
Abstract
A limited number of studies have investigated the biodiversity in deep continental hyperthermal aquifers and its influencing factors. Here, we present the first description of microbial communities inhabiting the Pannonian and Triassic hyperthermal aquifers from the Western Plain of Romania, the first one being considered a deposit of “fossilized waters,” while the latter is embedded in the hydrological cycle due to natural refilling. The 11 investigated drillings have an open interval between 952 and 3432 m below the surface, with collected water temperatures ranging between 47 and 104 °C, these being the first microbial communities characterized in deep continental water deposits with outflow temperatures exceeding 80 °C. The abundances of bacterial 16S rRNA genes varied from approximately 105–106 mL−1 in the Pannonian to about 102–104 mL−1 in the Triassic aquifer. A 16S rRNA gene metabarcoding analysis revealed distinct microbial communities in the two water deposits, especially in the rare taxa composition. The Pannonian aquifer was dominated by the bacterial genera Hydrogenophilus and Thermodesulfobacterium, together with archaeal methanogens from the Methanosaeta and Methanothermobacter groups. Firmicutes was prevalent in the Triassic deposit with a large number of OTUs affiliated to Thermoanaerobacteriaceae, Thermacetogenium, and Desulfotomaculum. Species richness, evenness, and phylogenetic diversity increased alongside with the abundance of mesophiles, their presence in the Triassic aquifer being most probably caused by the refilling with large quantities of meteoric water in the Carpathian Mountains. Altogether, our results show that the particular physico-cheminal characteristics of each aquifer, together with the water refilling possibilities, seem to determine the microbial community structure.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Subsurface ecosystems are among the least investigated environments mainly due to their low accessibility, even though they may harbor more than 50% of the Earth’s biomass [1]. As previous studies have focused mainly on the deep subseafloor sediments [2,3,4] and on hyperthermal areas such as hot springs or hydrothermal vents [5,6,7], the microbial diversity and ecology in deep hyperthermal aquifers under the continental crust remain largely unexplored [3, 8]. Additionally, these extreme habitats may be considered a frontier for the study of biodiversity, giving the opportunity to explore the microbial adaptation to difficult conditions, thus improving our understanding of the physiological limits of life [9]. While hot springs and hydrothermal vents are extreme, but generally unstable environments due to large oscillations in energy supplies, the thermal stability of geothermal aquifers enables the development of thermophilic microbial communities in harsh environments even on geological timescales [10].
As the continental subsurface is highly varied geologically, so is the microbial abundance and diversity [11]. The biodiversity in the deep continental habitats has only recently been assessed through next generation sequencing technologies [12, 13], even though these methods have already been used in the last decade to characterize the diversity of marine subsurface environments [14, 15]. Several active microbial communities with low diversity have been described in the groundwater feeding the Lidy hot springs, USA, where hydrogen-consuming methanogens were dominant [16], and in the deep fracture-derived groundwater from South Africa, an environment dominated by a single phylotype, a thermophilic sulfate reducer from the Firmicutes phylum [17]. A more complex and diverse community, dominated by the Methanospirillum, Thermodesulfovibrio, and Hydrogenobacter genera, was described in a thermal aquifer associated with the Great Artesian Basin, Australia, a thermal environment with the oldest water component estimated to be around 2 million years old, and a temperature of 64 °C [10]. Thus, all current studies describing microbes and prokaryotic communities functioning at temperatures higher than 80 °C had originated from hydrothermal vents, deep sub-sea floor sediments, and hot springs [11, 18], but none from the deep continental subsurface aquifers.
According to other studies that were focused on three deep unconnected aquifers from the Fennoscandian shield, with a maximum depth of 455 m and water temperatures of 12–14 °C, the connectivity to the surface was observed to be the main driver of the microbial composition [8, 19]. Unfortunately, usually due to low accessibility, this hypothesis has not been tested in the case of deep continental hyperthermal aquifers located at 1–3 km below the surface. Aquifers of this type, with up to 2 million years old water components and different degrees of connectivity to the surface ecosystem, have been encountered in Romania [20]. Here, due to a campaign that started in the 1960s in search of fossil fuel resources, over 250 wells were drilled, with depths between 800 and 3500 m. This led to the identification of many geothermal areas, most of them located in the Western part of the country, associated with the Pannonian basin [21]. These drillings grant the possibility to investigate the prokaryotic biodiversity in underexplored continental subsurface water deposits with a temperature that in some cases exceeds 80 °C [20].
This study presents the first description of microbial diversity in two hyperthermal aquifers (i.e., Pannonian and Triassic), located in the Western Plain of Romania and characterized by contrasting physico-chemical parameters and connectivity to the surface. The Pannonian aquifer is considered a confined water deposit, i.e., deposit of “fossilized waters,” while the Triassic one is embedded in the hydrological cycle due to natural refilling. In the latter case, microbial communities may be directly affected by the organic carbon input derived from photosynthetically driven ecosystems, as other authors concluded for similar environments [22]. In contrast, it is considered that the Pannonian aquifer is depleted in fresh organic carbon due to its complete isolation from the surface. Such pristine ecosystems rely on scarce energy from the sediments or the surrounding rocks [8, 22, 23]. To the best of our knowledge, the studies on the aquifers from the Fennoscandian shield are the only ones to investigate the role of natural refilling in shaping deep subsurface microbial communities [8, 12]. Thus, the aims of this study were (i) to describe the microbial diversity patterns and the community structure in 11 boreholes tapping the two hyperthermal aquifers with upwelling water temperatures ranging from 47 to 104 °C and (ii) to investigate the impact of physico-chemical parameters of the water and the role of connectivity to the surface as drivers of biodiversity in the deep aquifers within this geographic area.
