
Abstract
Microbial symbionts are increasingly recognized as playing a critical role in organismal health across a wide range of hosts. Amphibians are unique hosts in that their skin helps to regulate the exchange of water, ions, and gases, and it plays an active role in defense against pathogens through the synthesis of anti-microbial peptides. The microbiome of amphibian skin includes a diverse community of bacteria known to defend against pathogens, including the global pandemic lineage of Batrachochytrium dendrobatidis associated with mass amphibian die-offs. The relative influence of host phylogeny and environment in determining the composition of the amphibian skin microbiome remains poorly understood. We collected skin swabs from montane amphibians in Mexico and Guatemala, focusing on two genera of plethodontid salamanders and one genus of frogs. We used high throughput sequencing to characterize the skin bacterial microbiome and tested the impact of phylogeny and habitat on bacterial diversity. Our results show that phylogenetic history strongly influences the diversity and community structure of the total bacterial microbiome at higher taxonomic levels (between orders), but on lower scales (within genera and species), the effect of habitat predominates. These results add to a growing consensus that habitat exerts a strong effect on microbiome structure and composition, particularly at shallow phylogenetic scales.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Amphibian skin represents a unique environment to study symbiosis between vertebrate hosts and microbiota. Not only does it regulate exchange of gases, ions, and water in all amphibians [1] but it also plays a key role in host defense. The skin’s function is particularly important in terrestrial lungless salamanders (Plethodontidae), the most diverse group of salamanders representing 66% of all known species [2], as it is their sole means of gas exchange. Our understanding of the amphibian skin microbiome is advancing rapidly through the use of culture-free amplicon sequencing that can identify bacteria present at even low relative abundances. In particular, the community of bacteria found on the skin of amphibians is increasingly understood to play a critical role in defense against pathogens. Symbiotic skin bacteria may provide resistance to pathogens either by producing metabolites that directly impede pathogen growth, or by stimulating the host immune system [3]. Some bacteria have been shown to impede the growth of a pathogenic fungus, Batrachochytrium dendrobatidis (“Bd”) [4, 5] and increase survival of susceptible individuals [6].
Bd epizootics (in which infection with Bd leads to the frequently lethal disease, chytridiomycosis) have decimated populations of amphibians on multiple continents, resulting in hundreds of suspected extinctions [7]. Recently, a closely related chytrid fungus was discovered, Batrachochytrium salamandrivorans (“Bsal”) [8]. This pathogen has been shown to be lethal to some species of salamanders, but it has not yet been detected in Central or North American salamanders, which account for more than half the world’s salamander species [9]. Conservation strategies involving bioaugmentation or manipulation of naturally occurring skin bacteria against pathogens provide one of the most promising strategies to combat the multiple infectious disease threats that face amphibians [3].
Although amphibian skin microbiomes are strongly influenced by the immediate environment in which they are found [10], the uniqueness of amphibian skin compared to the surrounding habitat matrix appears to select for rare environmental bacteria [11], and most studies to date show that the skin microbiome represents a non-random assemblage of the bacteria found in the environment. Most studies have focused either on the microbiome of a single species of amphibian [10], or on communities of co-occurring species [11,12,13,14]. Major differences have been found between skin microbiomes of different species in community-based studies, but host species are often so distantly related (different families or orders) that it is hard to understand how strongly, and at what level, phylogenetic differences influence the microbiome. Several recent studies have examined multiple species within a genus, and have found that environmental factors or habitat seem to exert a stronger influence on microbiome than host phylogeny. In a diverse assemblage of frogs from across Madagascar that included multiple genera, Bletz et al. [15] found that host ecomorphology (related to habitat use) explained more variation in microbiome composition and richness than phylogeny or site-specific factors. In a study of the salamander genus Plethodon, Muletz-Wolz [16] found no significant effect of host species on microbiome structure within sites, but did detect differences in microbiome structure between geographic sites and along an environmental gradient within a single species. Similarly, Bird et al. [17] found that environmental factors rather than genetic distance between host clades primarily accounted for differences in microbiome composition between several clades of the polytypic salamander Ensatina eschscholtzii. In contrast, Prado-Irwin et al. [18] found that four populations of the plethodontid salamander Ensatina eschscholtzii xanthoptica, which were geographically separated and found in diverse habitats, nonetheless shared a core set of bacterial taxa, suggesting the relationship between host and microbiome may be conserved even at shallow phylogenetic scales. It remains to be seen if these findings can be generalized to other groups of amphibians, and at exactly what level of relatedness host-specific factors exert an influence on microbiome structure.
Here, we incorporate measures of phylogenetic difference across multiple taxonomic levels, sample sites, and environments to disentangle the influences of evolutionary history, geographic space, and environment on the species composition and community structure of the amphibian skin bacterial microbiome. We use both host genetic distance based on a mtDNA dataset, as well as distances derived from an analysis of a SNP dataset generated via high throughput sequencing, to study the degree to which phylogeny/population structure, habitat, and geographic separation impact the microbiome of two genera of plethodontid salamanders (Bolitoglossa, 3 spp.), Pseudoeurycea, 3 spp.) in Guatemala and Chiapas, Mexico. We compare these results to microbiomes of hylid frogs (Plectrohyla, 4 spp.) found in the same region, thus testing phylogenetic influences from the level of order to populations of the same species. Because amphibian declines are driven in part by the chytridiomycosis pandemic [19], we also explore the impact of Bd and Bsal presence on microbiome composition and structure. Our results show that phylogenetic history plays a strong role in influencing the diversity and community structure of the microbiome at higher taxonomic levels of hosts (frogs vs. salamanders), while habitat effects predominate at more shallow phylogenetic scales.
