
Abstract
Aminoglycosides are potent bactericidal antibiotics targeting the bacterial ribosome, where they bind to the A-site and disrupt protein synthesis. They are particularly active against aerobic, Gram-negative bacteria and act synergistically against certain Gram-positive organisms. Aminoglycosides are used in the treatment of severe infections of the abdomen and urinary tract, bacteremia, and endocarditis. They are also used for prophylaxis, especially against endocarditis. Bacterial resistance to aminoglycosides continues to escalate and is widely recognized as a serious health threat. This might be the reason for the interest in understanding the mechanisms of resistance. It is now clear that the resistance occurs by different mechanisms such as prevention of drug entry, active extrusion of drugs, alteration of the drug target (mutational modification of 16S rRNA and mutational modification of ribosomal proteins), and enzymatic inactivation through the expression of enzymes, which covalently modify these antibiotics. Enzymatic inactivation is normally due to acetyltransferases, nucleotidyltransferases, and phosphotransferases. In this review, we focus on the recent concept of molecular understanding of aminoglycoside action and resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The aminoglycosides are a family of molecules containing a molecular nucleus, an aminocyclitol ring that can be streptidine or 2‐deoxystreptamine and two or more amino sugars linked by glycosidic bonds to the nucleus. The first aminoglycoside, streptomycin, was discovered by Waksman, Schatz, and Bugie in 1944 and was isolated from Streptomyces griseus. Most aminoglycosides are naturally occurring substances produced by actinomycetes of either the genus Streptomyces or Micromonospora. Amikacin, netilmicin, dibekacin, isepamicin, and arbekacin, which are semisynthetic derivatives from the genus Streptomyces are labeled ‘‐cins’, whereas those originating from the genus Micromonospora (neomycin, tobramycin, paramomycin) are labeled ‘‐mycins’ (Giamerllou 1986). The bactericidal activity of aminoglycosides is primarily exerted by inhibition of protein synthesis. Aminoglycoside molecules bind to the bacterial 30S ribosomal subunit rendering the ribosome unavailable for translation, which results in cell death (Kotra et al. 2000). The importance of the interaction of the antibiotic molecule with the ribosome has been shown using mutants or by chemical modification of ribosomal components. Those derivatives that had a reduced affinity for the aminoglycoside molecule exhibited reduced levels of susceptibility. Other major metabolic perturbations caused by aminoglycosides, which could be secondary effects, include translation, membrane damage (altered membrane composition and permeability), altered cellular ionic concentrations, and disturbances in the synthesis of DNA and RNA (Fourmy et al. 1996). Aminoglycosides are active primarily against aerobic Gram‐negative bacilli and Gram‐positive cocci. However, it is well‐known that bacteria develop resistance to antibiotics and the frequency of multiresistant isolates has alarmingly increased (Mingeot‐Leclercq et al. 1999). As the problem of global antibiotic resistance continues to worsen, aminoglycosides have assumed increasing importance in clinical practice. Their broad antimicrobial spectrum, rapid bactericidal action, and ability to act synergistically with other drugs have made them especially useful in the treatment of serious nosocomial infections. However, as with other drugs, their overuse and misuse lead to the development of resistance in important microbial pathogens (Zembower et al. 1998). The detailed studies on several aminoglycoside‐modifying enzymes have been limited. Recently, we have purified streptomycin adenylyltransferase from recombinant Escherichia coli (Jana et al. 2005). Some mechanistic and mutational studies have been carried out and the crystal structures of two acetyltransferases, a nucleotidyltransferase, and a phosphotransferase have been reported (Davies and Wright 1997a). Genes encoding aminoglycoside-modifying enzymes are often located on plasmids, which permit cell-to-cell dissemination of the aminoglycoside resistance trait. Furthermore, several of these genes are also included in transposons and integrons, which result in their rapid dissemination at molecular level (Mingeot‐Leclercq et al. 1999). Structural studies of the aminoglycoside modifying enzymes are limited to interpretations of primary sequence information. The crystal structures of aminoglycoside phosphotransferase [APH (3’)], aminoglycoside acetyltransferase [AAC (6’)], and aminoglycoside nucleotidyltransferase [ANT (4’)] are known (Sakon et al. 1993; Wybenga-Groot et al. 1999; Burk et al. 2001). However, the exact biochemical functions of the conserved motifs in these enzymes have yet to be confirmed by more detailed structural studies. The molecular architectures of aminoglycoside-modifying enzymes are of special interest as there are few reports on 3-D structure of these enzymes. Furthermore, the elucidation of the active site of modifying enzymes can provide insight into the mechanism of nucleotide phosphate transfer and may provide a template for drug design. There is a growing literature on the various aspects of aminoglycoside antibiotic. Therefore, we decided the present review covers recent literature dwelling upon the molecular understanding of aminoglycoside action and resistance.
Chemical structure
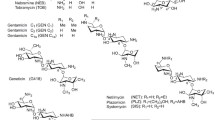
As a class of antibiotics, aminoglycosides have a backbone structure consisting of an aminocyclitol ring saturated with amine and hydroxyl substitutions. In the majority of clinically useful aminoglycosides, this aminocyclitol moiety is streptamine or 2-deoxystreptamine (Fig. 1). Streptomycin, possessing a streptidine molecule, is the only exception. The aminocyclitol nucleus is connected through glycosidic linkages to various amino sugars (aminoglycosides) (Spelman et al. 1989). The aminoglycosides can be conveniently divided into three structural types based on the position of their glycosidic linkages. These structural types include the 4,6-disubstituted 2-deoxystreptamines containing most of the clinically useful aminoglycosides such as gentamicin, tobramycin, amikacin, and netilmicin, the 4,5-disubstituted 2-deoxystreptamines (neomycin and paromomycin), and others (streptomycin and spectinomycin) (Fig. 2). Spectinomycin, although often considered an aminoglycoside, does not contain an amino sugar. Thus, several investigators have suggested that the term aminocyclitol be used to describe this entire group of agents rather than the less precise term aminoglycoside (Ristuccia and Cunha 1982). The aminoglycoside structure is important in understanding their chemical properties. These are basic, strongly polar compounds that are positively charged (cationic). They are highly soluble in water, relatively insoluble in lipids, and have enhanced antimicrobial activity in alkaline rather than acidic environments. As a result, aminoglycosides are minimally absorbed from the gut and penetrate the blood brain barrier poorly. The cationic nature of the aminoglycosides contributes to their antimicrobial activity. Because of their positive charge, they are able to bind negatively charged lipopolysaccharide of the bacterial cell wall and a variety of intracellular and cell membrane anionic molecules such as DNA, RNA, and phospholipids. Unfortunately, their positive charge at physiological pH also contributes to their toxicities, e.g., nephrotoxicity, ototoxicity, and neuromuscular blockade. Aminoglycosides are metabolically stable compounds that are excreted unchanged in the urine.