Materials and Methods
Sampling and Sample Processing
Eleven specific boreholes were sampled in 2015, five for the Pannonian Aquifer (sample codes P1 to P5) and six for the Triassic Aquifer (sample codes T1 to T6), each borehole being characterized by distinct location, hydrogeological and hydrochemical features (Fig. 1; Table 1and 2). Since each drilling was performed (between 1968 and 1982), water has continuously flowed at a rate of 5–26 L s−1 (Table 2), keeping the boreholes free from contamination (as suggested by Hubalek et al. [8]). Duplicate upwelling water samples were collected in sterile glass bottles of two liters each. The samples were transported on ice to the microbiology laboratory of the Institute of Biological Research (Cluj-Napoca, Romania) and were processed within the same day. Three fractions from each of the collected waters (Table S1) were vacuum filtered using 0.22 μm pore size sterile nitrocellulose filters (Fioroni, France) that were stored at −20 °C until DNA extraction.

Geographic location of the sampling sites. Five Pannonian (P) and six Triassic (T) drillings were targeted
DNA Extraction
Three filters were obtained for each sample, and the total DNA was extracted from the filters using the Chelex® 100 Resin (Bio-Rad) based protocol [24]. Briefly, 5% Chelex® 100 solution was prepared in TE buffer (100 mM Tris, 1 mM EDTA, pH 8). The filters were cut into small pieces using sterile scissors and 200 μl of the 5% Chelex® 100 solution was added to the filters in 1.5 mL Eppendorf tubes along with 5 μl Proteinase K (20 mg ml−1). The tubes were incubated overnight at 56 °C. Next, the tubes were vortexed and incubated again for 8 min at 100 °C followed by centrifugation at 15,000×g for 3 min. Approximately 150 μl of the supernatant was carefully transferred to a new Eppendorf tube, the replicates of the same sample being pooled. Next, in order to remove any traces of Chelex® 100 solution that may inhibit downstream reactions, the combined supernatant of each sample was subjected to a second DNA extraction and cleaning step using the ZR Soil Microbe DNA MiniPrep™ (ZymoResearch, USA). The manufacturer instructions were followed, starting with the step of supernatant transfer to a Zymo-Spin™ IV Spin Filter. Finally, the DNA was eluted in 35 μl Elution Buffer. A negative control for DNA extraction was included, in which a sterile polycarbonate filter was passed through all the steps of DNA extraction. The concentration of DNA extracted from both the samples and the negative controls was determined with Qubit® Fluorometer and the Qubit® dsDNA BR Assay Kit (Thermo Fisher Scientific, USA) (data not shown).
Quantitative Real-Time PCR
Quantitative real-time PCR (qPCR) was performed to evaluate the abundance of prokaryotic cells in the water samples by targeting the SSU rRNA gene using both the universal and bacterial specific primer pairs PRK341F/PRK806R and BACT1369F/PROK1492R, respectively [25,26,27]. qPCR reactions were performed in triplicate using the SsoFast Eva Green Supermix (Bio-Rad, Hercules, CA, USA) on the CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad). The program consisted of an initial denaturation at 98 °C for 120 s, followed by 45 cycles of 10 s at 98 °C and an annealing/elongation step performed at 55 °C for 30 s. The reaction mixtures contained the following components: 7 μl 1X Sso Fast EvaGreen SuperMix (Bio-Rad), 0.4 μM of the forward and reverse primers, and 10 ng of DNA and RNase/DNase-free water to a final volume of 14 μl.
The gene copy number was calculated by comparing the amplification results to a standard tenfold serial dilution of known quantities of recombinant plasmids (108−102) carrying the targeted gene. The plasmids were prepared by cloning (InsTAclone™ PCR cloning kit, Thermo Scientific, USA) the PCR amplification products of positive controls (Escherichia coli JM109) into vectors and transformation into competent cells. Plasmids were extracted with GeneJet Plasmid Miniprep Kit (Thermo Scientific, USA). The amplicons were validated through Sanger sequencing and BLASTn [28] similarity search using the GenBank database [29]. The copy number of standard plasmids was calculated from the plasmid (2886 bp) plus the insert lengths and an average molecular mass of 660 Da for each base pair. Nucleic acid extraction control (DNA concentration was below the level of detection—i.e., 0.1 ng μL−1), as well as negative qPCR controls were analyzed in the same run. A Ct cutoff value corresponding to the lowest Ct reported for the nucleic acid extraction controls was used, and the average number of gene copies detected in these controls was subtracted from the sample values. The inhibition effect of each sample DNA was evaluated by mixing 1 μl of standard DNA (104 copies) with 10 ng of each sample and running the qPCR reactions in triplicate. The amplification efficiency of the standard DNA alone (104 copies) was compared with the efficiency of the standard DNA combined with every sample, and no significant inhibition effect was observed. As the abundance results using both primer pairs were similar (data not shown), only the bacterial BACT1369F/PROK1492R amplification results were discussed.