Materials and Methods
Sample Collection
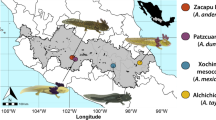
Our sampling strategy focused on obtaining individuals for comparisons at various levels of taxonomic scale, from different orders to different populations of the same species. We sampled in three main habitat types in western Guatemala and Chiapas, Mexico (with locality numbers in brackets; Fig. 1): pine-oak forest (San Juan Ixcoy [locality 5] and San Francisco El Retiro [4], Guatemala), cloud forest (La Cumbre [3] and San Rafael Pie de la Cuesta [9, 10], Guatemala; La Union Juárez [7] and Cerro Mozotal [1, 2], Chiapas), and fir forest (Todos Santos Cuchumatan [6] and Flores de Ixchiguan [8], Guatemala). Sample and locality information are given in Online Resource 1. Sites were classified for habitat qualitatively by the dominant tree type, and by the presence of abundant epiphytes and moss in cloud forest. Sample collection was conducted over the course of 2 weeks in October 2013. Each live animal was handled with a fresh pair of gloves. Animals were rinsed with 50 mL sterile water to remove transient bacteria [20, 21] and swabbed over the entire skin surface for 30 s. Swabs were transferred to sterile microcentrifuge tubes and stored at − 80 °C until processing. A second swab for Bd and Bsal detection was collected using standard protocols [22], air dried, and stored at 4 °C until processing.

Map of localities where amphibian skin microbiomes were sampled, with major mountain ranges indicated. Numbers refer to sampling localities given in Online Resource 1
Detection of Bd and Bsal
DNA was extracted using PrepMan Ultra (Thermo-Fisher Scientific, Waltham, MA, USA) [23]. Bd and Bsal infection intensities were quantified using quantitative polymerase chain reaction (qPCR) [24, 25] using an Applied Biosystems 7300 Real Time PCR system (Foster City, CA, USA). Samples were run singly, and separate reactions were used for Bd and Bsal detection quantification. The Bd standards used were created using a strain of the highly virulent global pandemic lineage (Bd-GPL1) collected in 2009 in the southern Sierra Nevada (CJB7). Samples were considered positive for Bd or Bsal if the output values were greater than 0 and amplification curves were sigmoidal. Zoospore equivalent (ZE) scores were calculated by multiplying the raw qPCR output by 80 to account for the subsampling and dilution that occurred during DNA extraction [26, 27].
Microbiome Sample Preparation and Bioinformatics
Skin microbial communities were analyzed using 16S amplicon sequencing, as described in SI Methods. Briefly, DNA was extracted from swabs using the PowerSoil DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA), and the hypervariable V3-V4 region of the bacterial 16S gene was amplified in two phases using region-specific (amplicon PCR) and dual-indexed (indexing PCR) primers, respectively. PCR product was purified, pooled, and sequenced on an Illumina MiSeq using a MiSeq Reagent Kit v3 (300 bp paired-end reads) (Illumina, Inc., San Diego, CA, USA). Base calling and demultiplexing were performed using MiSeq Reporter (Illumina, Inc.), and bioinformatic analyses were conducted using QIIME [28]. Paired-end reads were joined, and the resulting sequences were quality filtered at a threshold of Q20. Sequences were aligned using PyNAST [29] and the Greengenes core reference alignment version 13_8 [30]. Taxonomy was assigned using an open reference operational taxonomic unit (OTU) picking strategy [31] and the Greengenes Database version 13_8 [32,33,34]. Given the unexpected dominance of Chlamydiales in the skin microbiome of some individuals, taxonomic assignment of Chlamydiales sequences was confirmed using BLASTN 2.7.0 nr [35]. An approximately maximum-likelihood phylogenetic tree was generated using FastTree 2.1.3 [36]. We quantified species richness using Chao1 [37], species evenness defined as (Shannon entropy) / log2(number of observed OTUs), and phylogenetic diversity using Faith’s phylogenetic diversity (PD) [38]. Beta diversity was calculated using weighted and unweighted UniFrac distances [39] and visualized using principal coordinates analysis (PCoA) in Emperor [40].
Host Genetics
Genomic DNA was extracted using a guanidine thiocyanate extraction protocol for one individual per population for both Bolitoglossa and Pseudoeurycea. Fragments of the mitochondrial large subunit ribosomal RNA (16S) and cytochrome b (cytb) were amplified via PCR and sequenced (Voucher information and Genbank accession numbers given in Online Resource 2). TN93 genetic distances from concatenated 16S/cytb sequences were calculated using Mega v6 [41]. To generate the multilocus nuclear dataset, we used transcriptome sequencing for a limited representative subset of Bolitoglossa and Pseudoeurycea species (six individuals each genus) to design probes for exon capture. We enriched 1250 (Bolitoglossa) or 1246 (Pseudoeurycea) exonic markers using custom Agilent 1M capture arrays, and sequenced these markers on a single lane of an Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA, USA). Illumina sequence data were deposited in the NCBI Short Read Archive (Accession numbers in Online Resource 2). Exon capture data filtration, de novo assemblies, alignment, and variant identification followed an established bioinformatics pipeline described in Bi et al. [42]. Pairwise multilocus distance matrices for individuals of Bolitoglossa and Pseudoeurycea were calculated based on genotype likelihoods using ngsDist [43]. Details of PCR protocols, library preparation, and analysis of Illumina sequence data are given in Online Resource 3.