Backbone structures of the aminoglycosides

Representative structures of clinically useful aminoglycosides
Mechanism of action
The antimicrobial action of aminoglycosides is facilitated by their chemical structure. These antibiotics can be considered as polycationic species. Because they are polycationic, they show binding affinity for negatively charged residues in the outer membrane of Gram-negative bacilli and in nucleic acids. Their bactericidal activity is due to inhibition of bacterial protein synthesis through binding to prokaryotic 16S rRNA and disruption of the integrity of the bacterial cell membrane. The uptake process is self-promoted involving the drug-induced disruption of Mg2+ bridges between adjacent lipopolysaccharide molecules. They show their bactericidal activity through a multistep process.
Binding
First, aminoglycosides bind electrostatically to negatively charged residues in the outer membrane of Gram-negative bacteria in a passive, nonenergy dependent process (Hancock et al. 1991). Then they diffuse through outer membrane porin channels and enter the periplasmic space. Their subsequent transport across the cytoplasmic membrane requires metabolic energy from the electron transport system in an oxygen-dependent process. This phase of transport has been termed energy dependent phase I (EDP-I) and is the rate limiting step. The oxidative energy production required for transport explains why aminoglycosides are much less active in an anaerobic environment. EDP-I can also be inhibited by divalent cations, reduced pH, and hyperosmolarity. In the cytosol, aminoglycosides bind to the 30S subunit of ribosomes through an energy dependent process (EDP-II) (Bryan and Kwan 1983).
Misreading
The binding does not prevent the formation of the initiation complex of peptide synthesis, it perturbs the elongation of the nascent chain by impairing the proofreading process controlling translational accuracy. The aberrant proteins may be inserted into the cell membrane, leading to altered permeability and further stimulation of aminoglycoside transport (Melancon et al. 1992).
3-D complex
It is well-known that aminoglycosides bind to the bacterial ribosome and inhibit protein synthesis. The precise mechanisms of their antimicrobial activity are still a subject of study. The ribosome is a complex structure comprising three RNA molecules and more than 50 proteins. The complex catalyzes protein synthesis with the assistance of several guanosine 5c-triphosphate hydrolyzing protein factors. Aminoglycoside antibiotics bind to the 30S ribosomal subunit, which plays a crucial role in providing high-fidelity translation of genetic material. Recently, atomic structures for both the large and the small ribosomal subunits and high-resolution crystal structures of the 30S subunit with streptomycin, spectinomycin, paromomycin, and hygromycin B have been solved (Ban et al. 2000). Together with several available nuclear magnetic resonance (NMR) structures for the ribosomal constituents (Fourmy et al. 1998), these structures provide valuable information not only on processes during translation but also on molecular mechanisms of interaction of aminoglycosides with the bacterial ribosome. The 16S rRNA from E. coli is well-studied among the rRNA subunits, particularly the interactions of various aminoglycoside antibiotics with the 16S rRNA (Moazed and Noller 1987) and their effects on the process of translation of mRNA into polypeptides have been scrutinized (Noller 1991). Similar rRNA structures exist in other organisms, such as yeast and Tetrahymena. Treatment of rRNA with an aminoglycoside protects several nucleic acid bases in rRNA from chemical modification, implying that these molecules possess high affinities for certain sites in rRNA. This mode of binding was likened by Noller (1991) to that of enzyme inhibitors, which usually bind to the active sites of enzymes and interfere with their activities. Different classes of aminoglycoside antibiotics bind to different sites on the rRNA, depending on the structural complementarity between the two. For example, neomycin, paromomycin, gentamicin, and kanamycin are believed to bind to the A-site on the 16S rRNA in E. coli in a similar fashion and were shown to protect bases A1408 and G1494 in chemical footprinting experiments (Noller 1991). Four bases, A1408, A1492, A1493, and G1494, in the rRNA A-site interact with tRNA, although with different affinities. The binding of the aforementioned aminoglycosides to the A-site in the decoding region (i.e., the site of codon and anticodon recognition) interferes with the accurate recognition of cognate tRNA by rRNA during translation (Noller 1991). These interactions are also thought to interfere with the translocation of tRNA from the A-site to the peptidyl–tRNA site (P-site). Puglisi et al. (1998) recently provided structural evidence on the mode of interactions of paromomycin, a representative aminoglycoside of the neomycin class, with a 27-nucleotide RNA template that was designed to mimic the A-site region of the 16S rRNA in E. coli. The design of the RNA template was based on previous knowledge that paromomycin interacts with the C1407 · G1494 base pair, A1408, A1493, and U1495 and that these bases are absolutely necessary for high-affinity binding (Recht et al. 1996). Another structural study was performed on binding of tobramycin to an RNA aptamer (Cate et al. 1999). The RNA aptamer that was used in this study was a 26-nucleotide stem-loop RNA. There are four mismatched pairs, U7 · G20, G8 · U19, G9 · A18, and U11 · U16, in this RNA aptamer that are part of the zippered hairpin loop. Tobramycin binds in this groove partially encapsulated by the surface of the deep groove and the guanine base of the residue G15. In this complex, ring I of tobramycin sits on the floor of the deep groove. One of the amino groups on ring II of tobramycin interacts with the phosphate backbone in the deep groove, and the other amino group is exposed to the solvent. Ring III is positioned in the center of the deep groove, with hydroxyl groups directed toward the floor of the groove. The conformation of the RNA aptamer described above was suggested to be similar to those of the hairpin loops in tRNA and rRNA (Jiang and Patel 1998). The A-site makes weak contacts with the mRNA and tRNA, implying that this region plays a role in recognition of appropriate tRNA via subtle changes in the free energy. The binding of aminoglycoside near this site may affect the delicate process of interactions between codon and anticodon (Xi and Arya 2005). It was also proposed that the presence of an aminoglycoside stabilizes the complex of mRNA and tRNA at the A-site, which in turn affects the process of translation (Cate et al. 1999). It is difficult to surmise all the effects of aminoglycosides on the rRNA structure, and further structural studies with the aminoglycosides bound to the complexes, such as the 70S rRNA, will be helpful in elucidating and understanding the subtle changes that lead to the antibiotic actions of aminoglycosides. A number of investigators have used synthetic probes to understand the interactions between RNA templates and aminoglycosides. It has been suggested that aminoglycosides bind to more than one target site in the ribozyme (Michael et al. 1999). Recently, several aminoglycoside antibiotics such as neomycin B, tobramycin, and kanamycin A have been dimerized either symmetrically or asymmetrically by using a “tether,” and their binding affinities were compared to those of the monomeric parent aminoglycosides (Michael et al. 1999). It was suggested that, if there were multiple binding sites on the RNA, the dimerized aminoglycosides should bind with a higher affinity than the parent antibiotic, provided that multiple binding sites are accessible. It was indeed observed that the dimerized aminoglycosides bind to the Tetrahymena ribozyme 20 to 1,200-fold better than the parent aminoglycosides. One explanation for the higher binding affinity could be the increased number of positively charged amino groups on the dimerized aminoglycoside, but this effect seems to be synergistic with the entropic advantage gained by dimerization (Welch et al. 2005). It also indicated that the presence of multiple high-affinity binding sites for aminoglycoside antibiotics in an RNA molecule bulge in the RNA sequence is necessary to allow binding of aminoglycosides (Cho et al. 1998). By using a specific stem-loop derivative of the RNA aptamer, a series of chemical interference, chemical modification, and mutation studies was performed to understand the structural requirements for binding of tobramycin to the RNA aptamer. This aminoglycoside appeared to interact mainly with the nucleic acid bases in the RNA aptamer but not with the phosphate backbone. The presence of a bulge, however, was proposed to be important for the high-affinity binding of tobramycin in a stoichiometric ratio, and it was concluded that a bulge creates a cavity for interactions of the aminoglycoside and the nucleic acid base (Pilch et al. 2005). This analogy can be applied to other RNA sites, such as the hammerhead region and the A-site, where a cavity is present due to the noncanonical base-pairing, loops, or bulges that create a suitable site for the aminoglycosides to interact with the anionic phosphate groups and the nucleic acid bases.
Therapeutic uses
Aminoglycosides display bactericidal, concentration-dependent killing action and are active against a wide range of aerobic Gram-negative bacilli. They are also active against staphylococci and certain mycobacteria. Aminoglycosides are effective even when the bacterial inoculum is large, and resistance rarely develops during the course of treatment. These potent antimicrobials are used as prophylactic agents and in the treatment of a variety of clinical situations (Table 1) (Rybak and Whitworth 2005). Gentamicin is the aminoglycoside used most often because of its low cost and reliable activity against Gram-negative aerobes (Rougier et al. 2004). However, local resistance patterns should influence the choice of therapy. In general, gentamicin, tobramycin, and amikacin are used in similar circumstances, often interchangeably. Tobramycin may be the aminoglycoside of choice for use against Pseudomonas aeruginosa because it has shown greater in vitro activity. Nevertheless, the clinical significance of this activity has been questioned. Amikacin is particularly effective when used against bacteria that are resistant to other aminoglycosides because its chemical structure makes it less susceptible to inactivating enzymes. Depending on local patterns of resistance, amikacin may be the preferred agent for serious nosocomial infections caused by Gram-negative bacilli. In clinical practice, they possess many desirable properties, the most important of which may be rapid bactericidal activity against a wide range of pathogens (Peloquin et al. 2004). The advantages and disadvantages of aminoglycosides are listed in Table 2. The major limitations of these agents include a relatively low therapeutic index with both nephrotoxicity and ototoxicity. They are not absorbed orally due to their cationic nature and thus must be given parenterally by either an intravenous or intramuscular route. Streptomycin is the drug of choice against Francisella tularensis (tularemia), Yersinia pestis (plague), and as an alternative choice for the treatment of brucellosis. It is also the most active aminoglycoside against Mycobacterium tuberculosis including many multidrug resistant strains (Magnet and Blanchard 2005).
Aminoglycoside resistance
Most resistance to aminoglycosides is caused by inactivation by intracellular bacterial enzymes. Because of structural differences, amikacin is not inactivated by the common enzymes that inactivate gentamicin and tobramycin. Therefore, a large proportion of the Gram-negative aerobes that are resistant to gentamicin and tobramycin are sensitive to amikacin. In addition, with increased use of amikacin, a lower incidence of resistance has been observed compared with increased use of gentamicin and tobramycin (Watanabe et al. 2004). P. aeruginosa may show adaptive resistance to aminoglycosides. This occurs when formerly susceptible populations become less susceptible to the antibiotic as a result of decreased intracellular concentrations of the antibiotic. This decrease may result in colonization, slow clinical response, or failure of the antibiotic despite sensitivity on in vitro testing (Yao et al. 2004). Aminoglycosides are often combined with a beta-lactam drug in the treatment of Staphylococcus aureus infection. This combination enhances bactericidal activity, whereas aminoglycoside monotherapy may allow resistant staphylococci to persist during therapy and cause a clinical relapse once the antibiotic is discontinued (Davies and Wright 1997b). Infective endocarditis that is due to enterococci with high levels of resistance to aminoglycosides is becoming increasingly common. All enterococci have low-level resistance to aminoglycosides because of their anaerobic metabolism. In the treatment of bacterial endocarditis, a beta-lactam drug is also used synergistically to facilitate aminoglycoside penetration into the cell. When high-level resistance occurs, it is typically due to the production of inactivating enzymes by the bacteria. Because of the increasing frequency of this resistance, all enterococci should be tested for antibiotic susceptibility (Dworkin 1999). As with all antibiotics, resistance to aminoglycosides is becoming increasingly prevalent. Repeated use of aminoglycosides, especially when only one type is employed, leads to an increased incidence of resistance (Fluit and Schmitz 1999). Nevertheless, resistance to aminoglycosides requires long periods of exposure or very large inoculums of organisms and occurs less frequently than with other agents, such as third-generation cephalosporins, which are also effective against Gram-negative organisms (Neu 1992). The aminoglycoside resistance genes are derived from bacterial genes, which encode enzymes involved in normal cellular metabolism (Pezzella et al. 2004). The selective pressure of aminoglycoside usage causes mutations, which alter the expression of these enzymes, resulting in the ability to modify aminoglycosides. Bacteria can acquire foreign DNA by the mechanisms of transduction, transformation, and conjugation. This is facilitated by two types of genetic elements, self-transferable conjugative plasmids, and transposons (Ahmed and Shimamoto 2004).