Microbial Community Analysis
Libraries for the V3-V4 regions of the 16S rRNA gene were prepared for each sample together with a negative DNA extraction control and a positive control consisting of E. coli genomic DNA. For PCR amplification, the primer pair PRK341F/PRK806R [27] was used, modified by the addition of Illumina-specific adaptors. The PCR reaction for each sample (25 μl) contained 1X HOT FIREPol® PCR mix (Solis BioDyne, Estonia), 200 nM uniquely tagged forward and reverse primers, 1 μl of sample DNA and 18 μl water. The reaction conditions were 95 °C for 15 min, followed by 25 cycles of 95 °C for 30s, 50 °C for 30s, 72 °C for 45 s, ending with 72 °C for 7 min. The concentration of amplicons was measured with the Qubit®dsDNA HS Assay Kit using the Qubit® Fluorometer and equal amounts of amplicons for each sample (45 ng) were pooled into a normalized library. The library concentration was measured using the PerfeCta® NGS Quantification Kit for Illumina (Quanta BioSciences, USA) and the pooled library was diluted in Tris pH 8.5 to a final 4 nM concentration. The sequencing step was performed on a MiSeq platform (Illumina, USA) using V3 sequencing chemistry with 300 bp paired-end reads.
Amplicon Analysis
Raw sequence data (SRA accession number SRP076082) was processed and quality filtered through a combination of Usearch v8 and QIIME pipelines [30, 31]. Briefly, QIIME was used to extract the barcodes from the sequence data, to join the forward and reverse Illumina reads and to demultiplex the sequence data. Singleton removal and quality control filtering were performed using the Usearch v8 pipeline, by discarding sequences with less than 350 nucleotides and those with more than 0.2 total expected errors. Both de novo and reference chimera checking were carried out in Usearch v8, using the latest version of the Greengenes database (‘13_8’) as a reference [32], and the resulting Operational Taxonomic Unit (OTU) table was converted into the Biological Observational Matrix (BIOM) format [33]. Taxonomy was assigned using the default classifier in QIIME (Greengenes) against the updated ‘13_8’ version of the Greengenes database, and the mitochondrial and plastidial sequences were filtered out of the OTU table. Additionally, the OTUs found in the negative controls (10 OTUs, data not shown) were removed from the final OTU-table.
Statistical Data Analysis
For estimating alpha-diversity, the PD-whole tree, Shannon and Simpson diversity indices, and the Chao1 richness estimator were calculated using QIIME [30]. The Unweighted and Weighted Unifrac distances were calculated to evaluate the diversity among samples. Microbial community differences between aquifers were examined using the ANOSIM test and the Unweighted UniFrac similarity [34] in QIIME [30]. A Mantel test with 999 permutations was run (‘mantel’ function of the ‘vegan’ package in R) in order to investigate the correlation between the phylogenetic diversity patterns revealed by the Unweighted and Weighted Unifrac distances and all of the measured physico-chemical parameters that were first log transformed [35]. The dissimilarity indices for the physico-chemical parameters were computed using the Euclidean distances with the ‘vegdist’ function of the ‘vegan’ package in R. PCoA plots were generated in R using the ‘cmdscale’ function of the ‘calibrate’ package and the Unweighted and Weighted Unifrac distances. The environmental factors were fitted onto an ordination with the ‘envfit’ function of the ‘vegan’ package in R, being scaled by their correlation to the distance matrix. Only the physico-chemical parameters that exert a significant influence (p < 0.05) on the beta-diversity were plotted on the PCoA graphic.
To estimate the abundance of indigenous microbial species in the investigated samples, a classification of the OTUs as thermophiles or mesophiles was performed using the information available in the taxa description (data not shown). To strengthen this classification, the most abundant 100 OTUs were checked for their occurrence in thermal or hyperthermal environments through BLASTn [28] similarity search in the GenBank database [29]. The isolation source of the first 5 hit results was identified for each OTU and for it to be considered thermophilic, at least 3 hits should have been from thermal habitats. Also, as the strong correlation between the increase of the GC content and the optimal growth temperature appears to be universal for prokaryotes [3, 36, 37], the GC content of the amplified portion of the 16S rRNA gene was calculated for each OTU representative sequence. A Student’s t test was performed to check if there is a clear distinction in the GC content for the OTUs classified as thermophiles or mesophiles. Additionally, the default parameters of the MOLE-BLAST tool [38] were used for a particular unassigned OTU sequence with high abundance, in order to find the closest relatives in the GenBank database [29].
Prediction of Functionality
For functionality prediction with PICRUSt [39], the OTUs were picked using an open reference approach, and de novo OTUs were removed, keeping in the final OTU-table only the OTUs that had matching Greengenes IDs (‘13_8’). Data in the BIOM OTU-table was normalized using the 16S rRNA gene copy number for each OTU. The prediction of functional genes’ abundances was inferred from the normalized abundances of each OTU. In order to characterize the PICRUSt accuracy, the weighted nearest sequenced taxon index (NSTI) was computed for all samples, describing the extent to which microorganisms from each sample are related to sequenced genomes.