Climatic and Geographic Distance Matrix Calculation
In addition to our qualitative habitat classification, we used macroclimatic data and Euclidian distance between localities to investigate the relative impact of climate and spatial separation on microbiome differences between sites. Unique salamander occurrence records for the Maya geological block of Chiapas and western Guatemala were compiled from VertNet (www.vertnet.org), the literature, and uncatalogued specimens collected by the authors. Occurrence records were checked against known species distributions to identify incorrectly georeferenced points. We extracted data for each occurrence point, as well as sites where microbiome samples were collected, from the 19 bioclim variables from the WorldClim database [44] using QGIS [45]. A principal components analysis (PCA) was conducted using correlations in R [46]. PC1 and PC2 explained 89 and 8% of the variance in the climate data, respectively. Variables loading most strongly on PC1 were Bio12 (mean annual precipitation; negatively), and less strongly with Bio16 (precipitation of the wettest quarter; negatively). Thus, PC1 is primarily an index of precipitation, with smaller values indicating higher precipitation annually and during the wet season. PC2 is almost entirely an index of temperature seasonality (Bio4), with smaller values of PC2 indicating sites with greater temperature seasonality. Euclidian distances between salamander microbiome sites were calculated using the principal components in R [46].
Statistical Analysis
The relative abundances of particular bacterial taxa were compared using ANOVA. Alpha diversity was compared between metadata categories using ANOVA. Beta diversity was compared using PERMANOVA with 999 permutations, or with a t test with Bonferroni correction for multiple comparisons. We assessed and compared bacterial species composition using unweighted UniFrac distances, and community structure using weighted UniFrac distances. We compared distance matrices (unweighted and weighted UniFrac distances, phylogenetic distances, geographic distances, and climate distances) using Mantel or partial Mantel tests with 999 permutations. Results were generally comparable for Mantel tests using the mitochondrial or nuclear datasets; here, we report p values using the multilocus nuclear dataset. We described a core microbiome for salamanders and for frogs by identifying OTUs that were present in at least 90% of the samples from each group. Indicator species analyses were conducted using the “indicspecies” package in R [47]. Species-level OTU tables were filtered prior to indicator species analysis in order to reduce the chance that the indicator species identified were environmental microbes rather than resident bacteria. OTUs were removed if they were present in fewer than two samples or if there were fewer than 100 reads overall, following Kueneman et al. (2013). OTUs significantly impacted by Bd infection were determined using a Kruskal-Wallis test.
Results
Microbiome Summary Statistics
After assembly and quality filtering, a total of 2,058,236 microbial DNA sequences were included in the microbiome analysis, representing 18,848 OTUs. After rarefying at an even sampling depth of 7036 sequences per sample (the lowest number of sequences obtained from any individual sample), 506,592 sequences remained, representing 300 orders of bacteria and 3 orders of archaea. An average of 322 OTUs/sample were detected on Plectrohyla (n = 23); 524 OTUs/sample on Bolitoglossa (n = 19); 681 OTUs/sample on Pseudoeurycea (n = 30).
Effects of Phylogeny Across Host Taxonomic Scale
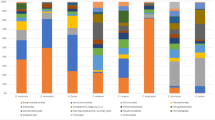
At the highest host taxonomic levels that we examined (order), host phylogeny had a strong impact on the composition and structure of total resident microbial communities (p = 0.001, PERMANOVA; Online Resource 4). Salamanders (order Caudata) hosted a more diverse skin microbiome compared with frogs (order Anura), with greater species richness (p = 0.008, Chao1, ANOVA), species evenness (p < 0.0001, ANOVA, equitability), and phylogenetic diversity (p = 0.0004, ANOVA, Faith’s phylogenetic diversity [PD]). Many of these differences were significant even when comparing only frogs and salamanders from the same habitat, cloud forest (frogs: n = 23, salamanders: n = 20; species composition: p = 0.001, PERMANOVA; community structure: p = 0.001, PERMANOVA; species richness: p = 0.138, Chao1, ANOVA, species evenness: p = 0.224, ANOVA, equitability, and phylogenetic diversity: p = 0.006, ANOVA, PD). Approximately half (11/23 individuals, 48%) of Plectrohyla hosted a comparatively simple skin microbiome composed primarily of Flavobacteriales (phylum: Bacteroidetes) and Actinomycetales (phylum: Actinobacteria) (Fig. 2). The relative abundance of these taxa was significantly lower in the skin microbiome of salamanders (Bolitoglossa, Pseudoeurycea; p < 0.0001, ANOVA). Twenty-one OTUs constituted the core microbiome of Plectrohyla, being present in at least 90% of frog samples (Table 1). This core microbiome dominated the microbial communities of most frog samples, with an average relative abundance of 74.9% (range 12.9–95.8%). The most abundant members of the core microbiome were Chryseobacterim (order: Flavobacteriales, average relative abundance 25.6%) and Dermacoccus (order: Actinomycetales, average relative abundance 15.4%).

Relative abundance of the 50 most abundant bacterial OTUs at the order level for 23 frogs and 49 salamanders captured in the wild, with the eight most abundant orders labeled. Each bar corresponds to a single individual, and bacterial orders are indicated by color. Species of amphibians are separated by white space between bars. Individual salamanders whose microbiome is dominated by Chlamydiales are marked by an asterisk
Members of the phylum Proteobacteria also dominated the skin microbiome of salamanders (30 Pseudoeurycea, 19 Bolitoglossa), and were significantly relatively less abundant among frogs (p = 0.02, ANOVA). The core microbiome of salamanders was composed of 15 OTUs, with an average relative abundance of 35.7% (range 3.8–73.2%) (Table 2). The most abundant members of the salamander core microbiome were Pseudomonas (order: Pseudomonadales, average relative abundance 10.8%), Methylobacterium (order: Rhizobiales, average relative abundance 6.9%), and family Enterobacteriaceae (order: Enterobacteriales, average relative abundance 6.6%). Three individual salamanders (one Bolitoglossa, two Pseudoeurycea) differed strikingly from the others sampled, with a microbiome dominated by Chlamydiales (phylum: Chlamydiae). Chlamydia accounted for 93 and 83% of the sequences derived from two individuals, while a third was dominated by Chlamydia (20%) and Candidatus Rhabdochlamydia (40%) (these three individuals are marked by asterisks in Fig. 2). At least one member of order Chlamydiales was detected in 45/50 salamanders sampled (90%), although the relative abundance was generally < 5% total sequences (median 0.3%). Sequence comparison using both Greengenes 13_8 [32] and BLASTN 2.7.0 nr [35] confirmed the taxonomic assignment of these Chlamydiales sequences.