Mechanism of resistance
The mechanisms of bacterial resistance to aminoglycosides have been the subject of numerous genetic and biochemical studies (Vakulenko and Mobashery 2003; Wright 2005). There are three general mechanisms of aminoglycoside resistance: (1) reduction of the intracellular concentration of the antibiotic within bacterial cells, usually via efflux of the agent out of the bacterial cell by either dedicated or general efflux pumps; (2) alteration of the molecular target of the antibiotic, usually as result of a spontaneous mutation in the gene encoding the target or substitution of the target’s function by an exogenous gene; and (3) enzymatic inactivation of the aminoglycoside (Walsh 2000; Azucena and Mobashery 2001; Walmsley 2001). It must be acknowledged that even for these broad classifications there are some resistance mechanisms that do not really fall neatly into any of those three categories. In addition, more than one resistance mechanism is at play (often in the same strain at the same time) in the case of some classes of drug. Given the wide diversity of resistance mechanisms and the genes encoding them, it would seem a fruitless enterprise to develop agents to circumvent their activity.
Active efflux pump
Aminoglycoside concentration was decreased inside a target cell by reduction of drug uptake, activation of drug efflux pump, or both (Hatch and Schiller 1998). This will affect the susceptibility of the strain to the whole class of aminoglycoside compounds and can be the cause of intrinsic or acquired resistance. Bacterial efflux pump is an energy-dependent (ATP) pump and is now recognized as a major cause of antibiotic resistance. This is particularly true for the multidrug-resistant opportunist pathogens responsible for nosocomial infections. Bacterial species constitutively expressing such transporters are intrinsically resistant to low levels of various antibiotics. However, mutations in the regulatory genes of the pumps or induction of expression in the presence of substrate, can lead to the overexpression of the originally constitutive or pump genes (Aires et al. 1999; Masuda et al. 2000). In the last several years, aminoglycosides were shown to be substrates for a number of multidrug efflux pumps, including members of the five superfamilies of bacterial transporters. The resistance nodulation cell division (RND) transporter superfamily plays an important role in Gram-negative bacteria. The transporters of the RND superfamily use the membrane proton-motive force as energy source. They are localized in the cytoplasmic membrane and in Gram-negative bacteria. They interact with a membrane fusion protein, located in the periplasmic space, and an outer membrane protein to form a continuous tripartite channel able to export substrates directly out of the cell (Westbrock-Wadman et al. 1999; Livermore 2002). Several RND proteins were shown to be involved in intrinsic and/or acquired, proton motive force-dependent, aminoglycoside resistance in various Gram-negative pathogens, including P. aeruginosa, Burkholderia pseudomallei, Acinetobacter baumannii, and E. coli (Poole 2005). Active efflux has been evidenced for neomycin, kanamycin, and hygromycin A in E. coli.
Target modification
16S rRNA methylation
Many aminoglycoside producing organisms express rRNA methylases, which are capable of modifying the 16S rRNA molecule at specific positions critical for the tight binding of the drug (Maravic 2004). A number of genes encoding such enzymes have been identified from several aminoglycoside producers. The corresponding rRNA methyltransferases form the aminoglycoside resistance family of methyltransferases. Kanamycin A and B are obtained from Streptomyces tenjimariensis and Streptomyces tenebrarius, respectively. They catalyze the modification of A1408 at the N1 position and confer high-level resistance to kanamycin, tobramycin, sisomicin, and apramycin, but not gentamicin. Gentamicin A is obtained from the gentamicin producer Micromonospora purpurea and kasugamycin is obtained from S. tenebrarius. They catalyze the modification of G1405 at the N7 position and conferring high-level resistance only to the 4,6-disubstituted deoxystreptamines including gentamicin (Doi et al. 2004). Methylation of these nucleotides presumably abolishes the intermolecular contacts that they make with the drug. There are also reports available on genes encoding a 16S rRNA methyltransferase in 2003 and 2004, which described the characterization of methyltransferase genes in clinical isolates of human Gram-negative pathogens. The rmtA and rmtB genes were found in clinical isolates of P. aeruginosa and Serratia marcescens, respectively. These strains were found in Japan, where arbekacin has been used extensively since 1990. The two genes share 82% sequence identity and the encoded Rmt enzymes confer high-level resistance [minimal inhibitory concentrations (MICs) >1024 μg/ml] to almost all clinically useful aminoglycosides including arbekacin (Galimand et al. 2003). The considerable primary sequence similarity was observed between the Rmt proteins and the 16S rRNA methylases of Actinomycetes. They show the high G + C content of the gene (55%). This suggests a possible gene transfer from the producing organisms to Gram-negative pathogens. Another 16S rRNA methylase was characterized from Klebsiella pneumoniae. The structural gene, armA, was located on plasmid containing several other resistant genes including those conferring resistance to beta-lactams, trimethoprim, sulfonamides, and other aminoglycoside resistance determinants (Galimand et al. 2005).
Ribosomal mutations
Aminoglycoside resistance can also occur by mutation of the ribosomal target. It is clinically relevant only for streptomycin in M. tuberculosis. Mycobacterium is the only genus of eubacteria with species that contain a single copy of the ribosomal operon. This implies that a single mutation can lead to the production of a homogeneous population of mutant ribosomes and, thus, can result in resistance (Meier et al. 1994). The mutations in the rrs gene, encoding the 16S rRNA and associated with streptomycin resistance in M. tuberculosis, affect two highly conserved regions. These are the 530 loop and the nucleotide 912, resulting in a decrease in affinity for streptomycin. Mutations in genes encoding ribosomal proteins can also alter the activity of aminoglycosides. Notably, mutations in protein S12 are the other major cause of streptomycin resistance in M. tuberculosis (Yamane et al. 2005).