Results and Discussion
Environmental Settings
The structure of the Pannonian aquifer is formed on the Pannonian basal sand horizon, and the collecting rocks consist of sand and poorly consolidated sandstones. Stable (18O, 13C) and radioactive (14C, 3H) isotope analysis, as well as the undetected levels of tritium, revealed a very slow dynamic of these waters, which are a mixture of two components with different ages [20]. The oldest component consists of ~2 million years old brackish water, while the youngest one includes ~35,000 years old meteoric water. Thus, since the modern water supply is missing, the Pannonian deposit is considered fossilized. The collecting rocks of the Triassic aquifer are represented by dolomitic limestones of Triassic age and the radiometric dating established that the oldest water component is approx. 26,000 years old. As opposed to the previous aquifer, the latter has a dynamic nature and is a part of the hydrological cycle due to natural refilling with large quantities of meteoric waters caused by karstic phenomena in the Carpathian Mountains [20]. Over the last decades, intensive studies [20, 40] have been conducted for the chemical characterization of these thermal waters, and a stable chemical composition was observed. According to Table 1, the thermal waters are neutral to slightly alkaline, with pH values ranging between 6.7 and 8.3. The relative high values of chemical oxygen demand (COD) in the Pannonian aquifer waters (8.4–16 mg L−1) and in one of the Triassic deposits (sample T2–20 mg L−1), might indicate water pollution with organic matter. Both aquifers are characterized by bicarbonate waters, the Na+ (1100–4120 mg L−1) and K+ (24.9–99.2 mg L−1) being dominant cations in the Pannonian basin, whereas in the Triassic aquifer the Ca2+ (103–259 mg L−1) and Mg2+ ions (15.7–61.6 mg L−1) prevail [40]. Na+ and Cl− have high concentrations in the Pannonian aquifer probably as a result of prolonged water-rock contact, these rocks being formed in coastal or lagoon conditions that allowed the accumulation of easily dissolvable chloride rocks. Also, the high HCO3 − values here are probably caused by the stagnant nature of these waters, causing HCO3 − accumulation and SO4 2− reduction. The high concentration of Ca2+ and Mg2+, as well as HCO3 − and SO4 2− in the Triassic aquifer waters may be caused by the presence of limestones, dolomites, and anhydrites (CaSO4) [20, 40].
Abundance of Bacterial 16S rRNA Genes
Total 16S rRNA gene copy numbers were calculated from qPCR results (Fig. S1). In the Pannonian aquifer, the 16S rRNA gene copy number varied between 1.3 × 105 and 1.4 × 106 mL−1, higher values being observed at 55 °C (sample P3), while in the Triassic aquifer the values ranged between 1.05 × 102 and 1.2 × 104 mL−1, with the lowest values being detected at 92–104 °C. The bacterial abundances appear to decline slowly with depth and with the increase in water temperature, an aspect generally reported in similar works [11, 12, 41]. The 16S rRNA gene copy numbers were close to those from 180 to 2300 m deep subsurface fracture fluids from Outokumpu Deep Drill Hole, eastern Finland [12, 42], or from depths of 2000–3500 m in Witwatersrand, South Africa [43], although neither of them are hyperthermal environments. Abundances in the range of 105–106 cells mL−1, as those observed in the Pannonian basin, were also encountered in the ~2 million years old geothermal aquifer associated with the Great Artesian Basin, Australia, with upwelling water temperature of 64 °C [10]. The low abundances of 16S rRNA genes, especially at the highest depths and temperatures of the Triassic aquifer, may be derived from the reduced energy fluxes in thermal aquifers and the slow lifecycle of subsurface microorganisms, making the characterization of these communities a more difficult process [11].
Prokaryotic Community Structure
The primers used for libraries preparation [27] were searched against SILVA reference database (Release 128) and a coverage of 84.1% for bacteria and 58.5% for archaea was obtained. Following filtering, sequences were clustered using the 97% sequence identity threshold into 43 to 121 OTUs per sample (Table S2). As the two aquifers have a different level of connectivity to the surface ecosystems, an important step in discussing the diversity of microorganisms was to estimate the abundance of indigenous and/or thermophilic microbial species in our samples. For this purpose, the first most abundant 100 OTUs were classified as mesophiles or thermophiles. Their abundance corresponds to 92–99% of the sequencing libraries, creating a relevant overview of the microbial diversity. Additionally, a Student’s t test confirmed that there is a clear distinction in the GC content for the OTUs classified as thermophiles (52–68%) or mesophiles (49–58%) (t = 8.2329, p < 0.05).
Therefore, in the Pannonian aquifer, 95–99% of the sequencing libraries were composed of thermophile and hyperthermophile OTUs, which validated this deposit as a pristine environment (Fig. S2). On the other hand, the Triassic aquifer contained 28–98% indigenous species, the contamination with less mesophiles being observed in T3 and T4. The presence of contaminant species is not unexpected, as this aquifer receives large quantities of meteoric waters and takes part in the hydrological cycle [20].
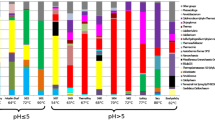
The taxonomy data revealed that the prokaryotic communities in the investigated aquifers were represented by 24 different phyla, 13 of them with abundance above 1% (Fig. 2). The most abundant phylum in the Pannonian aquifer was Proteobacteria (Alpha-, Beta-, Gamma-, Delta- and Epsilonproteobacteria), with 31.9–93.9% of the sequencing libraries, while in the Triassic waters both Proteobacteria (6.4–95.8%) and Firmicutes (2.8–57.5%) were prevalent. These phyla are usually dominant in the terrestrial subsurface environments, Alpha-, Beta-, and Gammaproteobacteria being commonly found in shallow aquifers, while the sulfate-reducing, spore-forming taxa of the Firmicutes are more often dominant in deeper environments [9, 41, 44, 45]. Thus, even though these two water deposits are geographically close, they have distinct prokaryotic communities. Thus, out of the 224 OTUs found in the 11 samples, only 5 of the dominant OTUs were shared among them, i.e., Rhodocyclaceae, Thermoanaerobacteriaceae, Thermodesulfovibrio, Archaeoglobus, and Acinetobacter, highlighting that the Pannonian and Triassic subsurface waters were inhabited by distinct microbial communities. This statement was also supported by the Kruskal-Wallis test performed for all OTU abundances, resulting in 10 dominant OTUs with significant different mean abundances (p < 0.05) in the two aquifers (Fig. 3).

