
Abstract
Protegrin pore formation is believed to occur in a stepwise fashion that begins with a nonspecific peptide interaction with the negatively charged bacterial cell walls via hydrophobic and positively charged amphipathic surfaces. There are five known nature protegrins (PG1-PG5), and early studies of PG-1 (PDB ID:1PG1) shown that it could form antiparallel dimer in membrane mimicking environment which could be a first step for further oligomeric membrane pore formation. Later, we solved PG-2 (PDB ID:2MUH) and PG-3 (PDB ID:2MZ6) structures in the same environment and for PG-3 observed a strong dαα NOE effects between residues R18 and F12, V14, and V16. These “inconsistent” with monomer structure NOEs appears due to formation of an additional antiparallel β-sheet between two monomers. It was also suggested that there is a possible association of protegrins dimers to form octameric or decameric β-barrels in an oligomer state. In order to investigate a more detailed oligomerization process of protegrins, in the present article we report the monomer (PDB ID: 2NC7) and octamer pore structures of the protegrin-5 (PG-5) in the presence of DPC micelles studied by solution NMR spectroscopy. In contrast to PG-1, PG-2, and PG-3 studies, for PG-5 we observed not only dimer NOEs but also several additional NOEs between side chains, which allows us to calculate an octamer pore structure of PG-5 that was in good agreement with previous AFM and PMF data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Innate immune systems response on the pathogens was formed by evolution and habitation in different environments necessitates the production of defensive molecules which are important for survival. Some of these significant components of the innate immune systems of numerous animal species, where they act as a first line of defense against invading pathogens produced by the host cell hinder the growth of or kill the invading organisms, and they have been named “antimicrobial peptides (AMPs)” (Niu et al. 2015; Sharma et al. 2016).
Evolved resistance mechanism of pathogens is one of the major cause for development a new antibiotics for effective treatment of bacterial infections and results in hospital-acquired infections. AMPs could be potentially useful as antibiotics due to their properties such as broad-spectrum antibacterial activity, high selectivity, disruption of bacterial cell membranes, being more active against pathogens compared to host cells, and reduced possibility for the development of bacterial drug resistance (Kohanski et al. 2007; Peschel and Sahl 2006; Zasloff 2002). In contrast to most antibiotics, most AMPs display hydrophobic and positively charged amphipathic surfaces, which facilitates their binding to the negatively charged bacterial cell walls. AMPs seem to have a generalized mechanism of action targeted towards basic structural features of pathogens, so structural studies of AMPs are crucial for the investigation of their folding and function at high resolution and for their structure modification in order to increase their potency and selectivity (Ma et al. 2015). The most widely accepted mechanism of AMP action is their interaction with pathogenic membranes. AMPs contain at least two types of positively charged residues [arginine (Arg) and lysine (Lys)] that enable them to directly act on the cell wall and phospholipid membranes of microorganisms, which are negatively charged (Borkar et al. 2015). Finally, an accumulation of the AMPs on the membrane surface causes displacement of native Ca2+ and Mg2+ ions. AMPs also have the hydrophobic portion of the peptide, which interacts with hydrophobic components of the membrane and together with the positive charges is responsible for the disruption of the membrane, leading to bacterial death (Epand and Vogel 1999; Hancock 1997; Nguyen et al. 2011; Powers and Hancock 2003). There are differences in the composition of the membrane between prokaryotic and eukaryotic membranes. Eukaryotic membranes are mostly enriched in neutral lipids (phosphocholine, phosphatidylethanolamine, and sphingomyelin) and sterols (cholesterol and ergosterol) (Sharma et al. 2016; Yeaman and Yount 2003). Bacterial membranes are more electronegative, owing to the presence of lipids that impart a net-negative charge and to be believed that a non-specific, non-receptor-mediated interaction between AMPs and their target membranes is electrostatic in nature owing to their mutually opposite net charges, leading to attraction (Bessalle et al. 1990; Sharma et al. 2016; Wade et al. 1990).