Enzymatic modification
Enzymatic modification is one of the most important mechanisms of aminoglycoside resistance (Hayashi et al. 1997; Hotta et al. 1996; Shaw et al. 1993), resulting in a loss of antibacterial activity due to a diminished affinity for the ribosomal A-site target (Llano-Sotelo et al. 2002). There are three classes of these enzymes: aminoglycoside acetyltransferases, aminoglycoside nucleotidyltransferases, and aminoglycoside phosphotransferases (Fig. 3).

Aminoglycoside-modifying enzymes and their substrates. A amikacin, Dbk dibekacin, G gentamicin, Gmb gentamicin B, I isepamicin, K kanamycin A, N netilmicin, S sisomicin, T tobramycin
Aminoglycoside acetyltransferases
AACs catalyze the regioselective acetylation of one of the four amino groups of aminoglycoside antibiotic. Acetylation reduces the affinity of these compounds for the acceptor tRNA site on the 30S ribosome by four orders of magnitude. The acetylation of aminoglycosides occurs after the random binding of both acetyl-CoA and amino group of aminoglycosides to the enzyme and was proposed to proceed via a direct nucleophilic attack by the amine on the thioester (Levings et al. 2005). The AACs are classified based on their regiospecificity of acetyl transfer on the aminoglycoside structure. For example, the AAC(6’) N-acetylate aminoglycoside on the amine group that is frequently found on position 6’ of the aminohexose linked to position 4 of the central 2-deoxystreptamine ring, while AAC(3) N-acetylate linked to position 3 of the 2-deoxystreptamine ring (Wright and Serpersu 2004). The first to be identified was the 178 amino acid AAC(2’)-Ia from Providencia stuartii. The AAC(3) family of aminoglycoside acetyltransferases is one of the largest. It includes four major types, I–IV, based on the pattern of aminoglycoside resistance that they confer (Sunada et al. 1999; Draker and Wright 2004). The first aminoglycoside-modifying enzyme to be purified to homogeneity was the E. coli R-plasmid-encoded gentamicin acetyltransferase. This allowed for the first studies of the substrate specificity of these enzymes. The bifunctional AAC(6’)-Ie APH(2")-Ia enzyme (Hegde et al. 2001) confers broad spectrum and high-level aminoglycoside resistance in enterococci and staphylococci. It differs from the two AAC(6’) described above in its genetic localization and catalytic mechanism. The structural gene of the enzyme is generally found on transposable elements and frequently carried on R plasmids (Vetting et al. 2004). These mobile supports account for the intergenus transfer of the resistant determinant, originally isolated from Enterococcus faecalis. The enzyme is monomeric in solution and the acetyltransferase activity exhibits exceptionally broad substrate specificity for aminoglycosides including fortimcin A and aminoglycosides possessing a hydroxyl group at the 6’-position. Three-dimensional structures for four members of the class have been reported (Hon et al. 1997; Burk et al. 2004). These show structural homology to the GCN5 superfamily of acyltransferases (Vetting et al. 2005). There are no common active site catalytic residues among all AACs. Analysis of the active site region where aminoglycosides bind, though, reveals a highly negatively charged surface that serves as a docking platform for these basic antibiotics.
Aminoglycoside nucleotidyltransferases
The ANTs represent the smallest class of aminoglycoside-inactivating enzymes. The clinically important aminoglycosides, such as gentamicin and tobramycin, are both modified by ANT(2"). The gene encoding this enzyme is widely distributed among pathogenic bacteria and its local prevalence is clearly selected by aminoglycoside usage in different clinical environments. There are 10 ANTs identified to date. These are of both chromosomally encoded and plasmid-encoded enzymes (Boehr et al. 2004). The ant(2") and ant(3") genes encoding adenylyltransferases are often identified on mobile genetic elements in resistant Gram-negative organisms. The ant(4’), ant(6), and ant(9) genes are also found on plasmids or integrated into transposons in Gram-positive organisms. These enzymes catalyze the reaction between Mg–ATP and aminoglycoside to form the O-adenylylated aminoglycoside and the magnesium chelate of inorganic pyrophosphate. Enzymes that regioselectively adenylylate the 6 and 3" positions of the streptomycin and the 9 and 3" positions of the spectinomycin have been identified. The reactions catalyzed by the ANT(2") and ANT(4’) are most significant and have been the most thoroughly mechanistically and structurally studied (Gates and Northrop 1988). The two investigators found the kinetic mechanism to be sequential and an ordered mechanism of substrate binding, with nucleotide (ATP) binding before aminoglycoside. The structure of aminoglycoside substrates bound to the enzyme has been characterized by NMR methods, although a 3-D structure of the entire enzyme remains elusive. The 3-D structure of only one ANT has been reported, that of ANT(4’) from S. aureus (Pedersen et al. 1995). The enzyme functions as a dimer, with the active site at the interface and with both monomers contributing residues to stabilize the substrates. The positioning of the substrates supports independent mechanistic evidence for direct attack of the nucleophilic hydroxyl on the α-phosphate of ATP.