Habitat and Climate Correlate with Differences in Species Composition at Finer Host Taxonomic Scales
At finer host taxonomic scales (below rank of order), habitat differences were associated with differences in species composition and community structure of the plethodontid salamander skin microbiome. Principal coordinates analysis using unweighted UniFrac distances yielded strong clustering by habitat type (pine-oak, fir, or cloud forest) among plethodontid salamanders, with a distinct set of microbial communities associated with the pine-oak habitat type (Fig. 3). This habitat clustering was significant in terms of species composition (p = 0.001, PERMANOVA) and community structure (p = 0.028, PERMANOVA). No differences between species or habitat type were apparent in a principal coordinates analysis of the weighted UniFrac distances (Fig. 3). Using indicator species analysis, we identified 34 operational taxonomic units (OTUs) strongly associated with the pine-oak habitat, while just 6 indicator OTUs were shared between pine-oak and another habitat type (Fig. 4; see Online Resource 7 for complete results). Meanwhile, fir and cloud forest had a higher number of overlapping indicator OTUs: 33 were shared, while just 15 and 12 were specific to fir and cloud forest, respectively.

Principal coordinates analysis (PCoA) using a weighted and b unweighted UniFrac distances between microbiome samples collected in three types of habitat. Habitat type (pine-oak, fir, and cloud forest) is indicated by shape of points and host species by point color

Indicator species analysis by habitat type shows that salamanders from pine-oak forest share many indicator species not found in fir and cloud forest, which are similar to each other. Left-hand vertical labels indicate indicator OTUs for each habitat type. Each row represents a unique bacterial OTU (species level classification), and each column represents an individual salamander. Color scale represents OTU abundance as a percent of rarefied sequencing reads
Phylogeny did continue to play a role within host genera and species, however. Pseudoeurycea and Bolitoglossa salamanders across habitats differed significantly at the host genus level in terms of their skin microbial species composition (p = 0.004, PERMANOVA) and community structure (p = 0.018, PERMANOVA), but not species richness (p = 0.518, Chao1, ANOVA), species evenness (p = 0.607, equitability, ANOVA), or phylogenetic diversity (p = 0.231, PD, ANOVA). At the species level, salamanders also differed significantly in terms of their skin microbial species composition (p = 0.001, PERMANOVA), but not community structure (p = 0.156 PERMANOVA), species richness (p = 0.514, Chao1, ANOVA), species evenness (p = 0.691, equitability, ANOVA), or phylogenetic diversity (p = 0.45, PD, ANOVA). Indicator species analysis demonstrates that 12 species-level OTUs were significantly associated with Bolitoglossa, while 34 OTUs were significantly associated with Pseudoeurycea (Fig. 5; see Online Resource 8 for complete results).

Indicator species analysis by salamander genus, showing differences in indicator OTUs between Bolitoglossa and Pseudoeurycea. Each row represents a unique OTU (species level classification), and each column represents an individual salamander. Color scale represents OTU abundance as a percent of rarefied sequencing reads. Left hand labels indicate the phylum of each indicator OTU
Mantel tests further substantiated the correlation between host genetic distance within each salamander genus and unweighted UniFrac distance (Pseudoeurycea: p = 0.001; Bolitoglossa: p = 0.038). However, the results of partial Mantel tests suggest that this correlation may be confounded with climate or geographic distance. For Pseudoeurycea, a composite climate distance derived from 19 bioclim variables (Online Resource 5) was also significantly correlated with unweighted UniFrac distance (p = 0.002, Mantel test), and for Bolitoglossa, unweighted UniFrac distance was significantly correlated with geographic distance between populations (p = 0.01, Mantel test). When climate and geographic distance were used as control matrices in partial Mantel tests for Pseudoeurycea and Bolitoglossa, respectively, the relationship between genetic distance and unweighted UniFrac distance was no longer significant (Pseudoeurycea: p = 0.287; Bolitoglossa: p = 0.714). The habitat types that we classified qualitatively based on dominant trees and epiphyte abundance are separated in climatic space (Online Resource 5), showing that differences in macroclimate between sites correspond to differences in habitat. Thus, we suggest that, at finer taxonomic scales among plethodontid salamanders, climate/habitat differences and spatial distance outweigh host-specific factors (measured by genetic distance) in determining the composition of the skin microbiome.
No Effect of Bd Infection in Hylid Frogs
Only 4/49 salamanders sampled tested positive for Bd, while 12/23 hylid frogs tested positive for Bd; therefore, we focused our analysis on hylids in order to increase statistical power. Almost all Bd infection levels were low to moderate, with a median of 18 ZE and a maximum of 1073.6 ZE; depending on a variety of factors, including host species and the Bd strain used in preparing qPCR standards, mortality often ensues at levels approximately an order of magnitude higher than the highest infection levels observed in our samples [27]. Infection with Bd was not associated with significant differences in species composition (p = 0.111, PERMANOVA), community structure (p = 0.15, PERMANOVA), species richness (0.656, Chao1, ANOVA), species evenness (p = 0.426, equitability, ANOVA), or phylogenetic diversity (0.6114, PD, ANOVA). However, differences in the relative abundance of certain taxa were significantly associated with Bd infection status (see Online Resource 6): these included Alicyclobacillus (phylum: Firmicutes), and Methylobacterium, Sphingomonas, Curvibacter, and an undescribed genus in family Caulobacteraceae (phylum: Proteobacteria) (all p < 0.05, Kruskal-Wallis test). All individuals included in the study tested negative for Bsal, the newly discovered chytrid fungus which is lethal to some salamanders [8, 48].
