
Abstract
A thoroughly characterized breeding colony of 172 pedigreed rhesus macaques was used to analyze exon 2 of the polymorphic Mamu-DPB1, -DQA1, -DQB1, and -DRB loci. Most of the monkeys or their ancestors originated in India, though the panel also included animals from Burma and China, as well as some of unknown origin and mixed breeds. In these animals, mtDNA appears to correlate with the aforementioned geographic origin, and a large number of Mamu class II alleles were observed. The different Mamu-DPB1 alleles were largely shared between monkeys of different origin, whereas in humans particular alleles appear to be unique for ethnic populations. In contrast to Mamu-DPB1, the highly polymorphic -DQA1/DQB1 alleles form tightly linked pairs that appear to be about two-thirds population specific. For most of the DQA1/DQB1 pairs, Mamu-DRB region configurations present on the same chromosome have been ascertained, resulting in 41 different -DQ/DRB haplotypes. These distinct DQ/DRB haplotypes seem to be specific for monkeys of a determined origin. Thus, in evolutionary terms, the Mamu-DP, -DQ, and -DR regions show increasing instability with regard to allelic polymorphism, such as for -DP/DQ, or gene content and allelic polymorphism, such as for -DR, resulting in population-specific class II haplotypes. Furthermore, novel haplotypes are generated by recombination-like events. The results imply that mtDNA analysis in combination with Mhc typing is a helpful tool for selecting animals for biomedical experiments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Occupying habitats stretching from eastern Afghanistan and Pakistan in the west to the East China Sea in the east, the rhesus monkey has the widest geographic range of any non-human primate. The territories range from sea levels to >3,000 m and from semi-desert scrub to humid green forest (Melnick et al. 1993), and the monkeys vary widely in accordance with their extensive distribution (Fooden 2000). Despite this unusually large and ecologically heterogeneous range, however, rhesus macaques have been divided into very few morphological subspecies, and an unambiguous division of subspecies seems to be virtually impossible (Fooden 2000; Melnick et al. 1993). However, mtDNA analysis, has revealed the interpopulation and intraspecific diversity of the rhesus macaque species (Fooden 2000; Hayasaka et al. 1996; Melnick et al. 1993; Morales and Melnick 1998; Tosi et al. 2000). Phylogenetic trees based on mtDNA restriction enzyme analysis of rhesus macaques from Pakistan, India, China, and Burma reveal a major east-west division (Melnick et al. 1993), and indicate that Indian/Pakistan monkeys evolved away from the Chinese and Burmese animals. This division may have been caused by a glacial ice barrier in the Brahmaputra River valley during the Pleistocene. Comparable results have been reported from the sequence analyses of different fragments of mtDNA, whereas from Y-chromosome topology no or only slight evidence of paraphyly among rhesus macaques [Macaca mulatta (abbreviated as Mamu)] has been shown (Melnick et al. 1993; Tosi et al. 2000, 2002; Verschoor, personal communication). Based on the extreme philopatry of female rhesus macaques, the discrepancies between mtDNA and Y-chromosome topologies are possibly explained by either Y-chromosome introgression or mitochondrial differential lineage sorting (Tosi et al. 2000). It is also plausible that SRY gene mutations are rarely permitted, because this part of the Y chromosome is subject to differential selection forces and is therefore less variable than the mtDNA 12S region.
The rhesus macaque is the most commonly used non-human primate model in pre-clinical, immunological research on transplantation, as well as on chronic and infectious diseases. In humans, as in rhesus macaques, the major histocompatibility complex (MHC) gene products play an essential role in adaptive immunology. MHC class I and II molecules are cell surface glycoproteins that present peptides of intra- or extra-cellular origin to CD8+ and CD4+ T cells, respectively. The most striking feature of the vertebrate MHC complex is the extensive polymorphism of some of its genes, leading to an unprecedented variety of gene products. T cells recognize self- and foreign-antigens in the context of MHC class I and II molecules, and the T-cell repertoire is shaped by these peptide-MHC complexes. Differential peptide binding of distinct MHC molecules can lead to different T-cell activation and, as a consequence, particular MHC molecules/alleles are associated with susceptibility or resistance to infectious diseases (Balla-Jhagjhoorsingh et al. 1999; Bontrop 2001; Boyson et al. 1996; Evans et al. 1999; Furchner et al. 1999; Horton et al. 2001; Muhl et al. 2002; Reichstetter et al. 1999; Sauermann et al. 2000). On one hand, activation of T cells which recognize self-peptides especially in the context of MHC class II molecules seems to be a risk factor for autoimmune diseases (Brok et al. 2000; Nepom and Kwok 1998; Slierendregt et al. 1995a; t Hart et al. 2001). On the other hand, foreign allogeneic donor-peptides presented by host MHC class I and II molecules can lead to graft rejection or graft-versus-host reaction after transplantation (Balner and Van Rood 1971; Jonker et al. 1998; Reichstetter et al. 1999; Wood et al. 2001).