Aminoglycoside phosphotransferases
Aminoglycoside kinases are known as aminoglycoside phosphotransferases. These are widely distributed among bacterial pathogens. Phosphorylation of the antibiotics results in a dramatic effect on their ability to bind to their target on the A-site of the ribosome. The genes encoding these enzymes are frequently found on multidrug resistance R plasmids, transposons, and integrons. APHs are classified based on their regiospecificity of phosphoryl transfer, their substrate specificity, and the specific gene sequence in question. APHs catalyze the regiospecific transfer of the γ-phosphoryl group of ATP to one of the hydroxyl substituents present on the aminoglycoside. They include a large number of aminoglycoside-modifying enzymes and are most relevant to clinical resistance to aminoglycosides in Gram-positive organisms (McKay et al. 1996). The APH (3’) family is especially ubiquitous and has been widely used as resistance marker in molecular biology research (example, the neo cassette). The best-studied APH is APH (3’)-IIIa, which has both 3’ and 5"-regiospecific phosphoryl transfer capacities. The enzyme is primarily found in Gram-positive cocci such as staphylococci and enterococci, and confers resistance to a broad range of aminoglycosides but not to gentamicin or tobramycin (McKay et al. 1996). Both antibiotics lack the critical 3’-hydroxyl groups that accept the phosphate group donated by ATP. The kinetic analysis demonstrated that the enzyme requires formation of a ternary complex with ATP and the aminoglycoside substrate and like all other kinases, divalent cations are essential for enzyme activity. The steady-state kinetic mechanism was shown to be sequential on the basis of the intersecting pattern of the lines observed in the reciprocal plot (McKay and Wright 1995; Roestamadji et al. 1995). The 3-D structure of the enzyme revealed a remarkable similarity with Ser, Thr, and Tyr protein kinases, which was not evident from the primary amino acid sequence (Nurizzo et al. 2004). Other aminoglycoside kinases include the spectinomycin-modifying enzyme APH(9) and APH(3") (StrA) and APH(6) (StrB), both of which modify streptomycin. The bifunctional enzyme, AAC(6’)–APH(2") is widely distributed among pathogenic bacteria and confers high level resistance to virtually all aminoglycosides except streptomycin and spectinomycin. The AAC(6’) domain of this bifunctional enzyme has overlapping aminoglycoside modification capacity with APH(2") domain, and aminoglycosides can be doubly modified (Boehr et al. 2005). As a result, this enzyme has shown very high MICs.
Implications for drug development
The rise of antibiotic resistance is a public health concern that has led to increased interest in studying the ways in which bacteria avoid the effects of antibiotics. Enzymatic inactivation by several families of enzymes has been observed to be the predominant mechanism of resistance to aminoglycoside antibiotics. Recently, a few reports have become available on the 3-D atomic structure of the aminoglycoside-modifying enzymes, such as kanamycin phosphotransferase and kanamycin nucleotidyltransferase. Relatively little information is known about their exact biochemical mechanism from their 3-D structures. The biochemical mechanisms of resistance and the substrate specificity and catalytic efficiency of these enzymes need to be investigated. The challenge is to determine the 3-D structures of three classes of modifying enzymes by X-ray crystallography and to understand the molecular basis for aminoglycoside resistance modification from their 3-D structures. This information could lead to the development of effective and potent inhibitors that will reverse antibiotic resistance. To date, not a single compound has been tested clinically that has reversed aminoglycoside resistance. Time has come to rethink about counterresistance. There are less arguments against the need for new aminoglycosides and strategies to design novel aminoglycoside-modifying enzyme inhibitors to avoid the emergence and dissemination of resistant bacteria. Recently, we mentioned a few strategies that can circumvent antibiotic resistance (Jana and Deb 2005). Current research holds out the promise that effective inhibitors of aminoglycoside-modifying enzymes may eventually restore the usefulness of aminoglycoside antibiotics. With the synthesis of inactivating enzyme-resistant analogs and the introduction of newer, less toxic antimicrobial agents, aminoglycosides continue to serve a useful role in the treatment of serious enterococcal and Gram-negative bacterial infections.
References
Ahmed AM, Shimamoto T (2004) A plasmid-encoded class 1 integron carrying sat, a putative phosphoserine phosphatase gene and aadA2 from enterotoxigenic Escherichia coli O159 isolated in Japan. FEMS Microbiol Lett 235:243–248
Aires JR, Kohler T, Nikaido H, Plesiat P (1999) Involvement of an active efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob Agents Chemother 43:2624–2628
Azucena E, Mobashery S (2001) Aminoglycoside-modifying enzymes: mechanisms of catalytic processes and inhibition. Drug Resist Updat 4:106–117
Ban N, Nissen P, Hansen J, Moore PB, Steitz TA (2000) The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289:905–920
Boehr DD, Daigle D, Wright GD (2004) Domain–domain interactions in the aminoglycoside antibiotic resistance enzyme AAC(6’)-APH(2"). Biochemistry 43:9846–9855
Boehr DD, Farley AR, LaRonde FJ, Murdock TR, Wright GD, Cox JR (2005) Establishing the principles of recognition in the adenine-binding region of an aminoglycoside antibiotic kinase [APH(3’)-IIIa]. Biochemistry 44:12445–12553
Bryan LE, Kwan S (1983) Roles of ribosomal binding, membrane potential, and electron transport in bacterial uptake of streptomycin and gentamicin. Antimicrob Agents Chemother 23:835–845
Burk DL, Hon WC, Leung AK, Berghuis AM (2001) Structural analyses of nucleotide binding to an aminoglycoside phosphotransferase. Biochemistry 40:8756–8764