After the initial interaction with membranes, the AMPs undergo self-association, multimerization, and peptide–peptide and/or peptide-lipid associations to form structures like Barrel Stave, Toroidal pore, and Carpet mechanisms (Panchal et al. 1996; Shai 1999; Matsuzaki et al. 1994). A very interesting class of AMPs is protegrins, which can form ion channels or pores upon interacting with target membranes, which cause the pathogen killing (Capone et al. 2010; Sokolov et al. 1999; Steinberg et al. 1997; Yang et al. 2000). Protegrins are small peptides about 16–18 amino acid residues in length, isolated from porcine leukocytes with a high content of positively charged arginine (Arg) and cysteine (Cys) residues, has a β-hairpin structure that is stabilized by disulfide bonds linking Cys-6 and Cys-15, and Cys-8 and Cys-13 (Capone et al. 2010; Langham et al. 2008). Protegrins have a broad spectrum of activity against both Gram-positive and Gram-negative bacteria, as well as the fungus Candida albicans, HIV-1 virus, and obtained a microbicidal activity against both log- and stationary-phase cultures of methicillin-resistant Staphylococcus aureus (MRSA) and Pseudomonas aeruginosa (Brogden 2005; Chen et al. 2000; Fahrner et al. 1996; Kokryakov et al. 1993; Mosca et al. 2000; Steinberg et al. 1997). There are five known naturally occurring porcine protegrins: PG-1 (RGGRL5CYCRR10RFCVC15VGR18), PG-2 (RGGRL5CYCRR10RFCIC15V16), PG-3 (RGGGL5CYCRR10RFCVC15VGR18), PG-4 (RGGRL5CYCRG10WICFC15VGR18), and PG-5 (RGGRL5CYCRP10RFCVC15VGR18), with a high content of cysteine (Cys) and several positively charged arginine (Arg) residues.
Protegrins studies have revealed a number of the key steps in their action: protegrin monomers dimerize in various types of lipid environment; protegrin peptides interact strongly with lipid bilayer membranes, particularly those that contain anionic lipids; and protegrins form pores in lipid bilayers, which results in uncontrolled ion transport and may be a key factor in bacterial death (Bolintineanu and Kaznessis 2011; Niu et al. 2015). Recent evidence has suggested that increased amounts of such cytotoxic peptides like amyloids (Aβ) are not only toxic to its host target cells but also possess antimicrobial activity due to the ability to form pores in lipid bilayers and biological membranes and have a possible antimicrobial effect, similary as PG-1 form amyloid-β peptide like fibrils (Jang et al. 2011; Usachev et al. 2014).
The most studied is the protegrin-1 (PG1) (PDB ID: 1PG1, 1ZY6) and supposed that it forms ion channels in rich zwitterionic membranes, octameric β-barrels, and tetrameric arcs with anionic lipid bilayer membranes, which mimic the inner membrane of Gram-negative bacteria (Langham et al. 2008; Mani et al. 2006). Roumestand et al. 1998 reported that PG-1 adopts a dimeric structure when it binds to dodecylphosphocholine micelles (DPC), which is frequently used as a model membrane mimetic (Usachev et al. 2015a, b) and suggested the possibility of an association between several dimers. Later, molecular dynamic simulation, atomic force microscopy (AFM), and electrical conductance studies showed that PG-1 forms lipid-dependent ion channels or pores similar to amyloid channels and other β-sheet pore formers, including bacterial toxins and other AMPs (Capone et al. 2010). However, even for PG-1 there are many experimental and theoretical studies, for another protegrins (PG-2, PG-3, PG-4, PG-5) structural studies are almost have not been investigated and biological functions among the different protegrins is also still limited. Oligomerization and pore formation processes of PG-2-PG-5 are also poorly investigated. In contrast to PG-1, for PG-2 (PDB ID:2MUH) in the presence of DPC micelles, no intramolecular NOEs were observed (Usachev et al. 2015a). Partially this may be due to the close values of the V14 and V16 CHα chemical shifts. For PG-3, we demonstrated that it forms an antiparallel NCCN dimer with a possible association of these dimers to form octameric or decameric β-barrels (Usachev et al. 2015b). In order to investigate other nature protegrins structure and for more detailed process of protegrins oligomerization and pore formation which was observed for PG-1 (Roumestand et al. 1998; Vivcharuk and Kaznessis 2010a) and PG-3 (Usachev et al. 2015b), in the present paper we used high-resolution NMR spectroscopy for structure determination of PG-5 (RGGRL5CYCRP10RFCVC15VGR18) in the presence of perdeuterated DPC micelles. Further research of other protegrins structure and their oligomerization into pores would unravel the mechanism of action of AMPs, which would be helpful in the development of pharmaceutically potent super antibiotics. A detailed structural and mechanistic knowledge of ion channel/pore forming peptides is required in order to tap the maximum use of these agents (Sharma et al. 2016).