As in humans, the class II region of the rhesus macaque is divided into -DP, -DQ, and -DR. The classical class II molecules are transmembrane heterodimers, composed of an α- and β-chain subunit, encoded by A and B genes, respectively, most of which show an equal or even higher degree of polymorphism in rhesus monkeys than in humans. Most of the sequence variation is confined to exon 2 of Mamu-DPB1, -DQA1, -DQB1, and -DRB genes. So far, 16 Mamu-DPB1, 18 -DQA1, 40 -DQB1, and 128 -DRB alleles have been reported (IMGT/NHP sequence database; Robinson et al. 2003). In contrast to humans, the Mamu-DPA1 gene is invariant and -DPB1 only moderately variable (Slierendregt et al. 1995b), whereas the HLA-DPA1 gene is oligomorphic and -DPB1 highly polymorphic. Mamu-DQA1/DQB1 pairs are tightly linked, even more than in humans, giving rise to only a small number of different DQA1/DQB1 combinations (Doxiadis et al. 2001; Otting et al. 2002; Vigon and Sauermann 2002).
The most striking feature of the Mamu class II region, however, is its high level of -DRB region configuration polymorphism, displaying diversity with regard to number and combination of loci, but with only limited allelic polymorphism within a given configuration (Doxiadis et al. 2000; Khazand et al. 1999; Lobashevsky et al. 1999; Otting et al. 2000). In extended studies of pedigreed rhesus macaques, at least 31 Mamu-DRB region configurations could be described, coupled to only a few DQA1/DQB1 pairs and vice versa (Doxiadis et al. 2001; Khazand et al. 1999). In biomedical studies, intensive use is made of rhesus macaques having widespread origins. Hence, the need exists for this animal’s MHC to be well characterized for a comparison of differences in their polymorphisms, diversity, and haplotype composition. Accordingly, this study involving pedigreed rhesus macaques of different origins describes a comparative analysis of exon 2 of the polymorphic Mamu-DPB1, -DQA1, -DQB1, and -DRB class II loci.
Material and methods
Animals
In this study, 172 rhesus macaques from the self-sustaining colony at the Biomedical Primate Research Centre (BPRC) were analyzed. Most were born in captivity, but the founder animals came from different supplier centers and zoos. The origin of most of the rhesus macaques has been documented. All animals were serologically typed for their Mamu-A, -B, and -DR antigens.
DNA isolation and direct sequencing of DQA1, DQB1, and DPB1 exon 2
Genomic DNA was extracted from EDTA blood samples or from immortalized B lymphocytes by a standard salting-out procedure. Samples were subjected to polymerase chain reaction (PCR) in a 50-μl reaction mixture containing 500 ng DNA, 250 μM each dNTP, 1×PCR buffer II (Applied Biosystems, Roche, N.J., USA), 2.5 mM MgCl2, 1.25 units Taq polymerase (Applied Biosystems), and 0.4 μM of each primer. The following primers were used for amplifying exon 2:
- 5′DQA1(GH26 + M13):
-
TGTAAACGACGCCAGTGTGTAAACTTGTACCAG
- 3′DQA1 (GH27, BamHI):
-
CACGGATCCGGTAGCAGCGGTAGAGTTG
- 5′DQB1 (+ M13):
-
TGTAAACGACGGCCAGTTCCCCGCAGAGGATTTCGTG
- 3′DQB1(XbaI):
-
TGCTCTAGAGGGCGACGACGACGCCTCACCTC
- 5′DPB1(+ M13):
-
TGTAAACGACGGCCAGTGAGAGTGGCGCCTCCGCTCAT
- 3′DPB1 (XbaI):
-
CCCTCTAGAGCCCGGCCCAAAGCCCTCACTC.
The cycling parameters consisted of an initial denaturation step of 2 min, 94 °C, followed by three cycles of 90 s for each step at 94 °C, 56 °C (DQA1) or 60 °C (DQB1/DPB1), and 72 °C, then 32 cycles of 30 s for each step at the same temperatures. A final extension step was performed at 72 °C for 7 min.
The PCR products were purified using the QIAquick gel extraction kit (QIAGEN GmbH, Germany) according to the manufacturer’s recommendations.
The purified DNA was sequenced on the ABI 3100 genetic analyzer (Applied Biosystems, Foster City, Calif.) in accordance with the manufacturer’s guidelines, using 200–500 ng purified DNA, 0.2 μM primer, 1 μl BigDye terminator (Applied Biosystems), and 1×dilution buffer (400 mM Tris-HCl, 10 mM MgCl2) in a total of 10 μl. Sequences were analyzed using HETERO for heterozygote analysis based on sequence chromatograms and ALLELE for allele assignment (Rozemuller et al. 1993, 2001; Versluis et al. 1993).