Burk DL, Ghuman N, Wybenga-Groot L-E, Berghuis AM (2004) X-ray structure of the AAC(6’)-Ii antibiotic resistance enzyme at 1.8 A resolution; examination of oligomeric arrangements in GNAT superfamily members. Protein Sci 12:426–437
Cate JH, Yusupov MM, Yusupova GZ, Earnest TE, Noller HF (1999) X-ray crystal structure of 70S ribosome functional complexes. Science 285:2095–2104
Cho J, Hamasaki K, Rando RR (1998) The binding site of a specific aminoglycoside binding RNA molecule. Biochemistry 37:4985–4992
Davies J, Wright GD (1997a) Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol 5:63–70
Davies J, Wright GD (1997b) Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol 5:234–240
Doi Y, Yokoyama K, Yamane K, Wachino J, Shibata N, Yagi T, Shibayama K, Kato H, Arakawa Y (2004) Plasmid-mediated 16S rRNA methylase in Serratia marcescens conferring high-level resistance to aminoglycosides. Antimicrob Agents Chemother 48:491–496
Draker KA, Wright GD (2004) Molecular mechanism of the enterococcal aminoglycoside 6’-N-acetyltransferase’: role of GNAT-conserved residues in the chemistry of antibiotic inactivation. Biochemistry 43:446–454
Dworkin RJ (1999) Aminoglycosides for the treatment of gram-negative infections: therapeutic use, resistance and future outlook. Drug Resist Updat 2:173–179
Fluit AC, Schmitz FJ (1999) Class 1 integrons, gene cassettes, mobility, and epidemiology. Eur J Clin Microbiol Infect Dis 18:761–770
Fourmy D, Recht MI, Blanchard SC, Puglisi JD (1996) Structure of the A-site of E. coli. 16 S rRNA complexed with an aminoglycoside antibiotic. Science 274:1367–1371
Fourmy D, Yoshizawa S, Puglisi JD (1998) Paromomycin binding induces a local conformational change in the A-site of 16 S rRNA. J Mol Biol 277:333–345
Galimand M, Courvalin P, Lambert T (2003) Plasmid-mediated high-level resistance to aminoglycosides in Enterobacteriaceae due to 16S rRNA methylation. Antimicrob Agents Chemother 47:2565–2571
Galimand M, Sabtcheva S, Courvalin P, Lambert T (2005) Worldwide disseminated armA aminoglycoside resistance methylase gene is borne by composite transposon Tn1548. Antimicrob Agents Chemother 49:2949–2953
Gates CA, Northrop DB (1988) Substrate specificities and structure-activity relationships for the nucleotidylation of antibiotics catalyzed by aminoglycoside nucleotidyltransferase 2"-I. Biochemistry 27:3820–3825
Giamerllou H (1986) Aminoglycosides plus beta-lactams against Gram-negative organisms. Am J Med 80:126–137
Hancock RE, Farmer SW, Li ZS, Poole K (1991) Interaction of aminoglycosides with the outer membranes and purified lipopolysaccharide and OmpF porin of E. coli. Antimicrob Agents Chemother 35:1309–1314
Hatch RA, Schiller NL (1998) Alginate lyase promotes diffusion of aminoglycosides through the extracellular polysaccharide of mucoid Pseudomonas aeruginosa. Antimicrob Agents Chemother 42:974–977
Hayashi SF, Norcia LJ, Seibel SB, Silvia AM (1997) Structure-activity relationships of hygromycin A and its analogs: protein synthesis inhibition activity in a cell free system. J Antibiot (Tokyo) 50:514–521
Hegde SS, Javid-Majd F, Blanchard JS (2001) Overexpression and mechanistic analysis of chromosomally encoded aminoglycoside 2’-Nacetyltransferase (AAC(2’)-Ic) from Mycobacterium tuberculosis. J Biol Chem 276:45876–45881
Hon WC, McKay GA, Thompson PR, Sweet RM, Yang DS, Wright GD, Berghuis AM (1997) Structure of an enzyme required for aminoglycoside antibiotic resistance reveals homology to eukaryotic protein kinases. Cell 89:887–895
Hotta K, Zhu CB, Ogata T, Sunada A, Ishikawa J, Mizuno S, Kondo S (1996) Enzymatic 2’-N-acetylation of arbekacin and antibiotic activity of its product. J Antibiot (Tokyo) 49:458–464
Jana S, Deb JK (2005) Molecular targets for design of novel inhibitors to circumvent aminoglycoside resistance. Curr Drug Targets 6(3):353–361
Jana S, Karan G, Deb JK (2005) Purification of Streptomycin adenylyltransferase from a recombinant Escherichia coli. Protein Expr Purif 40:86–90
Jiang L, Patel DJ (1998) Solution structure of the tobramycin-RNA aptamer complex. Nat Struct Biol 5:769–774
Kotra LP, Haddad J, Mobashery S (2000) Aminoglycoside: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob Agents Chemother 44:3249–3256
Levings RS, Partridge SR, Lightfoot D, Hall RM, Djordjevic SP (2005) New integron-associated gene cassette encoding a 3-N-aminoglycoside acetyltransferase. Antimicrob Agents Chemother 49:1238–1241
Livermore DM (2002) Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin Infect Dis 34:634–640
Llano-Sotelo B, Azucena EF, Kotra LP, Mobashery S, Chow CS (2002) Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem Biol 9:455–463
Magnet S, Blanchard JS (2005) Molecular insights into aminoglycoside action and resistance. Chem Rev 105:477–497
Maravic G (2004) Macrolide resistance based on the Erm-mediated rRNA methylation. Curr Drug Targets Infect Disord 4:193–202
Masuda N, Sakagawa E, Ohya S, Gotoh N, Tsujimoto H, Nishino T (2000) Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-OprM efflux pumps in Pseudomonas aeruginosa. Antimicrob Agents Chemother 44:3322–3327
McKay GA, Wright GD (1995) Kinetic mechanism of aminoglycoside phosphotransferase type IIIa: evidence for a Theorell–Chance mechanism. J Biol Chem 270:24686–24692
McKay GA, Roestamadji J, Mobashery S, Wright GD (1996) Recognition of aminoglycoside antibiotics by the enterococcal/staphylococcal aminoglycoside 3’-phosphotransferase type IIIa: role of substrate amino groups. Antimicrob Agents Chemother 40:2648–2650
Meier A, Kirschner P, Bange F-C, Vogel U, Bottger E-C (1994) Genetic alterations in streptomycin-resistant Mycobacterium tuberculosis: mapping of mutations conferring resistance. Antimicrob Agents Chemother 38:228–233
Melancon P, Tapprich WE, Brakier-gingras L (1992) Single-base mutations at positions 2661 of E. coli. 23 S rRNA increase efficiency of translational proofreading. J Bacteriol 174:7896–7901