Prokaryotic community composition at the phylum, class and order level. The relative abundances of dominant (> 1% of the sequencing library) bacterial and archaeal taxonomical groups in the 11 geothermal water samples collected from the Pannonian (P) and Triassic (T) aquifers

Heatmap highlighting 10 major OTUs with significantly (p < 0.05) different mean abundances in the geothermal aquifers. The color key value is the log10 of the OTU mean in the two geothermal aquifers
Among Proteobacteria, the Rhodocyclaceae family, a diverse group including thermophiles, sulfur-oxidizing chemoautotrophs, anaerobes, and methylotrophs [46], was abundant in samples P1, P2 and P5, with one OTU specific to the Pannonian aquifer (Fig. 3). Within this family of microorganisms, the thermophilic, facultatively chemolithoautotrophic hydrogen-oxidizing organisms affiliated to Hydrogenophilus sp. [47], seemed to be thriving in the Pannonian water deposit. Members of this genus were previously isolated from geothermal sites in Japan [48], a hot spring in Graendalur, Iceland [49], Yellowstone National Park, USA [50] and in the geothermal aquifer from Australia that has a similar age and thermal regime to the Pannonian aquifer [10]. Moderate thermophilic species of the Moraxellaceae and Halothiobacillaceae families were dominant in P3 and P4, samples with the lowest temperature (55 and 47 °C, respectively). Interestingly, Deltaproteobacteria was, for the first time, observed as a dominant class in a deep subsurface continental aquifer. Mainly represented by the Thermodesulfobacterium genus, Deltaproteobacteria was an abundant class in P5, constituting 27% of the sequencing library. The Thermodesulfobacterium genus was previously described in hot spring sediments from Yellowstone National Park, Wyoming, and in a 1000 m deep water sample from a mesothermic petroleum reservoir in North Slope, Alaska, USA [51, 52]. Some studies reported that Proteobacteria comprises a greater proportion of the community immediately after drilling as a result of contamination [11, 44], yet declines after a few months, as water becomes dominated by indigenous microbes and the equilibrium is restored. But, in other cases, the Proteobacteria phylum appears to truly prevail [11, 53], this probably being the case of the Pannonian aquifer also, which contains between 95 and 99% thermophilic and hyperthermophilic taxa. The communities encountered in remote and deep aquifers are shaped by limitation of resources, competition and predation, but ultimately the microbial composition relies on stochastic events that determine what groups of organisms inhabit that specific environment in the moment of isolation in the first place [8]. This presumption may explain, to some extent, the abundance of Proteobacteria in the “fossilized” water of the Pannonian aquifer.
Firmicutes was a dominant phylum in the Triassic aquifer, comprising between 2.8 and 57.5% of the sequencing libraries. Thus, even if the microbial communities here may be disturbed by meteoric water refilling, large populations of specific subsurface and thermophilic taxa, such as Thermoanaerobacteriaceae, Desulfotomaculum spp., Thermacetogenium spp. [1, 54, 55] were observed. Regarding this aquifer, one of its specific OTUs presents a high sequence similarity to Caldicellulosiruptor saccharolyticus (Fig. 3), an anaerobic thermophilic species that possesses cellulolytic capability. These organisms have been isolated from neutral or slightly alkaline geothermal springs in Iceland [56], California [57], and Russia [58] and have a great biotechnological potential being able to degrade a wide spectrum of carbohydrates [58]. Desulfotomaculum genus, particular to this water deposit (Fig. 3), was often encountered in a variety of deep fracture water communities. These thermophilic organisms are able to form heat-resistant endospores that can survive in dry conditions, in extreme heat and pressure or oxic stress for several months or years [11, 59]. As vegetative cells of the Desulfotomaculum genus were found to be active at temperatures below 85 °C, whereas spores were able to survive at temperatures above 100 °C even after serial autoclaving exposures [60, 61], it may be speculated that this genus is part of the active microbial population in sample T3 (72 °C), but is most likely represented by endospores in sample T5 (104 °C). Additionally, an OTU related to Ammonifex thiophilus, an extreme thermophilic organism isolated from the geothermal area of Uzon Caldera, Kamchatka, Russia, was observed only in the Triassic aquifer. It has a facultative chemolithoautotrophic metabolism, using hydrogen and formate as electron donors and thiosulfate, sulfate or elemental sulfur as electron acceptors, producing hydrogen sulfide [58].
The Archaea (up to 31.7%) domain is usually encountered in deep continental waters [1, 44], members of the Euryarchaeota phylum being considered indigenous groundwater organisms, whereas OTUs affiliated to Crenarchaeota may represent in some cases drilling fluid contaminants [45, 54]. However, Desulfurococcales and Thermoproteales orders within Crenarchaeota phylum are probably indigenous taxa in the Triassic aquifer, being extreme thermophiles and “sulfur-dependent” organisms [62, 63]. Sulfate-reducing microbes from Euryarchaeota were major components in T4 and P2 samples, being represented by different species of the Archaeoglobus genus, microorganisms that have a major role in the biogeochemical sulfur cycle [64, 65]. Even though sulfate had a much lower concentration in the Pannonian than in the Triassic aquifer, never exceeding 25 mg L−1, one uncultured Archaeoglobus species seems to be encountered only in this aquifer, while another OTU assigned to the same genus was shared between aquifers. Additionally, acetogenic and hydrogenotrophic methanogens from the Methanosaeta and Methanothermobacter genera [10, 54] were abundant in the P1 and P5 samples.