Denaturing gradient gel electrophoresis (DGGE) of DRB exon 2 and sequencing
Separation of Mamu-DRB alleles with DGGE, dilution of DNA out of visualized bands, and subsequent direct sequencing of the alleles was performed as described earlier (Doxiadis et al. 2000; Knapp et al. 1997) with the sequencing primers 5′DRBseq and 3′DRBseq (Khazand et al. 1999). For cloning and sequencing purposes, exon 2 was amplified in a PCR reaction as described for DQA1/DQB1/DPB1, but in a final volume of 100 μl, with the following primers:
- 5′DRB (SalI):
-
CCGGTCGACTGTCCCCCCAGCACGTTTC
- 3′DRB (XbaI):
-
TCTAGATCACCTCGCCGCTGCACTGT.
Purification of PCR products, restriction enzyme digestion, cloning, and isolation of ssDNA were performed as described (Kenter et al. 1992). Cycle sequencing reactions were as summarized above. The data were analyzed using the program “Navigator” (Applied Biosystems, Roche, N.J.).
MtDNA analysis
Amplification of part of the mitochondrial 12S rRNA gene was performed essentially according to published methods (Kocher et al. 1989). Briefly, 200–500 ng of mtDNA, isolated from fresh EDTA blood, immortalized PBLs, or serum, was used as template in a final reaction volume of 50 μl, containing 1×PCR buffer + BSA (Applied Biosystems), 2 mM MgCl2, 200 μM each dNTP, 2 units AmpliTaq Gold (Applied Biosystems). The cycling reaction started with an activation step at 95 °C for 15 min, followed by 35 cycles at 95 °C for 20 s, 55 °C for 20 s, 72 °C for 40 s, and a final elongation step at 72 °C for 5 min.
The PCR products were purified and sequenced as described above for DQA1/DQB1/DPB1 and analyzed using the “Navigator” program.
Phylogenetic analysis
Multiple sequence alignments were created using the ClustalW Sequence Alignment program included in the MacVector 6.0 software package (Oxford Molecular). Phylogenetic analysis of the 12S rRNA nucleotide sequences was performed using PAUP version 4.0b10 (Swafford 2002). Pairwise distances were calculated using the Kimura-2 parameter, and the neighbor-joining method was used to create a phylogram. Confidence estimates of the groupings were calculated using the bootstrap method, generated from 1,000 replicates.
Results and discussion
Animal origin and mtDNA genotype
From a pedigreed rhesus macaque colony of approximately 900 animals, 172 individuals were selected. Indication of their origin or the origin of their ancestors was given by the respective import facilities. The founder animals showed morphological differences for pelage, body and head length and shape, and weight. However, as the intra-species morphological differences of rhesus macaques can be greater than the differences between animals of different origins (Fooden 2000), morphological criteria are not sufficient to determine the origin of rhesus macaques. Therefore, mtDNA sequences were analyzed from nearly all animals to obtain further information about their origin. For the mtDNA analysis, we chose a segment of the 12S ribosomal RNA coding sequence, which is generally accepted to show species-related variability (van der Kuyl et al. 1995, 2000). For those animals of uncertain origin, paternal and maternal ancestors, if available, were additionally subjected to phylogenetic analysis.
The resulting phylogenetic tree has three main branches; one represents monkeys from the western part of the macaque habitat, namely, Indian monkeys, while the second represents sequences of animals from the eastern region (Fig. 1). Whereas the 12S mtDNA of Indian-origin monkeys form a nearly uniform cluster, the second branch is divided into at least two clades: one of Chinese-origin monkeys and the other of Burmese-origin macaques. Both clades are more diverse than the Indian cluster. In addition, the third branch forms an outgroup of all animals analyzed (Fig. 1, K96 with children L99 and 9176, as well as L93, L95, and L65). Considering that branching distance in a mtDNA phylogenetic tree is related to geographic distances (Zhang and Shi 1993), these rhesus macaques seem to have originated a considerable distance away from the habitats of the other rhesus macaques included in this study. Since an SRY gene sequence analysis examined so far shows no differences at all (data not shown), molecular data, which could be informative concerning monkey origins, are restricted, as mtDNA is inherited only via the maternal lineage. Thus, conclusions about the origin of the respective rhesus macaques must be drawn with caution. For example, whether monkeys L93 and L95 are thoroughbred remains uncertain, because the father was not available for testing. Rhesus macaques BB10 and BB113, however, were proven to be Chinese/Indian mixed-breed monkeys in keeping with the Chinese origin of the father, 3945, although in the phylogenetic tree their mtDNA cluster together with Indian monkeys (Fig. 1). With these reservations in mind, we designated 110 animals included in this study to be of Indian origin, 24 of Burmese, and three of Chinese, in addition to 26 Chinese/Indian and four Burmese/Indian mixed-breeds and five mixed-breeds of Indian and unknown origin. Animals of non-Indian origin included in the study are listed in Table 1.