Michael K, Wang H, Tor Y (1999) Enhanced RNA binding of dimerized aminoglycosides. Bioorg Med Chem 7:1361–1371
Mingeot-Leclercq MP, Glupczynski Y, Tulkens PM (1999) Aminoglycosides: activity and resistance. Antimicrob Agents Chemother 43:727–737
Moazed D, Noller NF (1987) Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 327:389–394
Neu HC (1992) The crisis in antibiotic resistance. Science 257:1064–1073
Noller HF (1991) Ribosomal RNA and translation. Annu Rev Biochem 60:191–227
Nurizzo D, Shewry SC, Perlin MH, Brown SA, Dholakia JN, Fuchs RL, Deva T, Baker EN, Smith CA (2004) The crystal structure of aminoglycoside-3’-phosphotransferase-IIa, an enzyme responsible for antibiotic resistance. J Mol Biol 327:491–506
Pedersen LC, Benning MM, Holden HM (1995) Structural investigation of the antibiotic and ATP-binding sites in kanamycin nucleotidyl transferases. Biochemistry 34:13305–13311
Peloquin CA, Berning SE, Nitta AT, Simone PM, Goble M, Huitt GA, Iseman MD, Cook JL, Curran-Everett D (2004) Aminoglycoside toxicity: daily versus thrice-weekly dosing for treatment of mycobacterial diseases. Clin Infect Dis 38:1538–1544
Pezzella C, Ricci A, DiGiannatale E, Luzzi I, Carattoli A (2004) Tetracycline and streptomycin resistance genes, transposons, and plasmids in Salmonella enterica isolates from animals in Italy. Antimicrob Agents Chemother 48:903–908
Pilch DS, Kaul M, Barbieri CM (2005) Defining the basis for the specificity of aminoglycoside–rRNA recognition: a comparative study of drug binding to the A sites of Escherichia coli and human rRNA. J Mol Biol 346:119–134
Poole K (2005) Efflux-mediated antimicrobial resistance. J Antimicrob Chemother 56:20–51
Puglisi JD, Fourmy D, Recht ML (1998) Binding of neomycin-class aminoglycoside antibiotics to the A-site of 16 S rRNA. J Mol Biol 277:347–362
Recht MI, Fourmy D, Blanchard SC, Dahlquist KD, Puglisi JD (1996) RNA sequence determinants for aminoglycoside binding to an A-site rRNA model oligonucleotide. J Mol Biol 262:421–436
Ristuccia AM, Cunha BA (1982) The aminoglycosides. Drug Discov Today 66:303–312
Roestamadji J, Grapsas I, Mobashery S (1995) Loss of individual electrostatic interactions between aminoglycoside antibiotics and resistance enzymes as an effective means to overcoming bacterial drug resistance. J Am Chem Soc 117:11060–11069
Rougier F, Claude D, Maurin M, Maire P (2004) Aminoglycoside nephrotoxicity. Curr Drug Targets 4:153–162
Rybak LP, Whitworth CA (2005) Ototoxicity: therapeutic opportunities. Drug Discov Today 10:1313–1321
Sakon J, Liao HH, Kanikula AM, Benning MM, Rayment I, Holden HM (1993) Molecular structure of kanamycin nucleotidyl transferase determined to 3 Å resolution. Biochemistry 32:11977–11984
Shaw KJ, Rather PN, Hare RS, Miller GH (1993) Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev 57:138–163
Spelman DW, McDonald M, Spice WJ (1989) Aminoglycoside antibiotic agents: a review. Therapeutics 151:346–349
Sunada A, Nakajima M, Ikeda Y, Kondo S, Hotta K (1999) Enzymatic 1-N-acetylation of paromomycin by an actinomycete strain # 8 with multiple aminoglycoside resistance and paromomycin sensitivity. J Antibiot (Tokyo) 52:809–814
Vakulenko SB, Mobashery S (2003) Versatility of aminoglycosides and prospects for their future. Clin Microbiol Rev 16(3)430–450
Vetting MW, Magnet S, Nieves E, Roderick SL, Blanchard JS (2004) A bacterial acetyltransferase capable of regioselective N-acetylation of antibiotics and histones. Chem Biol 433:212–226
Vetting MW, Sde Carvalho LP, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS (2005) Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys 433:212–226
Walmsley M (2001) The structure and function of drug pumps. Trends Microbiol 9:71–79
Walsh C (2000) Molecular mechanisms that confer antibacterial drug resistance. Nature 406:775–781
Watanabe A, Nagai J, Adachi Y, Katsube T, Kitahara Y, Murakami T, Takanz M (2004) Targeted prevention of renal accumulation and toxicity of gentamicin by aminoglycoside binding receptor antagonists. J Control Release 95:423–433
Welch KT, Virga KG, Whittemore NA, Ozen C, Wright E, Brown CL, Lee RE, Serpersu EH (2005) Discovery of non-carbohydrate inhibitors of aminoglycoside-modifying enzymes. Bioorg Med Chem 13:6252–6363
Westbrock-Wadman S, Sherman D-R, Hickey M-J, Coulter SN, Zhu YQ, Warrener P, Nguyen LY, Shawar RM, Folger KR, Stover CK (1999) Characterization of a Pseudomonas aeruginosa efflux pump contributing to aminoglycoside impermeability. Antimicrob Agents Chemother 43:2975–2983
Wright GD (2005) Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv Drug Deliv Rev 57:1451–1470
Wright E, Serpersu EH (2004) Isolation of aminoglycoside nucleotidyltransferase(2")-Ia from inclusion bodies as active, monomeric enzyme. Protein Expr Purif 35:373–380
Wybenga-Groot LE, Draker K, Wright GD, Berghuis AM (1999) Crystal structure of an aminoglycoside 6’-N-acetyltransferase: defining the GCN5-related N-acetyltransferase superfamily fold. Structure Fold Des 7:497–507
Xi H, Arya DP (2005) Recognition of triple helical nucleic acids by aminoglycosides. Curr Med Chem Anti Canc Agents 5:327–338
Yamane K, Wachino J, Doi Y, Kurokawa H, Arakawa Y (2005) Global spread of multiple aminoglycoside resistance genes. Emerg Infect Dis 11:951–953
Yao S, Sgarbi PW, Marby KA, Rabuka D, O’Hare SM, Cheng ML, Bairi M, Hu C, Hwang S-B, Hwang C-K (2004) Glyco-optimization of aminoglycosides: new aminoglycosides as novel anti-infective agents. Bioorg Med Chem Lett 14:3733–3738
Zembower TR, Noskin GA, Postelnick MJ, Nguyen C, Peterson LR (1998) The utility of aminoglycosides in an era of emerging drug resistance. Int J Antimicrob Agents 10:95–105
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jana, S., Deb, J.K. Molecular understanding of aminoglycoside action and resistance. Appl Microbiol Biotechnol 70, 140–150 (2006). https://doi.org/10.1007/s00253-005-0279-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-005-0279-0