Experimental
Materials
The synthesis and purification procedures of PG-5 peptide was done by Dr. Andrey Filippov (Chemistry of Interfaces Laboratory, Luleå University of Technology) and have been previously described (Usachev et al. 2015a). The purity of the peptide was estimated as better than 95 %. The sample was lyophilized and stored at a temperature of 193 K before use.
The NMR samples of PG-5 were prepared as previously described for PG-1 (30), PG-2 (Usachev et al. 2015a) and PG-3 (Usachev et al. 2015b) in DPC micelles. The peptide (4 mg) was solubilized in an aqueous solution (H2O or 2H2O, 500 μl) containing 20 mg perdeuterated DPC (molar ratio ~1:12). 3-(trimethylsilyl)-propionic-2,2,3,3-2H4 acid (TMSP-2,2,3,3-2H4) (98 % atom 2H, Aldrich) was added as an internal chemical shift standard for 1H NMR spectroscopy. Perdeuterated d38 DPC (98 % 2H) and TSP-d4 were purchased from Aldrich.
NMR spectroscopy and spatial structure calculations
NMR experiments were carried out at 293 K on Bruker Avance IIIHD 700-MHz spectrometer equipped with quadruple resonance (1H, 13C, 15N, and 31P) CryoProbe. The proton chemical shifts were referred to the TMSP-2,2,3,3-2H4.
Two-dimensional (2D) experiments (DQF-COSY, TOCSY, and NOESY) spectra were acquired in the phase-sensitive mode using the States-TPPI method and using a time-domain data size of 512 t 1 × 4096 t 2 complex points and 32 transients per complex t 1 increments. The water resonance was suppressed by “3-9-19” pulse sequence with gradients using flip-back pulse in COSY experiments (Piotto et al. 1992; Sklenar et al. 1993) and using excitation sculpting with gradients in TOCSY and NOESY experiments (Hwang and Shaka 1995). TOCSY spectra were obtained with a mixing time of 80 ms and NOESY spectra with a mixing time of 400 ms. Chemical shifts of PG-5 were deposited in the BioMagResBank database under the accession number 26009.
NMR spectra were processed by NMRPipe (Delaglio et al. 1995) and analyzed using SPARKY. Sequence-specific backbone resonance assignments and side-chain assignments for all residues were obtained using a combination of 2D COSY, TOCSY, and NOESY experiments. Inter-proton distances obtained from analysis of intensities of cross-peaks from NMR NOESY spectra were used as the primary data for the calculations by the molecular dynamics method (Efimov et al. 2016; Khodov et al. 2015, 2016). Following structural calculations, the ensemble of structures was subjected to restrained molecular dynamics using the Xplor-NIH (Schwieters et al. 2003; Ayupov and Akberova 2015). Individual structures were minimized, heated to 1000 K for 6000 steps, cooled in 100 K increments to 50 K, each with 3000 steps, and finally minimized with 1000 steps of the steepest descent, followed by 1000 steps of conjugate gradient minimization. A total of 1000 structures were calculated and 20 with minimal energy were chosen. None of the 20 structures had any violated nuclear Overhauser effect (NOE) distances. The atomic coordinates of final PG-5 monomer structure have been deposited in the Protein Data Bank (PDB code: 2NC7). To calculate the 3D structures of the PG-5 octamer pore the same protocol was used where the distance restraints were used as input in the standard distance geometry (DG)/simulated annealing (SA) refinement and energy minimization protocol (Schwieters et al. 2003). The program MolProbity (Chen et al. 2010; Davis et al. 2007) was used to assess the overall quality of the structures. The PG-5 peptide structures were visualized with CHIMERA (Pettersen et al. 2004).
Results and discussion
In this study, a 2D NMR 1H–1H COSY, 1H–1H TOCSY and 1H–1H NOESY methods of protein NMR spectroscopy approaches was used for sequential assignment of the backbone and sidechain resonances for antimicrobial peptide protegrin-5 in the presence of DPC micelles (detergent/protein ratio of ~1:12) as a membrane mimicking environment (Table 1). Final chemical shifts values were deposited in the BioMagResBank database (http://www.bmrb.wisc.edu) under accession number 26009.