Phylogenetic tree of mtDNA sequence coding for the 12S rRNA of selected rhesus macaques of different origins. Geographic clusters are marked by colors
MhcMamu haplotypes
Serologically defined Mamu-A, -B, -DR haplotypes
The 900 BPRC-bred rhesus macaques were pedigreed and 253 major histocompatibility (MHC) haplotypes could be defined based upon the combination of different Mamu-A, -B, and -DR serotypes. Since most of the animals originated in India, 231 of these MHC haplotypes were specific for Indian rhesus macaques. An additional 22 unique Mamu-A, -B, and -DR haplotypes were observed only in monkeys of Chinese origin. Nearly all the serologically defined MHC haplotypes seem to be origin specific. For rhesus macaques originating in Myanmar (Burma) and of unknown origin, serological typing was hampered because the typing sera used were generated by active immunization against the founder rhesus macaque population, which was mostly of Indian origin.
MhcMamu class II haplotypes defined by molecular methods
Haplotype definition was performed by segregation analysis, with one exception: one Mamu-DQ/DRB haplotype was defined by comparing the combination of alleles with those of a homozygous same-origin animal. In the case of haplotype determination of rhesus macaques from India and China, the molecular typing data provided additional information to the known, serologically defined haplotypes.
DP region
Mamu-DPA1 and -DPB1 genes encode the functional DP protein. Whereas the Mamu-DPA1 gene is monomorphic, the -DPB1 gene exhibits moderate allelic variation, with 16 alleles described so far (Slierendregt et al. 1995b; IMGT/NHP sequence database). From the selected panel, 161 rhesus macaques were analyzed for their Mamu-DPB1 alleles by oligotyping or direct sequencing (Doxiadis et al. 2001; Otting et al. 1998). Of the known 16 -DPB1 alleles, 11 could be detected in monkeys originating from India. Mamu-DPB1*05, -DBP1*09, and-DPB1*14 seem to be specific for Burmese monkeys, -DPB1*15 for Chinese, and -DPB1*01 and -DPB1*02 for Indian animals (Fig. 2). The frequency of the specific -DPB1 alleles varies in animals of Indian or Burmese origin, a phenomenon also observed for HLA-DPB1 allele frequencies in human populations (Fig. 2). By far the most frequent -DPB1 allele in Indian populations is -DPB1*10, whereas in Burmese rhesus macaques -DPB1*14 is nearly as frequent. However, most of the Mamu-DPB1 alleles are shared by animals of all different origins, which is in sharp contrast to humans, whose unique alleles are readily detected in novel ethnic populations. Moreover, these results confirm the low polymorphism and evolutionary stability of the DPB1 locus in rhesus macaques (Doxiadis et al. 2001; Hashiba 2000; Otting et al. 1998).

Frequencies of -DPB1 alleles in rhesus macaques of different origins. Origin-specific alleles are highlighted by the same colors as used in Fig. 1. N number of haplotypes; gf gene frequency; anumber of rhesus macaques was too small to calculate gene frequencies; n.d. not determined because of homozygosity or uncertainty of segregation
DQ region
To date, 20 Mamu-DQA1 and 40 -DQB1 alleles have been described (Otting et al. 2002; Vigon and Sauermann 2002; Robinson et al. 2003). In the present study, we analyzed approximately 150 animals by direct sequencing of exon 2 of their Mamu-DQA1 and -DQB1 alleles, respectively. Nineteen -DQA1 and 25 -DQB1 alleles could be detected and, in addition, one unreported -DQA1 and three -DQB1 alleles were discovered. Of those 28 Mamu-DQB1 alleles, 11 variants were observed in monkeys of both Indian as well as Burmese origin (Fig. 3). As observed for -DPB1, the -DQB1 allele frequencies differ in the rhesus macaque populations. DQB1*1801 is the most prominent allele in Indian rhesus macaques and the third most frequent in monkeys of Burmese origin, whereas for the Mamu-DQB1*0601 frequency the opposite is observed. Both alleles, however, are absent in our rhesus macaques from China. Although only a few Chinese monkeys and very few monkeys of unknown origin were analyzed, six origin-specific -DQB1 alleles could be detected in these populations. In contrast to Mamu-DPB1 alleles, which are mostly shared by animals of all different origins, more than half of the -DQB1 alleles, namely, 15 of 28 variants, are population specific (Fig. 3).

Frequencies of -DQB1 alleles in rhesus macaques of different origins. N number of haplotypes detected; gf gene frequency; anumber of rhesus macaques was too small to calculate gene frequencies; n.d. not determined because of homozygosity or uncertainty of segregation
For 27 of the 28 -DQB1 alleles observed in this study, the corresponding -DQA1 allele, present on the same haplotype, was determined, resulting in only 31 -DQA1/DQB1 combinations. This confirmed the extraordinarily strong linkage of Mamu-DQA1 with -DQB1 (Doxiadis et al. 2000) (Fig. 4). The population specificity observed for -DQB1 alleles was increased when the presence and frequency of DQA1/DQB1pairs were analyzed. More than two-thirds, namely 23, of the 31 DQA1/DQB1 combinations were seen in monkeys of one specific origin only. According to the frequency of Indian macaques within the panel, most of the origin-specific -DQ pairs were detected in rhesus macaques from India, but Chinese-, Burmese-, and unknown origin-specific combinations were present as well (Fig. 4). Members of the ancient lineage Mamu-DQB1*06 all form unique and mostly population-specific combinations with the -DQA1 lineage -DQA1*01. The same holds for the ancient -DQA1*05, which forms unique and mostly population-specific combinations with -DQB1 alleles, although these belong to different lineages.