Discussion
Our results indicate that at higher phylogenetic levels (e.g., from host order to genus), the species composition of skin bacterial communities was significantly affected by phylogenetic history (indicated by differences within taxonomic ranks). While microbiome studies in non-model organisms have only begun to be published fairly recently, interspecific or multi-population microbiome studies would benefit from incorporation of some measure of phylogenetic relatedness [49]. Although crude, our distance-based analysis within genera allowed us to disentangle lineage-specific effects from environmental and/or spatial effects.
Phylogenetic history appears to play only a minor role in skin microbiome assemblage at shallower scales (within genera and species) in our study groups. Differences in microbiome composition between Pseudoeurycea and Bolitoglossa could be due to many factors, including differences in microhabitat preference and skin chemistry. Guatemalan Pseudoeurycea are exclusively terrestrial, found inside logs and under cover objects, while the Bolitoglossa species of the lincolni-franklini group included in this study are found in both terrestrial microhabitats and in arboreal bromeliads. Additionally, these Pseudoeurycea are all drab while the Bolitoglossa are brightly colored (red/yellow blotches on a black background); such differences could be accompanied by chemical differences between the skin of the two genera. Given our results showing that habitat appears to have a strong effect on the microbiome of salamanders, microhabitat differences between genera or species could account for some of the variation observed in the microbiome.
Several recent studies exploring the factors most strongly correlated with the structure of the amphibian skin microbiome have also concluded that site and habitat play an important role (e.g., [15, 17, 50]), while others have detected a core bacterial microbiome across habitats and geographically separated populations, suggesting a stable mutualism between host and skin microbiome in some cases [18]. Our study contributes to this increasingly complex body of research in supporting a major role for habitat in determining microbiome structure at relatively shallow phylogenetic scales. If phylogenetic signal in relevant aspects of skin chemistry or physiology is high (i.e., if closely related species are similar in these aspects), we may expect to see differences in microbiome composition only between more distantly related species within similar environments. Testing relatively closely related species that exhibit differences in skin chemistry but inhabit similar environments (or that are sympatric) would provide one avenue to test this hypothesis.
In the few other studies comparing salamanders and frogs, salamanders (Ambystomatidae, Salamandridae) had less diverse microbiomes [11,12,13]. Although our sampling of salamanders and frogs within communities was not exhaustive, our results show that plethodontid salamander microbiome is more diverse than that of hylid frogs found in similar habitat. The salamanders included in most other published microbiome studies (with the exception of Fitzpatrick and Allison [10] and Muletz-Wolz et al. [50]) spend part of their lives in water bodies and were sampled in ponds; this major difference in microhabitat (aquatic vs. terrestrial) could be partly responsible for the discrepancy in salamander vs. frog microbiome diversity. By contrast, Plectrohyla are primarily terrestrial, although they breed in and are typically found along streams. Thus, aquatic vs. terrestrial habitat use cannot explain the differences in microbial diversity seen between Plectrohyla and salamanders in this study. Since all Plectrohyla included in our study were collected in a cloud forest habitat (while plethodontids were sampled in a variety of habitats), we were unable to compare the relative effects of taxonomy and habitat on the microbiome of hylids and plethodontids; however, our comparisons between hylids and plethodontids sampled in the same habitat (PERMANOVA, Mantel tests) suggest that many of the significant differences we detected are due to taxonomy, rather than habitat. In future studies, an experimental design including comprehensive sampling of different clades with species found in multiple habitats would provide an ideal system to test the relative impact of phylogenetic history and habitat on microbiome composition and structure.
Fitzpatrick and Allison [10] found a strong correlation between the microbiome of salamanders and their immediate environment, suggesting that the habitat/climate influence on microbiome may extend to even smaller spatial scales. On the other hand, some studies of amphibian skin microbiomes have found that many bacteria common on amphibian skin are rare in environmental samples, suggesting a stronger role for the host in influencing skin microbiome composition [11, 14]. Because environmental microbiome samples were not included in the current study, the extent to which substrate modulates the composition of the microbiome cannot be assessed. The fact that distance between sites is correlated with unweighted UniFrac distance in Bolitoglossa suggests that site-specific factors may mediate the species pool available to colonize these salamanders. The inclusion of environmental samples from different microhabitats in future studies would allow for a more rigorous test of the influence of habitat and microhabitat on amphibian skin microbiome structure in this system. Although perhaps not feasible in this system, transplant experiments would also allow for a direct experimental test of this hypothesis.