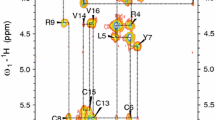
On the basis of sequential dNN(i,i+1), dαN(i,i+1), medium-range, and long-range NOEs (Fig. 1) and two disulfide bonds (Cys6-Cys13 and Cys8-Cys15) secondary structure of PG-5 peptide in DPC micelles was identified as a β-hairpin. These data are summarized in Table 2.

Fragments of the 2D NMR 1H–1H NOESY spectrum (mixing time 400 ms) acquired at 700 MHz with cryoprobe for the PG-5 in the presence of DPC micelles recorded at 293 K in H2O + D2O. a NH–CαH region, sequential NOE connectivity shown as dashed lines. b CαH–CαH region. Intermonomer V14CαH–V16CαH NOE signal marked by arrow. c Summary of the sequential and medium-range NOEs for the PG-5 in a solution of H2O + D2O with DPC micelles. The relative intensity of NOEs is represented by the thickness of the bars. When an unambiguous assignment was not possible due to peak overlap, the NOEs are drawn with gray shaded boxes. The two disulfide bonds are displayed in red
A total of 164 distance constraints derived from NOE experiment recorded in H2O and D2O were used for structure calculations by molecular dynamics method in the Xplor-NIH program (Schwieters et al. 2003) (Table 2). A total of 1000 structures were calculated and 20 with minimal energy were chosen. None of the 20 structures had any violated NOE distances. The final NMR ensemble of 20 structures has been deposited in the Protein Data Bank with the code 2NC7. Superimposed conformations of the minimized structures for the PG-5 in a solution of H2O + D2O with DPC micelles are shown in Fig. 2. The overall quality of the structures was assess by the program MolProbity (Chen et al. 2010; Davis et al. 2007). The Ramachandran analysis of the best 20 structures showed that all φ and ψ angles were localized to allowed (100 %) and favored (72.8 %) areas of the Ramachandran plot. The mean value of the overall backbone root-mean-squared deviation (RMSD) for backbone was 0.53 ± 0.21 Å and for heavy atoms 1.34 ± 0.27 Å.

Spatial structure of protegrin-5. a Overlay of the ensemble of 20 final energy-minimized XPLOR structures in stereo (PDB ID: 2NC7). The main and side chains are shown in black and light gray, respectively. b Ribbon diagrams of the lowest energy structure. Aliphatic amino acid residues are marked in red (Leu and Val), polar residue (Tyr) are marked in green, phenylalanine residue is marked in magenta, and positively charged residues (Arg) are marked in cyan. c Kyte–Doolittle hydrophobicity surface structural model for PG-5: from blue for the most hydrophilic, to white, to red for the most hydrophobic (Kyte and Doolittle 1982). An apolar cluster (Leu5, Phe12, Val14, and Val16) marked by a dotted line. The structures were drawn using Chimera [https://www.cgl.ucsf.edu/chimera/]
Analysis of PG-5 monomer structure shows that the side chains of Leu5, Phe12, Val14, and Val16 form an apolar cluster (Fig. 2c), the same as was previously shown for PG-2 and PG-3, and this area could bind to the bacterial cell walls via hydrophobic (Val, Phe, Leu residues) and positively charged (Arg residues) amphipathic surfaces (Aminova et al. 2013; Usachev et al. 2014, 2015a, b; Kolosova et al. 2016). As mentioned above, after the initial interaction with membranes, the AMPs undergo self-association, multimerization, and peptide–peptide and/or peptide-lipid associations to form structures like Barrel Stave, Toroidal pore, and Carpet mechanisms (Panchal et al. 1996; Shai 1999). Protegrin dimerization in the presence of DPC micelles was observed for PG-1 (Roumestand et al. 1998) and PG-3 (Usachev et al. 2015b) from 2D 1H–1H NOESY spectra, where there were observed several additional NOEs between side chains, which appear inconsistent with the β-sheet structure, and it was assumed that these NOEs appear due to formation of an additional antiparallel β-sheet between two monomers. In contrast to PG-2, such “inconsistent” NOEs were not observed due to only 16 residues in its length (no G17 and V18 residues) and close values of V14 and V16 CαH chemical shifts (Usachev et al. 2015a). Nevertheless, the dimeric structure by PG-2 in a lipid environment can take place