Mamu-DQA1/DQB1 combinations in rhesus macaques of different origins. DQA1/DQB1 pairs specific for rhesus macaques of a certain origin are highlighted by the same bright colors as in Fig. 1 and Fig. 2. Mamu-DQA1/DQB1 pairs present in monkeys of various origins are differentiated by composed colors (In Indian, Bu Burma, Ch China, Un Unknown)
DQ/DR region
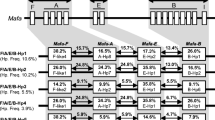
One hundred animals from the rhesus macaque panel were analyzed for their -DRB alleles by cloning and sequencing of exon 2 (Otting et al. 2000). The -DRB alleles of one additional animal were deduced by comparison with known alleles in DGGE (Doxiadis et al. 2001; Otting et al. 2000). Of the Mamu-DRB alleles known thus far (IMGT/NHP sequence database), 65 were detected and an additional four could be described. For 22 of the 31 different -DQ combinations (Fig. 4) the 32 accompanying -DRB configurations were determined, resulting in 41 distinct DQ/DRB haplotypes (Fig. 5). For monkeys from a certain geographic region, 29 of the 32 -DRB haplotypes are specific; however, all 41 of the DQ/DRB haplotypes are origin specific (Fig. 5). Most of the -DQ pairs are observed in combination with one or two different -DRB configurations. Only two DQA1/DQB1 pairs, DQA1*2601/DQB1*1801 and DQA1*2602/DQB1*1811, are seen with more than three -DRB region configurations. Twenty-three DQ/DRB haplotypes were detected in rhesus macaques originating in India, 12 in animals from Burma, three in those from China, and three in those of unknown origin. Some of these haplotypes are combinations of DQ pairs with unique, population-specific DRB region configurations. For example, the -DQA1*0503, -DQB1*2401 pair is linked to the -DRB region configuration -DRB3*0408, -DRB*W404 specifically in Chinese monkeys, whereas the same -DQ pair is coupled to the -DRB region configuration -DRB1*07031, -DRB4*0103, -DRBW*2505 in Burmese rhesus macaques (Fig. 5, nos. 40 and 41). Other origin-specific DQ/DRB combinations differ from those observed in monkeys from different habitats only by allelic -DRB variations (Fig. 5, nos.13 and 14). A third possibility for creating a variant DQ/DRB haplotype is by recombination of a given -DRB haplotype with different DQA1/DQB1 pairs (Fig. 6). Four -DRB region configurations are observed which combine with two different Mamu-DQ pairs. Only two -DRB haplotypes, Mamu-DRB1*0306, -DRB1*1003, and -DRB1*0406, -DRB5*0301, were observed in combination with more than two DQA1/DQB1 pairs.

Mamu-DQ/DRB haplotypes in rhesus macaques of different origins. DQ/DRB haplotypes are listed according to DQA1/DQB1 pairs. The origin of the rhesus macaques in which a certain DQ/DRB haplotype is detected is indicated by colors. aOnly -DRB6 alleles without the 63-bp deletion are summarized. bThe presence of DRB1*1003 on this haplotype has been proven only by segregation in one offspring of the family. Alleles mentioned in parentheses may also be present; + the presence of a specific allele of a certain lineage has not yet been confirmed (In India, Bu Burma, Ch China, Un unknown)

Identical Mamu-DRB region configurations in combination with different -DQA1/DQB1 pairs. Alleles in parentheses may also be present; + the presence of a specific allele of a known lineage has not yet been confirmed; ? the presence of a specific allele or lineage has not yet been confirmed (In India, Bu Burma, Ch China, Un unknown)
Class II haplotype stability
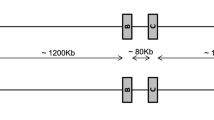
The polymorphic Mamu-DPB1 locus differs from its human counterpart in the degree and type of polymorphism (Bontrop et al. 1999; Doxiadis et al.2000, 2001; Hashiba 2000; Otting et al. 2000). The level of allelic variation with only 16 Mamu-DPB1 alleles described is much lower in rhesus macaques than in humans, whereas the situation is vice versa for variability indices. Therefore it is not surprising that rhesus macaques of origins as varied as India, China, and Burma show only small differences in the presence and frequency of -DPB1 alleles. Furthermore, the Mamu-DPA1 locus is described as being monomorphic, so the -DP region in rhesus macaques seems relatively stable. With the Mamu-DQ region, however, the situation is different. First, the amount of polymorphism for -DQA1, as well as -DQB1, is in rhesus macaques at least as high as in humans. Second, the number of Mamu-DQB1 alleles, and even more distinct, those of DQA1/DQB1 pairs that are specific for monkeys of a specific origin, is much higher than in the case of the -DPB1 locus, namely, two-thirds. This population specificity is increased when in addition the -DRB region is observed; nearly all of the -DRB haplotypes are specific for rhesus macaques of a certain origin. Unfortunateley, Mamu class II haplotype data of non-Indian rhesus macaques are restricted in this study because of the presence of mostly Indian monkeys within the BPRC’s breeding colony. As a result, only a limited number of haplotypes based on segregation studies have been deduced. However, analysis of exon 2 of -DRB from several Chinese monkeys by DGGE revealed, for most of the animals, unknown banding patterns. These preliminary data indicate that Mhc class II analysis of breeding colonies of non-Indian rhesus macaques will result in the definition of a large number of undescribed Mamu-DRB configurations and class II haplotypes. Furthermore, the combination of DQA1/DQB1 and -DRB alleles leads to 41 haplotypes, all of them population-specific. Recombination events not only between the DP and DQ/DR region, but also between DQ and DR seem to be one of the possibilities for the formation of a new haplotype (Fig. 6). Furthermore, the existence of the same -DRB alleles in two or more different DQ/DRB region configurations makes it plausible that new haplotypes were generated by unequal crossing-over events (Fig. 7).