Dominance of the microbiome of three individual salamanders by a bacterium of the order Chlamydiales is surprising, given that all known species of this order are obligate intracellular endosymbionts [51]. However, a recent study of the skin microbiome of Ensatina eschscholtzii xanthoptica, a plethodontid salamander in California, also found a small number of individuals with a high relative abundance of Chlamydiales [18]. It is not clear if the dominance of Chlamydia in these microbiomes is the result of an opportunistic increase in its relative abundance following microbiome disturbance, or if an increase in Chlamydia led directly to a decrease in bacterial species diversity. It is possible that this species of Chlamydiales is an amphibian pathogen, as several others are known to infect amphibians [52, 53]. Its presence at low levels in most salamanders, however, suggests that some other factor may disturb the microbiome and lead to near-dominance by Chlamydia. Even if this bacterium is not directly pathogenic, it may still have adverse effects on the host. While multiple species of bacteria are known to impede the growth of Bd [4, 54], and likely other pathogens, a diverse skin microbiome may be required for adequate defense against pathogens. Dominance of the skin microbiome by Chlamydia was correlated with strikingly low relative abundances of other bacteria in our dataset. Species of Chlamydia have greatly reduced genomes as a consequence of obligate intracellularity [55], and are thus unlikely to defend against pathogens to the same degree (if at all) as bacteria that would otherwise be present on the skin, leaving the host more open to infections from other pathogens. The prevalence and relative abundances of Chlamydia in other wild populations of amphibians, as well as its role as a pathogen or potential facilitator of pathogens, merits additional study.
Our results have implications both for possible future bioaugmentation conservation interventions and for synergistic interactions between pathogens, climate change, and habitat destruction. The role of symbiotic skin bacteria of amphibians in disease resistance is not yet clear. Most studies examining the relationship between the microbiome and Bd infection show that differences in microbiome composition and community structure affect the intensity and outcome of Bd infection [4, 6, 56, 57]. However, this relationship is still not fully understood. For example, an experimental study showed that Bd invasion altered the microbiome of Bd-susceptible frogs [58]. The interactions between Bd and the skin microbiome are likely modulated by factors such as the number and density of zoospores invading the skin and any prior history of host exposure to the pathogen. Our results suggest that, at shallower phylogenetic scales, habitat is a strong determinant of the amphibian skin microbiome. All of the sites we sampled were in montane forest and differences in climate between them, while real, are small compared to climatic differences with other habitats in the same region (Online Resource 5). This raises the possibility that global climate change or conversion/disturbance of forest could result in major skin microbiome changes for resident amphibians by radically altering local conditions, which could in turn affect host susceptibility to pathogens. Bd infection intensity and prevalence has been shown to vary between forested and deforested areas [59] and across different environments within species [60], and this interaction between land cover change and pathogen virulence could be influenced by environmentally induced changes in the microbiome. Controlled experiments with multiple environmental conditions are needed to assess this interaction.
Data Availability
Sanger sequence data for salamanders are deposited in Genbank (Accession numbers in Online Resource 2). Illumina sequence data for salamanders are deposited in the NCBI Sequence Read Archive (Bioproject PRJNA505751). 16S sequence data for microbiome samples are deposited in the NCBI Sequence Read Archive (Bioproject PRJNA505069).
References
Shoemaker V, Hillman S, Hillyard S et al (1992) Exchange of respiratory gases, ions, and water in terrestrial amphibians. In: Feder M, Burggren W (eds) Environmental physiology of the amphibians. University of Chicago Press, Chicago, pp 125–150
AmphibiaWeb (2017) University of California, Berkeley, CA, USA. http://amphibiaweb.org. Accessed 19 Apr 2017
Bletz MC, Loudon AH, Becker MH, Bell SC, Woodhams DC, Minbiole KPC, Harris RN (2013) Mitigating amphibian chytridiomycosis with bioaugmentation: characteristics of effective probiotics and strategies for their selection and use. Ecol. Lett. 16:807–820. https://doi.org/10.1111/ele.12099
Becker MH, Walke JB, Cikanek S, Savage AE, Mattheus N, Santiago CN, Minbiole KPC, Harris RN, Belden LK, Gratwicke B (2015) Composition of symbiotic bacteria predicts survival in Panamanian golden frogs infected with a lethal fungus. Proc. R. Soc. B Biol. Sci. 282:20142881. https://doi.org/10.1098/rspb.2014.2881
Kueneman JG, Woodhams DC, Van Treuren W et al (2016) Inhibitory bacteria reduce fungi on early life stages of endangered Colorado boreal toads (Anaxyrus boreas). ISME J 10:934–944. https://doi.org/10.1038/ismej.2015.168
Harris RN, Brucker RM, Walke JB, Becker MH, Schwantes CR, Flaherty DC, Lam BA, Woodhams DC, Briggs CJ, Vredenburg VT, Minbiole KPC (2009) Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J 3:818–824. https://doi.org/10.1038/ismej.2009.27
Skerratt LF, Berger L, Speare R, Cashins S, McDonald KR, Phillott AD, Hines HB, Kenyon N (2007) Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. Ecohealth 4:125–134