due to its antimicrobial activity similarly as for other protegrins or for the human defensin HNP-3 (Hill et al. 1991). In the present paper, we also observed such “inconsistent” NOEs for PG-5 between V14CαH and V16CαH (Figs. 1b, 3b), G17CαH and C13NH, V16CγH and C13NH, R4CδH and Y7CδH, R4CδH and Y7CεH, R4CδH and C8NH, which are an indication that PG-5 also adopts an antiparallel dimer. Protegrin has been hypothesized to kill bacteria by permeabilizing their membranes due to the formation of ion channels in membranes. Several experimental and computational studies (Bolintineanu and Kaznessis 2011; Capone et al. 2010; Lazaridis et al. 2013) shown by potentials of mean force (PMF) calculations and atomic force microscopy (AFM) that protegrins could associate and insert into an anionic membrane forming an octameric or decameric β-barrels. According to PMF data (Lazaridis et al. 2013), octamers were more stable and exhibit a favorable binding energy to the pore so we used this model for our further calculations of pore structure for PG-5 based on the observed intermonomer NOEs (the final structure presented in Fig. 3). In contrast to the proposed structure of the octamer pore for PG-3 (Usachev et al. 2015b), in the present paper by molecular dynamic calculations based on the experimental NOEs we calculated a PG-5 octamer pore structure, which was is in good agreement with previous PMF calculations for PG-1 (Lazaridis et al. 2013).

Structure of the transmembrane pore of protegrin-5. a Top view. The pore is composed of eight identical protein chains. Lipid molecules are not shown. b Side-view of the pore. Intermonomer NOEs are shown in boxes. c Coulombic surface of the outer surface of the pore. The color gradient from red to blue represents negative (−10 kT) to positive electrostatic potentials (+10 kT). d Electrostatic potential in the lumen of the pore. The membrane region is represented in gray
Conclusions
Finally, we can summarize all available structural information for PG-1, PG-2, PG-3, and PG-5 to formulate a model of protegrin pore formation that is believed to occur in a stepwise fashion that begins with a nonspecific peptide interaction with the bacterial cell walls via hydrophobic and positively charged amphipathic surfaces (Fig. 4). Then the dimer formation by the protegrin occurs as the first step of further oligomerization. Dimerization is followed by oligomer formation and oligomers assemble into an octameric pore structure, which is responsible for the disruption of the membrane, leading to a bacterial death (Epand and Vogel 1999; Hancock 1997; Nguyen et al. 2011; Powers and Hancock 2003).

Model for pore formation by protegrin in biological membranes. Protegrin pore formation is believed to occur in a stepwise fashion that begins with a nonspecific peptide interaction with the bacterial cell walls via hydrophobic and positively charged amphipathic surfaces. Then the dimer formation occurs as the first step of further oligomerization. Dimerization is followed by oligomer formation and oligomers assemble into an octameric pore structure. Structural motifs that undergo membrane insertion are highlighted in grey
References
Aminova RM, Galiullina LF, Silkin NI, Ulmetov AR, Klochkov VV, Aganov AV (2013) Investigation of complex formation between hydroxyapatite and fragments of collagen by NMR spectroscopy and quantum-chemical modeling. J Mol Struct 1049:13–21
Ayupov RKh, Akberova NI (2015) Molecular dynamics of the pyridoxine derivative in the acetylcholinesterase active cavity. Res J Pharm Biol Chem Sci 6:1717–1722
Bessalle R, Kapitkovsky A, Gorea A, Shalit I, Fridkin M (1990) All-D-magainin—chirality, antimicrobial activity and proteolytic resistance. FEBS Lett 274:151–155
Bolintineanu DS, Kaznessis YN (2011) Computational studies of protegrin antimicrobial peptides: a review. Peptides 32:188–201
Borkar MR, Pissurlenkar RRS, Coutinho EC (2015) Mapping activity elements of protegrin antimicrobial peptides by HomoSAR. RSC Adv 5:78790–78798
Brogden KA (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3:238–250