Possible generation of new Mamu class II haplotypes. A question mark indicates that the presence of a specific allele has not yet been ascertained; + the presence of a certain allele of a known lineage has not yet been determined (In India, Bu Burma, Ch China, Un unknown)
Therefore, simple allotyping, such as for -DRB1 alone in humans, is not sufficient for rhesus macaques. A genetically well-characterized rhesus macaque colony will help to breed and define animals carefully for biomedical research in case of known susceptibility or resistance MHC alleles. Examples are given by the Mamu-A*01 allele, the gene product of which is associated with an improved control of a simian immunodeficiency virus infection, and the Mamu-A26 molecule that is highly protective against collagen-induced arthritis (CIA), the experimental form of rheumatoid arthritis (Allen et al. 2001; Mothe et al. 2003; Bakker et al. 1992). In addition, class II molecules are described in rhesus macaques as being involved in susceptibility to experimental autoimmune encephalomyelitis (EAE) in rhesus macaques, the experimental form of multiple sclerosis (MS) (Slierendregt et al. 1995a). The gene product of Mamu-DPB1*01, which was demonstrated to function as restriction element in myelin basic protein (MBP) peptide presentation, was in this study detected only in Indian monkeys. As well as in disease association studies, Mhc typing data are also crucial for transplantation studies. Furthermore, definition of the MHC class II haplotypes in rhesus macaques of different origins can help to sustain an outbred rhesus macaque colony, which will ensure by its diversity that the appropiate animals are available to solve future questions. Thus, armed with knowledge of the MHC, researchers can make an informed choice about which animals to use to elicit the most relevant answer to a specific biomedical question. The approach described will lead to a refinement of the studies and to the need for fewer animals in biomedical research.
References
Allen TM, Mothe BR, Sidney J, Jing P, Dzuris JL, Liebl ME, Vogel TU, O’Connor DH, Wang X, Wussow MC, Thomson JA, Altman JD, Watkins DI, Sette A (2001) CD8(+) lymphocytes from simian immunodeficiency virus-infected rhesus macaques recognize 14 different epitopes bound by the major histocompatibility complex class I molecule mamu-A*01: implications for vaccine design and testing. J Virol 75:738–749
Balla-Jhagjhoorsingh SS, Koopman G, Mooij P, Haaksma, TG, Teeuwsen VJ, Bontrop RE, Heeney JL (1999) Conserved CTL epitopes shared between HIV-infected human long-term survivors and chimpanzees. J Immunol 162: 2308–2314
Bakker NP, van Erck MG, Otting N, Lardy NM, Noort RC, ‘t Hart BA, Jonker M, Bontrop RE (1992) Resistance to collagen-induced arthritis in a nonhuman primate species maps to the major histocompatibility complex class I region. Exp Med 75:933–937
Balner H, Van Rood JJ (1971) Transplantation antigens in rhesus monkeys. Nature 232:121
Bontrop RE (2001) Non-human primates: essential partners in biomedical research. Immunol Rev 183:5–9
Bontrop RE, Otting N, de Groot NG, Doxiadis GG (1999) Major histocompatibility complex class II polymorphisms in primates. Immunol Rev 167:339–350
Boyson JE, Iwanaga KK, Golos TG, Watkins DI (1996) Identification of the rhesus monkey HLA-G ortholog. Mamu-G is a pseudogene. J Immunol 157:5428–5437
Brok HP, Uccelli A, Kerlero De Rosbo N, Bontrop RE, Roccatagliata L, de Groot NG, Capello E, Laman JD, Nicolay K, Mancardi GL, Ben-Nun A, Hart BA (2000) Myelin/oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis in common marmosets: the encephalitogenic T cell epitope pMOG24–36 is presented by a monomorphic MHC class II molecule. J Immunol 165:1093–1101
Doxiadis GG, Otting N, de Groot NG, Noort R, Bontrop RE (2000) Unprecedented polymorphism of Mhc-DRB region configurations in rhesus macaques. J Immunol 164:3193–3199
Doxiadis GG, Otting N, de Groot NG, Bontrop RE (2001) Differential evolutionary MHC class II strategies in humans and rhesus macaques: relevance for biomedical studies. Immunol Rev 183:76–85