Martel A, Spitzen-van der Sluijs A, Blooi M, Bert W, Ducatelle R, Fisher MC, Woeltjes A, Bosman W, Chiers K, Bossuyt F, Pasmans F (2013) Batrachochytrium salamandrivorans sp. nov. causes lethal chytridiomycosis in amphibians. Proc. Natl. Acad. Sci. U. S. A. 110:15325–15329. https://doi.org/10.1073/pnas.1307356110
Yap TA, Koo MS, Ambrose RF, Wake DB, Vredenburg VT (2015) Averting a north American biodiversity crisis. Science 349:481–482. https://doi.org/10.1126/science.aab1052
Fitzpatrick BM, Allison AL (2014) Similarity and differentiation between bacteria associated with skin of salamanders (Plethodon jordani) and free-living assemblages. FEMS Microbiol. Ecol. 88:482–494. https://doi.org/10.1111/1574-6941.12314
Walke JB, Becker MH, Loftus SC, House LL, Cormier G, Jensen RV, Belden LK (2014) Amphibian skin may select for rare environmental microbes. ISME J 8:2207–2217. https://doi.org/10.1038/ismej.2014.77
McKenzie VJ, Bowers RM, Fierer N et al (2012) Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J 6:588–596. https://doi.org/10.1038/ismej.2011.129
Kueneman JG, Parfrey LW, Woodhams DC, Archer HM, Knight R, McKenzie VJ (2013) The amphibian skin-associated microbiome across species, space and life history stages. Mol. Ecol. 23:1238–1250. https://doi.org/10.1111/mec.12510
Rebollar EA, Hughey MC, Medina D, Harris RN, Ibáñez R, Belden LK (2016) Skin bacterial diversity of Panamanian frogs is associated with host susceptibility and presence of Batrachochytrium dendrobatidis. ISME J 10:1682–1695. https://doi.org/10.1038/ismej.2015.234
Bletz MC, Archer H, Harris RN, McKenzie VJ, Rabemananjara FCE, Rakotoarison A, Vences M (2017) Host ecology rather than host phylogeny drives amphibian skin microbial community structure in the biodiversity hotspot of Madagascar. Front. Microbiol. 8:1–14. https://doi.org/10.3389/fmicb.2017.01530
Muletz-Wolz CR, Almario JG, Barnett SE et al (2017) Inhibition of fungal pathogens across genotypes and temperatures by amphibian skin bacteria. Front Microbiol 8:1551. https://doi.org/10.3389/fmicb.2017.01551
Bird AK, Prado-Irwin SR, Vredenburg VT, Zink AG (2018) Skin microbiomes of California terrestrial salamanders are influenced by habitat more than host phylogeny. Front. Microbiol. 9:442
Prado-Irwin SR, Bird AK, Zink AG, Vredenburg VT (2017) Intraspecific variation in the skin-associated microbiome of a terrestrial salamander. Microb. Ecol. 74:745–756. https://doi.org/10.1007/s00248-017-0986-y
Wake DB, Vredenburg VT (2008) Are we in the midst of the sixth mass extinction? A view from the world of amphibians. Proc. Natl. Acad. Sci. 105:11466–11473
Culp CE, Falkinham J, Belden LK (2007) Identification of the natural bacterial microflora on the skin of eastern newts, bullfrog tadpoles and redback salamanders. Herpetologica 63:66–71
Lauer A, Simon MA, Banning JL, André E, Duncan K, Harris RN (2007) Common cutaneous bacteria from the eastern red-backed salamander can inhibit pathogenic fungi. Copeia 2007:630–640
Cheng TL, Rovito SM, Wake DB, Vredenburg VT (2011) Coincident mass extirpation of neotropical amphibians with the emergence of the infectious fungal pathogen Batrachochytrium dendrobatidis. Proc. Natl. Acad. Sci. U. S. A. 108:9502–9507
Boyle DG, Boyle DB, Olsen V, Morgan JAT, Hyatt AD (2004) Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis. Aquat. Org. 60:141–148. https://doi.org/10.3354/dao060141
Hyatt AD, Boyle DG, Olsen V, Boyle DB, Berger L, Obendorf D, Dalton A, Kriger K, Hero M, Hines H, Phillott R, Campbell R, Marantelli G, Gleason F, Colling A (2007) Diagnostic assays and sampling protocols for the detection of Batrachochytrium dendrobatidis. Dis. Aquat. Org. 73:175–192. https://doi.org/10.3354/dao073175
Blooi M, Pasmans F, Longcore JE, Spitzen-van der Sluijs A, Vercammen F, Martel A (2013) Duplex real-time PCR for rapid simultaneous detection of Batrachochytrium dendrobatidis and Batrachochytrium salamandrivorans in amphibian samples. J. Clin. Microbiol. 51:4173–4177. https://doi.org/10.1128/JCM.02313-13
Briggs CJ, Knapp RA, Vredenburg VT (2010) Enzootic and epizootic dynamics of the chytrid fungal pathogen of amphibians. Proc. Natl. Acad. Sci. U. S. A. 107:9695–9700. https://doi.org/10.1073/pnas.0912886107
Vredenburg VT, Knapp RA, Tunstall TS, Briggs CJ (2010) Dynamics of an emerging disease drive large-scale amphibian population extinctions. Proc. Natl. Acad. Sci. U. S. A. 107:9689–9694. https://doi.org/10.1073/pnas.0914111107
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R (2010) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. https://doi.org/10.1093/bioinformatics/btp636
DeSantis TZ, Hugenholtz P, Larsen N et al (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072. https://doi.org/10.1128/AEM.03006-05
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. https://doi.org/10.1038/ismej.2011.139
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261–7526. https://doi.org/10.1128/AEM.00062-07
Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG, Angenent LT, Knight R, Ley RE (2012) Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J 6:94–103. https://doi.org/10.1038/ismej.2011.82
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7:203–214. https://doi.org/10.1089/10665270050081478
Price MN, Dehal PS, Arkin AP (2010) FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. https://doi.org/10.1371/journal.pone.0009490
Chao A, Chazdon RL, Colwell RK, Shen T-J (2005) A new statistical approach for assessing similarity of species composition with incidence and abundance data. Ecol. Lett. 8:148–159. https://doi.org/10.1111/j.1461-0248.2004.00707.x
Faith DP (1992) Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61:1–10. https://doi.org/10.1016/0006-3207(92)91201-3
