BJ (2003) Methionine biosynthesis and its regulation in Corynebacterium glutamicum: parallel pathways of transsulfuration and direct sulfhydrylation. Appl Microbiol Biotechnol 62:459–467
Lerner CG, Inouye M (1991) Pleiotropic changes resulting from depletion of Era, an essential GTP-binding protein in Escherichia coli. Mol Microbiol 5:951–957
Leuchtenberger W (1996) Amino acids—technical production and use. In: Roehr M (ed) Biotechnology, 2nd edn. VCH, Weinheim, pp 466–504
Liebl W, Bayerl A, Schein B, Stillner U, Schleifer KH (1989) High efficiency electroporation of intact Corynebacterium glutamicum cells. FEMS Microbiol Lett 65:299–304
Litvak Y, Selinger Z (2003) Bacterial mimics of eukaryotic GTPase-activating proteins (GAPs). Trends Biochem Sci 28:628–631
MacNeil DJ et al (1992) Complex organization of the Streptomyces avermitilis genes encoding the avermectin polyketide synthase. Gene 115:119–125
Molnar-Perl I (2001) Derivatization and chromatographic behavior of the o-phthaldialdehyde amino acid derivatives obtained with various SH-group-containing additives. J Chromatogr 913:283–302
Mondal S, Chatterjee SP (1994) Enhancement of methionine production by methionine analogue ethionine resistant mutants of Brevibacterium heali. Acta Biotechnol 14:199–204
Neidhardt FC, Ingraham JL, Schaechter M (1990) Physiology of the bacterial cell: a molecular approach. Sinauer Associates, Sunderland, Mass.
Neuhoff V, Arold N, Taube D, Ehrhardt W (1988) Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie brilliant blue G-250 and R-250. Electrophoresis 9:255–262
Nikiforova V, et al (2002) Engineering of cysteine and methionine biosynthesis in potato. Amino Acids 22:259–278
Rey D, Ruckert C, Kalinowski J, Puhlor A, Bathe B, Hutmacher K, Pfefferle W (2002) Nucleotide sequences which code for the metD gene. In: World Intellectual Property Organization. http://www.wipo.int/ipdl/en/search/pct/search-adv.jsp
Rey DA, Puhler A, Kalinowski J (2003) The putative transcriptional repressor McbR, member of the TetR-family, is involved in the regulation of the metabolic network directing the synthesis of sulfur containing amino acids in Corynebacterium glutamicum. J Biotechnol 103:51–65
Rosenberg IM (1996) Protein analysis and purification. Benchtop techniques. Birkhäuser, Boston
Ruckert C, Puhler A, Kalinowski J (2003) Genome-wide analysis of the l-methionine biosynthetic pathway in Corynebacterium glutamicum by targeted gene deletion and homologous complementation. J Biotechnol 104:213–228
Sahm H, Eggeling L, Eikmanns BJ, Kramer R (1996) Construction of l-lysine-, l-threonine-, or l-leucine-overproducing strains of Corynebacterium glutamicum. Ann NY Acad Sci 782:25–39
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
Sauer U (2001) Evolutionary engineering of industrially important microbial phenotypes. Adv Biochem Eng Biotechnol 73:129–169
Schaefer A, Tauch A, Jaeger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73
Schägger H, von Jagow G (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 166:368–379
Simic P, Sahm H, Eggeling L (2001) l-Threonine export: use of peptides to identify a new translocator from Corynebacterium glutamicum. J Bacteriol 183:5317–5324
Tani Y, Lim W-J, Yang H-C (1988) Isolation of l-methionine enriched mutant of a methylotrophic yeast, Candida boidinii No. 2201. J Ferment Bioeng 66:153–158
Voncken JW, et al (1995) Increased neutrophil respiratory burst in bcr-null mutants. Cell 80:719–728
Zhang J, Inouye M (2002) MazG, a nucleoside triphosphate pyrophosphohydrolase, interacts with Era, an essential GTPase in Escherichia coli. J Bacteriol 184:5323–5329
Acknowledgements
We thank L. Eggeling and G. Leblon for kindly providing strain C. glutamicum ATCC14752 and plasmid pCGL0040, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mampel, J., Schröder, H., Haefner, S. et al. Single-gene knockout of a novel regulatory element confers ethionine resistance and elevates methionine production in Corynebacterium glutamicum. Appl Microbiol Biotechnol 68, 228–236 (2005). https://doi.org/10.1007/s00253-005-1893-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-005-1893-6