Evans DT, Knapp LA, Jing P, Piekarczyk MS, Hinshaw V S, Watkins DI (1999) Three different MHC class I molecules bind the same CTL epitope of the influenza virus in a primate species with limited MHC class I diversity. J Immunol 162:3970–3977
Fooden J (2000) Systematic review of the rhesus macaque, Macaca mulatta (Zimmermann, 1780). Field Museum of Natural History, Chicago
Furchner M, Erickson AL, Allen T, Watkins DI, Sette A, Johnson PR, Walker CM (1999) The simian immunodeficiency virus envelope glycoprotein contains two epitopes presented by the Mamu-A*01 class I molecule. J Virol 73:8035–8039
Hashiba KMST (2000) Evolution of Catarrhini DPB1 exon 2 under intragenic recombination. In: Kasahara ME (ed) Major Histocompatibility Complex. Springer-Verlag, Tokyo, pp 386–397
Hayasaka K, Fujii K, Horai S (1996) Molecular phylogeny of macaques: implications of nucleotide sequences from an 896-base pair region of mitochondrial DNA. Mol Biol Evol 13:1044–1053
Horton H, Rehrauer W, Meek EC, Shultz MA, Piekarczyk, MS, Jing P, Carter DK, Steffen SR, Calore B, Urvater JA, Vogel TU, Wilson NA, Watkins DI (2001) A common rhesus macaque MHC class I molecule which binds a cytotoxic T-lymphocyte epitope in Nef of simian immunodeficiency virus. Immunogenetics 53:423–426
Jonker M, van de Hout Y, Neuhaus P, Ringers J, Kuhn EM, Bruijn JA, Noort R, Doxiadis G, Otting N, Bontrop RE, Claas FH, van Rood JJ (1998) Complete withdrawal of immunosuppression in kidney allograft recipients: a prospective study in rhesus monkeys. Transplantation 66:925–927
Kenter M, Otting N, Anholts J, Jonker M, Schipper R, Bontrop RE (1992) Mhc-DRB diversity of the chimpanzee (Pan troglodytes). Immunogenetics 37:1–11
Khazand M, Peiberg C, Nagy M, Sauermann U (1999) Mhc-DQ-DRB haplotype analysis in the rhesus macaque: evidence for a number of different haplotypes displaying a low allelic polymorphism. Tissue Antigens 54:615–624
Knapp LA, Cadavid LF, Eberle ME, Knechtle SJ, Bontrop RE, Watkins DI (1997) Identification of new Mamu-DRB alleles using DGGE and direct sequencing. Immunogenetics 45:171–179
Kocher TD, Thomas WK, Meyer A, Edwards SV, Paabo S, Villablanca FX, Wilson AC (1989) Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci USA 86: 6196–200
Lobashevsky A, Smith JP, Kasten-Jolly J, Horton H, Knapp L, Bontrop RE, Watkins D, Thomas J (1999) Identification of DRB alleles in rhesus monkeys using polymerase chain reaction-sequence-specific primers (PCR-SSP) amplification. Tissue Antigens 54:254–263
Melnick DJ, Hoelzer GA, Absher R, Ashley MV (1993) mtDNA diversity in rhesus monkeys reveals overestimates of divergence time and paraphyly with neighboring species. Mol Biol Evol 10:282–295
Morales JC, Melnick DJ (1998) Phylogenetic relationships of the macaques (Cercopithecidae: Macaca), as revealed by high resolution restriction site mapping of mitochondrial ribosomal genes. J Hum Evol 34:1–23
Mothe BR, Weinfurter J, Wang C, Rehrauer W, Wilson N, Allen TM, Allison DB, Watkins DI (2003) Expression of the major histocompatibility complex class I molecule Mamu-A*01 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol 77:2736–2740
Muhl T, Krawczak M, Ten Haaft P, Hunsmann G, Sauermann U (2002) MHC class I alleles influence set-point viral load and survival time in simian immunodeficiency virus-infected rhesus monkeys. J Immunol 169:3438–3446
Nepom GT, Kwok WW (1998) Molecular basis for HLA-DQ associations with IDDM. Diabetes 47:1177–1184
Otting N, Doxiadis GG, Versluis L, de Groot NG, Anholts J, Verduin W, Rozemuller E, Claas F, Tilanus MG, Bontrop RE (1998) Characterization and distribution of Mhc-DPB1 alleles in chimpanzee and rhesus macaque populations. Hum Immunol 59:656–664
Otting N, de Groot NG, Noort MC, Doxiadis GG, Bontrop RE (2000) Allelic diversity of Mhc-DRB alleles in rhesus macaques. Tissue Antigens 56:58–68
Otting N, de Groot NG, Doxiadis GG, Bontrop RE (2002) Extensive Mhc-DQB variation in humans and non-human primate species. Immunogenetics 54:230–239
Reichstetter S, Kwok WW, Kochik S, Koelle DM, Beaty JS, Nepom GT (1999) MHC-peptide ligand interactions establish a functional threshold for antigen-specific T-cell recognition. Hum Immunol 60:608–618