Capone R, Mustata M, Jang H, Arce FT, Nussinov R, Lal R (2010) Antimicrobial protegrin-1 forms ion channels: molecular dynamic simulation, atomic force microscopy, and electrical conductance studies. Biophys J 98:2644–2652
Chen J, Falla TJ, Liu HJ, Hurst MA, Fujii CA, Mosca DA, Embree JR, Loury DJ, Radel PA, Chang CC, Gu L, Fiddes JC (2000) Development of protegrins for the treatment and prevention of oral mucositis: structure-activity relationships of synthetic protegrin analogues. Biopolymers 55:88–98
Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D 66:12–21
Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) Nmrpipe—a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Efimov SV, Khodov IA, Ratkova EL, Kiselev MG, Berger S, Klochkov VV (2016) Detailed NOESY/T-ROESY analysis as an effective method for eliminating spin diffusion from 2D NOE spectra of small flexible molecules. J Mol Struct 1104:63–69
Epand RM, Vogel HJ (1999) Diversity of antimicrobial peptides and their mechanisms of action. BBA Biomembr 1462:11–28
Fahrner RL, Dieckmann T, Harwig SSL, Lehrer RI, Eisenberg D, Feigon J (1996) Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem Biol 3:543–550
Hancock REW (1997) Peptide antibiotics. Lancet 349:418–422
Hill CP, Yee J, Selsted ME, Eisenberg D (1991) Crystal-structure of defensin Hnp-3, an amphiphilic dimer—mechanisms of membrane permeabilization. Science 251:1481–1485
Hwang TL, Shaka AJ (1995) Water suppression that works—excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J Magn Reson Ser A 112:275–279
Jang H, Arce FT, Mustata M, Ramachandran S, Capone R, Nussinov R, Lal R (2011) Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys J 100:1775–1783
Khodov IA, Alper GA, Mamardashvili GM, Mamardashvili NZ (2015) Hybrid multi-porphyrin supramolecular assemblies: synthesis and structure elucidation by 2D DOSY NMR studies. J Mol Struct 1099:174–180
Khodov IA, Efimov SV, Klochkov VV, de Carvalho LAEB, Kiselev MG (2016) The importance of suppressing spin diffusion effects in the accurate determination of the spatial structure of a flexible molecule by nuclear Overhauser effect spectroscopy. J Mol Struct 1106:373–381
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810
Kokryakov VN, Harwig SSL, Panyutich EA, Shevchenko AA, Aleshina GM, Shamova OV, Korneva HA, Lehrer RI (1993) Protegrins—leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett 327:231–236
Kolosova OA, Usachev KS, Aganov AV, Klochkov VV (2016) Antimicrobial peptide protegrins interact with DPC micelles by apolar hydrophobic cluster: structural studies by high-resolution NMR spectroscopy. Bionanosci. doi:10.1007/s12668-016-0218-9
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132
Langham AA, Khandelia H, Schuster B, Waring AJ, Lehrer RI, Kaznessis YN (2008) Correlation between simulated physicochemical properties and hemolycity of protegrin-like antimicrobial peptides: predicting experimental toxicity. Peptides 29:1085–1093
Lazaridis T, He Y, Prieto L (2013) Membrane interactions and pore formation by the antimicrobial peptide protegrin. Biophys J 104:633–642
Ma Z, Wei DD, Yan P, Zhu X, Shan AS, Bi ZP (2015) Characterization of cell selectivity, physiological stability and endotoxin neutralization capabilities of alpha-helix-based peptide amphiphiles. Biomaterials 52:517–530
Mani R, Tang M, Wu X, Buffy JJ, Waring AJ, Sherman MA, Hong M (2006) Membrane-bound dimer structure of a beta-hairpin antimicrobial peptide from rotational-echo double-resonance solid-state NMR. Biochemistry 45:8341–8349
Matsuzaki K, Murase O, Tokuda H, Funakoshi S, Fujii N, Miyajima K (1994) Orientational and aggregational states of magainin 2 in phospholipid bilayers. Biochemistry 33:3342–3349
Mosca DA, Hurst MA, So W, Viajar BS, Fujii CA, Falla TJ (2000) IB-367, a protegrin peptide with in vitro and in vivo activities against the microflora associated with oral mucositis. Antimicrob Agents Chemother 44(7):1803–1808
Nguyen LT, Haney EF, Vogel HJ (2011) The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol 29:464–472
Niu MF, Chai SM, You XY, Wang WH, Qin CL, Gong Q, Zhang TT, Wan P (2015) Expression of porcine protegrin-1 in Pichia pastoris and its anticancer activity in vitro. Exp Ther Med 9:1075–1079