Overall, 11 OTUs remained unassigned showing the potential of the investigated aquifers to reveal novel species or genera. Interestingly, one of the unassigned sequence comprised more than 30% of the microbial community in sample P1, being similar (99% identity in the V3-V4 region of the 16S rRNA gene) to an uncultured bacterium, previously encountered only in a geothermal water fueling an oxbow lake from the central part of the Pannonian Basin, Hungary [66]. Following the MOLE-BLAST search, this OTU clustered together to other uncultured Clostridia (Fig. S3) involved in hydrocarbon degradation at high temperature [67, 68].
Prokaryotic Diversity Patterns
In order to compare the alpha-diversity indices among samples, the OTU-table was normalized to the lowest number of sequences (5000) through random re-sampling. The conditional uncovered probability [69] for the OTU-table ranged between 0.001 and 0.006 (Table S2), showing that the sequencing depth was high enough for a comprehensive description of biodiversity, this being revealed also by the rarefaction plots (Fig. S4). Unexpectedly, the overall species number (Table S2) increased with temperature, the Pearson correlation coefficient between the OTUs number and temperature being r = 0.747 (p < 0.05). These results were surprising considering that prokaryotic communities at higher temperatures are usually less complex than those at lower temperatures, hot waters being dominated by only a few genera that are adapted to temperature stress [11, 70, 71]. Some studies also reported a linear positive correlation between biodiversity/richness and temperature [72,73,74]. The results revealed a different picture when the proportion of thermophiles and mesophiles were analyzed separately. Thus, in the case of thermophiles and hyperthermophiles, their proportion showed a significant negative correlation with temperature (r = −0.65, p < 0.05) and depth (r = −0.61, p < 0.05), which is in accordance with the other studies. In our samples, especially in the Triassic aquifer, typical thermophilic and hyperthermophilic species are found together with ubiquitous, aerobic, mesophilic bacteria. A possible explanation may reside in the continuous water refilling of the Triassic aquifer [20] that creates a hot, “shallow” environment [8, 11]. In this context, the number of OTUs, as well as the Simpson, Shannon and the PD-whole tree indices (Fig. S5) were all strongly correlated with the proportion of mesophiles in our samples (r between 0.58–0.66, p < 0.05). The presence of mesophiles here probably led to an overall false positive increase in species richness, evenness and phylogenetic diversity along with the temperature and depth. Actually, the communities become more even and more dispersed (low cell abundances) because of temperature stress, nutrients limitation and seclusion [8]. For the Triassic samples, the rise of diversity was correlated with the abundance of mesophiles, which usually are not adapted to extreme temperature, and most likely are not able to take part with a specific role in this ecosystem. Nonetheless, further work is needed to establish if there is a contribution of mesophiles in the active microbial communities from the Triassic aquifer.
In order to observe the beta-diversity patterns, PCoA plots were generated using the Unweighted and Weighted Unifrac distance matrices. As it can be observed in Fig. 4a, the clustering pattern generated by the Weighted Unifrac distances was neither strongly correlated with temperature, depth, abundance of mesophiles, nor with any of the physico-chemical parameters. The P3 sample appeared distinct from all the others. In this sample, 81% of the sequencing library was composed by two particular OTUs that are minor components in other samples, belonging to the Moraxellaceae family and the Tepidimonas genus, previously encountered in a hot spring community from Papua New Guinea [75] and in a terrestrial sulfidic spring, USA [76]. P1 and P5 samples had in common a large population (14–21%) of Hydrogenophilus, while the T3 and T6 both contained ~30% of the sequences included in the Thermodesulfovibrio genus. A dominant OTU from the Acinetobacter genus was shared between the T2 (45.6%) and P2 (97%) samples, showing a 99% similarity to a sequence reported in a hot spring community from the South of Thailand (AB862146). Other cases of such ultimate low diversity, with one dominant species ecosystem, were encountered in porewater collected from 1.8 km below surface in the Cambrian Mt. Simon, Illinois Basin, where Halomonas sulfidaeris made 97–99.4% of the sequence library [53], or in the groundwater feeding Lady Hot Springs in Idaho, which was dominated by hydrogenotrophic methanogens (95% of the community) [11, 16].

a Plot of the first two principal coordinate axes for PCoA using Weighted UniFrac distance matrix. b The same plot using Unweighted UniFrac distances. The environmental factors that exert a significant influence (p < 0.05) on the beta-diversity were fitted onto the PCoA graphic
Interestingly, by using the Unweighted Unifrac distances, a clustering pattern corresponding to distinct aquifers was observed (Fig. 4b). Next, the ANOSIM test was run for statistical support, resulting in an r value of 0.752 (p = 0.004), which indicated that the grouping of samples in distinct aquifers is strong and statistically significant. As the qualitative distances reveal the differences in the minor components that are considerably obscured by the quantitative measures [77], it may be assumed that the mesophilic species that do not form abundant populations in the Triassic aquifer may explain the observed pattern. But unlike the case of alpha-diversity, the amount of mesophiles does not strongly correlate with the beta-diversity matrix. Instead, other parameters, that are specific for each aquifer, like temperature, depth, Na+, Ca2+, SO4 2−, pH and electric conductivity were significantly correlated with the diversity pattern (p < 0.05). Additionally, the Mantel test performed for the physico-chemical parameters and the Unweighted Unifrac matrix returned a significant r of 0.574 (p = 0.004). These results were not surprising since they highlight the major differences in physico-chemical parameters between aquifers. Thus, the particular physico-chemical variables and the water refilling, factors that are specific to each aquifer, most probably have a cumulative effect in shaping the community structure, especially in the rare taxa distribution.