Robinson J, Waller MJ, Parham P, de Groot N, Bontrop R, Kennedy LJ, Stoehr P, Marsh SG (2003) IMGT/HLA and IMGT/MHC: sequence databases for the study of the major histocompatibility complex. Nucleic Acids Res 31:311–314
Rozemuller EH, Bouwens AG, Bast BE, Tilanus MG (1993) Assignment of HLA-DPB alleles by computerized matching based upon sequence data. Hum Immunol 37:207–212
Rozemuller EH, Kropveld A, Kreyveld E, Leppers FG, Scheidel KC, Slootweg PJ, Tilanus MG (2001) Sensitive detection of p53 mutation: analysis by direct sequencing and multisequence analysis. Cancer Detect Prev 25:109–116
Sauermann U, Stahl-Hennig C, Stolte N, Muhl T, Krawczak M, Spring M, Fuchs D, Kaup FJ, Hunsmann G, Sopper S (2000) Homozygosity for a conserved Mhc class II DQ-DRB haplotype is associated with rapid disease progression in simian immunodeficiency virus-infected macaques: results from a prospective study. J Infect Dis 182:716–724
Slierendregt BL, Hall M, t Hart B, Otting N, Anholts J, Verduin W, Claas F, Jonker M, Lanchbury JS, Bontrop RE (1995a) Identification of an Mhc-DPB1 allele involved in susceptibility to experimental autoimmune encephalomyelitis in rhesus macaques. Int Immunol 7:1671–1679
Slierendregt BL, Otting N, Kenter M, Bontrop RE (1995b) Allelic diversity at the Mhc-DP locus in rhesus macaques (Macaca mulatta). Immunogenetics 41:29–37
Swafford DL (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods), version 4. Sinauer, Sunderland, Massachusetts
t Hart BA, Brok HP, Amor S, Bontrop RE (2001) The major histocompatibility complex influences the ethiopathogenesis of MS-like disease in primates at multiple levels. Hum Immunol 62:1371–1381
Tosi AJ, Morales JC, Melnick DJ (2000) Comparison of Y chromosome and mtDNA phylogenies leads to unique inferences of macaque evolutionary history. Mol Phylogenet Evol 17:133–144
Tosi AJ, Morales JC, Melnick DJ (2002) Y-chromosome and mitochondrial markers in Macaca fascicularis indicate introgression with Indochinese M. mulatta and a biogeogrphic barrier in the Isthmus of Kra. Int J Primatol 23:161–178
van der Kuyl AC, Kuiken CL, Dekker JT, Goudsmit J (1995) Phylogeny of African monkeys based upon mitochondrial 12S rRNA sequences. J Mol Evol 40:173–180
van der Kuyl AC, van Gennep DR, Dekker JT, Goudsmit J (2000) Routine DNA analysis based on 12S rRNA gene sequencing as a tool in the management of captive primates. J Med Primatol 29:309–317
Versluis LF, Rozemuller E, Tonks S, Marsh SG, Bouwens AG, Bodmer JG, Tilanus MG (1993) High-resolution HLA-DPB typing based upon computerized analysis of data obtained by fluorescent sequencing of the amplified polymorphic exon 2. Hum Immunol 38:277–283
Vigon N, Sauermann U (2002) Sequence-based typing techniques for rhesus macaque MhcMamu-DQB1 allow the identification of more than 35 alleles. Tissue Antigens 59:88–94
Wood KJ, Jones ND, Bushell AR, Morris PJ (2001) Alloantigen-induced specific immunological unresponsiveness. Philos Trans R Soc Lond B Biol Sci 356:665–680
Zhang YP, Shi LM (1993) Phylogenetic relationships of macaques as inferred from restriction endonuclease analysis of mitochondrial DNA. Folia Primatol (Basel) 60:7–17
Acknowledgements
The authors wish to thank D. Devine for editing the manuscript and Henk van Westbroek for preparing the figures. The work was financed in part by the European Union project IMGT-QLG2-CT-2000-01287 and the NIH project 1-R24-RR16038-01 (CFDA no. 93.306).
Author information
Authors and Affiliations
Corresponding author
Additional information
The sequences reported in this paper have been deposited in the EMBL database (accession nos. AJ534296–AJ534304, AJ 564564, and AJ557455–AJ557511)
Rights and permissions
About this article
Cite this article
Doxiadis, G.G.M., Otting, N., de Groot, N.G. et al. Evolutionary stability of MHC class II haplotypes in diverse rhesus macaque populations. Immunogenetics 55, 540–551 (2003). https://doi.org/10.1007/s00251-003-0590-9
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-003-0590-9