Panchal RG, Cusack E, Cheley S, Bayley H (1996) Tumor protease-activated, pore-forming toxins from a combinatorial library. Nat Biotechnol 14:852–856
Peschel A, Sahl HG (2006) The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat Rev Microbiol 4:529–536
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Piotto M, Saudek V, Sklenar V (1992) Gradient-tailored excitation for single-quantum NMR-spectroscopy of aqueous-solutions. J Biomol NMR 2:661–665
Powers JPS, Hancock REW (2003) The relationship between peptide structure and antibacterial activity. Peptides 24:1681–1691
Roumestand C, Louis V, Aumelas A, Grassy G, Calas B, Chavanieu A (1998) Oligomerization of protegrin-1 in the presence of DPC micelles. A proton high-resolution NMR study. FEBS Lett 421:263–267
Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM (2003) The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160:65–73
Shai Y (1999) Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. BBA Biomembr 1462:55–70
Sharma S, Sahoo N, Bhunia A (2016) Antimicrobial peptides and their pore/ion channel properties in neutralization of pathogenic microbes. Curr Top Med Chem 16:46–53
Sklenar V, Piotto M, Leppik R, Saudek V (1993) Gradient-tailored water suppression for H-1-N-15 HSQC experiments optimized to retain full sensitivity. J Magn Reson Ser A 102:241–245
Sokolov Y, Mirzabekov T, Martin DW, Lehrer RI, Kagan BL (1999) Membrane channel formation by antimicrobial protegrins. BBA Biomembr 1420:23–29
Steinberg DA, Hurst MA, Fujii CA, Kung AHC, Ho JF, Cheng FC, Loury DJ, Fiddes JC (1997) Protegrin-1: a broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob Agents Chemother 41:1738–1742
Usachev KS, Filippov AV, Khairutdinov BI, Antzutkin ON, Klochkov VV (2014) NMR structure of the Arctic mutation of the Alzheimer’s A beta(1-40) peptide docked to SDS micelles. J Mol Struct 1076:518–523
Usachev KS, Efimov SV, Kolosova OA, Filippov AV, Klochkov VV (2015a) High-resolution NMR structure of the antimicrobial peptide protegrin-2 in the presence of DPC micelles. J Biomol NMR 61:227–234
Usachev KS, Efimov SV, Kolosova OA, Klochkova EA, Aganov AV, Klochkov VV (2015b) Antimicrobial peptide protegrin-3 adopt an antiparallel dimer in the presence of DPC micelles: a high-resolution NMR study. J Biomol NMR 62:71–79
Vivcharuk V, Kaznessis Y (2010a) Free energy profile of the interaction between a monomer or a dimer of protegrin-1 in a specific binding orientation and a model lipid bilayer. J Phys Chem B 114:2790–2797
Vivcharuk V, Kaznessis YN (2010b) Dimerization of protegrin-1 in different environments. Int J Mol Sci 11:3177–3194
Wade D, Boman A, Wahlin B, Drain CM, Andreu D, Boman HG, Merrifield RB (1990) All-D amino acid-containing channel-forming antibiotic peptides. Proc Natl Acad Sci USA 87:4761–4765
Yang L, Weiss TM, Lehrer RI, Huang HW (2000) Crystallization of antimicrobial pores in membranes: magainin and protegrin. Biophys J 79:2002–2009
Yeaman MR, Yount NY (2003) Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 55:27–55
Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415:389–395
Acknowledgments
We thank Dr. Andrey Filippov for the peptide synthesized. The work was performed according to the Russian Government Program of Competitive Growth of Kazan Federal University; by the subsidy allocated to Kazan Federal University for the project part of the state assignment in the sphere of scientific activities; by RFBR, according to the research project No. 16-34-60001 mol_a_dk.
Author information
Authors and Affiliations
Corresponding author
Additional information
Database: Structural data are available in the Protein Data Bank/BioMagResBank databases under the accession numbers 2nc7/26009.
Rights and permissions
About this article

Cite this article
Usachev, K.S., Kolosova, O.A., Klochkova, E.A. et al. Oligomerization of the antimicrobial peptide Protegrin-5 in a membrane-mimicking environment. Structural studies by high-resolution NMR spectroscopy. Eur Biophys J 46, 293–300 (2017). https://doi.org/10.1007/s00249-016-1167-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-016-1167-5