Functionality Prediction in the Microbial Communities
The putative physiology for the classified OTUs was predicted with PICRUSt [39], and the weighted NSTI indices (Table S4) varied between 0.00076 (sample P2) and 0.18 (sample P4). These values show the average branch length that separates each OTU from a reference genome in every sample. Therefore, caution is required when interpreting these results, especially for P4 sample, as PICRUSt prediction are uncertain for environments containing a high proportion of unexplored microorganisms [78]. Figure 5 presents the percentage of genes that might be involved in the nitrogen, sulfur, and methane metabolisms, as well as those of the carbon fixation pathways in prokaryotes. Methanogenesis appeared more common in the Pannonian aquifer, where Methanosaeta species are able to utilize acetate as electron donor [79] and those that belong to Methanothermobacter genus can reduce carbon dioxide to methane using molecular hydrogen as electron donor [80]. As methanogens and sulfate-reducers normally compete for resources, the former group is dominant in anaerobic habitats where sulfate is limited, which is the case of the Pannonian deposit, and vice versa [81]. The organisms responsible for dissimilatory sulfate-reduction are most likely archaea from the Archaeoglobus genus in the Pannonian aquifer [59], while the high sulfate ion concentrations in the Triassic groundwaters favor the presence of clostridial (e.g., Desulfotomaculum) and non-clostridial sulfate-reducers (e.g., Thermodesulfovibrio, Thermodesulfobacteriaceae, Archaeoglobus). Desulfotomaculum species are capable of using hydrogen, fatty acids, alanine, or phenyl-substituted organic acids as electron donors for dissimilatory sulfate reduction, but it is believed that when temperatures exceed 80 °C most likely the dissimilatory sulfate reduction is performed by archaeal sulfate reducers, as those of the Archaeoglobus genus [59]. Sulfate is reduced by these organisms to hydrogen sulfide that can be further assimilated by other prokaryotes as a substrate for growth [64]. Regarding the carbon fixation pathways in prokaryotes, the reductive citric acid together with the dicarboxylate-hydroxybutyrate cycles seem to dominate in almost all communities, being characteristic for the microaerophiles and anaerobes, or to the strictly anaerobic hyperthermophilic archaea, respectively [82]. The reductive citric acid cycle was also found to be a dominant pathway for carbon fixation, being previously reported for a microbial community inhabiting the deep Outokumpu drill hole [12] or the deep groundwaters at Olkiluoto, Finland [79]. This cycle is considered one of the oldest autotrophic pathways that had evolved, being widespread in the deep-sea hydrothermal vents and the deep continental subsurface communities [83], while the Wood-Ljungdahl pathway was reported as dominant in other subsurface environments [79, 84]. Although present, this pathway seems not to be that widespread in the Triassic or the Pannonian aquifers. The simultaneous existence of different carbon-fixation pathways in the deep subsurface may imply that several strategies are used in such harsh conditions, so that multiple resources become available to the microbial communities [53].

The predicted percentage of genes involved in the nitrogen, methane and sulfur metabolism, as well as the carbon fixation pathways in prokaryotes, in the Pannonian and Triassic geothermal water samples, generated using PICRUSt
Conclusions
This study presented the first characterization of microbial biodiversity in the Pannonian and Triassic hyperthermal aquifers, with upwelling water temperatures ranging between 47–84 and 72–104 °C, respectively. Although in geographical proximity, the Pannonian aquifer is a highly stable hydrological system, probably independent from any water supplies from the surface, while the Triassic deposit is part of the hydrological cycle due to natural refilling. The abundance of prokaryotes showed a positive correlation with the richness, evenness and phylogenetic diversity indices. This could be explained by the presence of mesophilic bacteria in the Triassic aquifer, leading to a false positive increase in diversity. These findings suggest that communities at higher temperatures become more even and dispersed, and the microbial activity might decrease with temperature and depth, but future studies should be conducted to confirm this hypothesis. Furthermore, in the case of the Pannonian and Triassic aquifers, the particular physico-chemical parameters and levels of connectivity to the surface seem to shape the main differences in alpha- and beta-diversity patterns. Overall, this study provides valuable insights into the microbial diversity in these newly investigated environments, bringing important information on the ecology of these deep subsurface continental waters with temperatures higher than 80 °C. Further work is required to establish the part of the microbial community that is active in the subsurface, and also for the true functional diversity characterization.