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228–8235. https://doi.org/10.1128/AEM.71.12.8228
Vázquez-Baeza Y, Pirrung M, Gonzalez A, Knight R (2013) EMPeror : a tool for visualizing high-throughput microbial community data. Gigascience 2:2–5
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30:2725–2729. https://doi.org/10.1093/molbev/mst197
Bi K, Vanderpool D, Singhal S, Linderoth T, Moritz C, Good JM (2012) Transcriptome-based exon capture enables highly cost-effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics 13:403. https://doi.org/10.1186/1471-2164-13-403
Vieira FG, Lassalle F, Korneliussen TS, Fumagalli M (2016) Improving the estimation of genetic distances from next-generation sequencing data. Biol. J. Linn. Soc. 117:139–149. https://doi.org/10.1111/bij.12511
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 25:1965–1978. https://doi.org/10.1002/joc.1276
QGIS Development Team (2015) QGIS geographic information system. Open Source Geospatial Foundation Project. http://www.qgis.org/
R Development Core Team (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing Vienna, Austria
De Caceres M, Legendre P (2009) Associations between species and groups of sites: indices and statistical inference. Ecology 90:3566–3574
Martel A, Blooi M, Adriaensen C, van Rooij P, Beukema W, Fisher MC, Farrer RA, Schmidt BR, Tobler U, Goka K, Lips KR, Muletz C, Zamudio KR, Bosch J, Lotters S, Wombwell E, Garner TWJ, Cunningham AA, Spitzen-van der Sluijs A, Salvidio S, Ducatelle R, Nishikawa K, Nguyen TT, Kolby JE, van Bocxlaer I, Bossuyt F, Pasmans F (2014) Recent introduction of a chytrid fungus endangers Western Palearctic salamanders. Science 346:630–631
Easson CG, Thacker RW (2014) Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Front. Microbiol. 5:1–11. https://doi.org/10.3389/fmicb.2014.00532
Muletz-Wolz CR, Yarwood SA, Campbell Grant EH et al (2017) Effects of host species and environment on the skin microbiome of plethodontid salamanders. J. Anim. Ecol. 87:341–353. https://doi.org/10.1111/1365-2656.12726
Corsaro D, Valassina M, Venditti D (2003) Increasing diversity within Chlamydiae. Crit. Rev. Microbiol. 29:37–78. https://doi.org/10.1080/713610404
Densmore CL, Green DE (2007) Diseases of amphibians. ILAR J. 48:235–254. https://doi.org/10.1093/ilar.48.3.235
Martel A, Adriaensen C, Bogaerts S, Ducatelle R, Favoreel H, Crameri S, Hyatt AD, Haesebrouck F, Pasmans F (2012) Novel chlamydiaceae disease in captive salamanders. Emerg. Infect. Dis. 18:1020–1022. https://doi.org/10.3201/eid1806.111137
Woodhams DC, Alford RA, Antwis RE, Archer H, Becker MH, Belden LK, Bell SC, Bletz M, Daskin JH, Davis LR, Flechas SV, Lauer A, Gonzalez A, Harris RN, Holden WM, Hughey MC, Ibáñez R, Knight R, Kueneman J, Rabemananjara F, Reinert LK, Rollins-Smith LA, Roman-Rodriguez F, Shaw SD, Walke JB, McKenzie V (2015) Antifungal isolates database of amphibian skin-associated bacteria and function against emerging fungal pathogens. Ecology 96:595. https://doi.org/10.1890/14-1837.1
Sakharkar KR, Kumar Dhar P, Chow VVTK (2004) Genome reduction in prokaryotic obligatory intracellular parasites of humans: a comparative analysis. Int. J. Syst. Evol. Microbiol. 54:1937–1941. https://doi.org/10.1099/ijs.0.63090-0
Vredenburg V, Briggs C, Harris R (2011) Host pathogen dynamics of amphibian chytridiomycosis: the role of the skin microbiome in health and disease. In: Olson L, Choffnes E, Relman D, Pray L (eds) Fungal diseases: an emerging threat to human, animal, and plant health. National Academy Press, Washington, D.C., pp 342–355
Longo AV, Savage AE, Hewson I, Zamudio KR (2015) Seasonal and ontogenetic variation of skin microbial communities and relationships to natural disease dynamics in declining amphibians. R Soc Open Sci 2:140377. https://doi.org/10.1098/rsos.140377
Jani AJ, Briggs CJ (2014) The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc. Natl. Acad. Sci. U. S. A. 111:E5049–E5058. https://doi.org/10.1073/pnas.1412752111
Becker CG, Zamudio KR (2011) Tropical amphibian populations experience higher disease risk in natural habitats. Proc. Natl. Acad. Sci. U. S. A. 108:9893–9898. https://doi.org/10.1073/pnas.1014497108
Savage AE, Becker CG, Zamudio KR (2015) Linking genetic and environmental factors in amphibian disease risk. Evol. Appl. 8:560–572. https://doi.org/10.1111/eva.12264
Acknowledgments
Funding was provided by UC MEXUS Collaborative Grant #CN-13-614 to G. Parra-Olea and D.B. Wake; NSF #IOS-1258133, and the Belmont Forum project People, Pollution, and Pathogens (P3), NSF 1633948 to V.T. Vredenburg; NSF #DEB-1026396 to R. Bowie and D.B. Wake; and by a Fulbright Colombia “Estudiante Doctoral Colombiano” Scholarship to S.V. Flechas. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation or the National Institute of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Statement
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. The article does not contain any studies with human participants performed by any of the authors.
Electronic Supplementary Material
Online Resource 1
(DOCX 21 kb)
Online Resource 2
(DOCX 92 kb)
Online Resource 3
(DOCX 114 kb)
Online Resource 4
(PNG 74 kb)
Online Resource 5
(PNG 118 kb)
Online Resource 6
(DOCX 96 kb)
Online Resource 7
(XLSX 15 kb)
Online Resource 8
(XLSX 11 kb)
Rights and permissions
About this article

Cite this article
Ellison, S., Rovito, S., Parra-Olea, G. et al. The Influence of Habitat and Phylogeny on the Skin Microbiome of Amphibians in Guatemala and Mexico. Microb Ecol 78, 257–267 (2019). https://doi.org/10.1007/s00248-018-1288-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-018-1288-8