References
Nyyssönen M, Bomberg M, Kapanen A, Nousiainen A, Pitkänen P, Itävaara M (2012) Methanogenic and sulphate-reducing microbial communities in deep groundwater of crystalline rock fractures in Olkiluoto, Finland Geomicrobiol J. 29:863–878. doi:10.1080/014904512011635759
Inagaki F, Nunoura T, Nakagawa S, Teske A, Lever M, Lauer A, et al. (2006) Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean margin Proc Nat Acad Sci USA 103:2815–2820
Ragon M, Van-Driessche AES, García-Ruíz JM, Moreira D, Lopez-Garcia P (2013) Microbial diversity in the deep-subsurface hydrothermal aquifer feeding the giant gypsum crystal-bearing Naica Mine, Mexico Front. Microbiol. 4:37. doi:10.3389/fmicb201300037
Walsh EA, Birkpatrick JB, Rutherford SD, Smith DC, Sogin M, Hondt S (2016) Bacterial diversity and community composition from seasurface to subseafloor ISME J 10:979–989
Marteinsson VT, Hauksdóttir S, Hobel CFV, Kristmannsdóttir H, Hreggvidsson GO, Kristjánsson JK (2001) Phylogenetic diversity analysis of subterranean hot springs in Iceland Appl. Environ. Microbiol. 67:4242–4248
Summit M, Baross JA (2001) A novel microbial habitat in the mid-ocean ridge subseafloor Proc. Natl. Acad. Sci. U. S. A. 98:2158–2163
Kimura H, Asada R, Masta A, Naganuma T (2003) Distribution of microorganisms in the subsurface of the Manus Basin hydrothermal vent field in Papua New Guinea Appl. Environ. Microbiol. 69:644–648. doi:10.1128/AEM691644-6482003
Hubalek V, Wu X, Eiler A, Buck M, Heim C, Dopson M, et al. (2016) Connectivity to the surface determines diversity patterns in subsurface aquifers of the Fennoscandian shield ISME J 10:2556. doi:10.1038/ismej201636
Sahl JW, Schmidt R, Swanner ED, Mandernack KW, Templeton AS, Kieft TL, et al. (2008) Subsurface microbial diversity in deep-granitic-fracture water in Colorado Appl. Environ. Microbiol. 74:143–152. doi:10.1128/AEM01133-07
Kimura H, Sugihara M, Yamamoto H, Patel BK, Kato K, Hanada S (2005) Microbial community in a geothermal aquifer associated with the subsurface of the Great Artesian Basin, Australia Extremophiles 9: 407–414. doi:10.1007/s00792–005–0454-3
Kieft TL (2016) Microbiology of the Deep Continental Biosphere in Hurst CJ (ed) Their world: A diversity of microbial environments, Springer, Cham pp 225–249
Nyyssönen M, Hultman J, Ahonen L, Kukkonen I, Paulin L, Laine P, et al. (2014) Taxonomically and functionally diverse microbial communities in deep crystalline rocks of the Fennoscandian shield ISME J 8:126–138. doi:10.1038/ismej2013125
Lau MC, Cameron C, Magnabosco C, Brown CT, Schilkey F, Grim, et al. (2014) Phylogeny and phylogeography of functional genes shared among seven terrestrial subsurface metagenomes reveal N-cycling and microbial evolutionary relationships Front Microbiol 5:531. doi:10.3389/fmicb201400531
Sogin ML, Morrison HG, Huber JA, Mark-Welch D, Huse SM, Neal PR, et al. (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere” Proc. Natl. Acad. Sci. U. S. A. 103:12115–12120
Brazelton WJ, Nelson B, Schrenk MO (2012) Metagenomic evidence for H 2 oxidation and H 2 production by serpentinite-hosted subsurface microbial communities Front. Microbiol. 2:268. doi:10.3389/fmicb201100268
Chapelle FH, O’Neill K, Bradley PM, Methé BA, Ciufo SA, et al. (2002) A hydrogen-based subsurface microbial community dominated by methanogens Nature 415:312–315. doi:10.1038/415312a
Lin LH, Wang PL, Rumble D, Lippmann-Pipke J, Boice E, Pratt LM, et al. (2006) Long term biosustainability in a high energy, low diversity crustal biotome Science 314:479
Roussel EG, Bonavita MA, Querellou J, Cragg BA, Webster G, Prieur D, et al. (2008) Extending the sub-sea-floor biosphere Science 320:1046
Hengsuwan M, Heim C, Simon K, Hansen BT (2015) Long-term investigation of Sr-isotope and rare earth element fractionation processes within three major aquifers in the Äspö hard rock laboratory (Sweden) Geomicrobial J 32:243–254
Țenu A (1981) Zăcăminte de ape hipertermale din Nord-Vestul României. Editura Academiei Republicii Socialiste România, București,
Bendea C, Bendea G, Rosca M, Cucueteanu D (2013) Current status of geothermal energy production and utilization in Romania J Sustain Energy 4:1–9
Edwards KJ, Becker K, Colwell F (2012) The deep, dark energy biosphere: intraterrestrial life on Earth Annu. Rev. Earth Planet. Sci. 40:551–568
Lovley DR, Chapelle FH (1995) Deep subsurface microbial precesses Rev. Geophys. 33:365–381
Suenaga E, Nakamura H (2005) Evaluation of three methods for effective extraction of DNA from human hair J Chromatogr B Analyt Technol Biomed Life Sci 820:137–141. doi:10.1016/jjchromb200411028
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. John Wiley and Sons, Chichester, pp. 115–175
Suzuki M, Taylor L, DeLong E (2006) Quantitative analysis of small subunit rRNA genes in mixed microbial populations via 5′-nuclease assays Appl. Environ. Microbiol. 66:4605–4614
Yu Y, Lee C, Kim J, Hwang S (2005) Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction Biotechnol. Bioeng. 89:670–679
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool J. Mol. Biol. 215:403–410
Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2015) GenBank Nucleic Acids Res. 43:D30–D35. doi:10.1093/nar/gku1216
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. (2010) QIIME allows analysis of high-throughput community sequencing data Nat. Methods 7:335–336. doi:10.1038/nmethf303
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST Bioinformatics 26:2460–2461. doi:10.1093/bioinformatics/btq461
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie LE, Keller K, et al. (2006) Greengenes, a chimera-checked 16S rRNA gene database and worlbench compatible with ARB Appl. Environ. Microbiol. 72(7):5069–5072
McDonald D, Clemente JC, Kuczynski J, Rideout JR, Stombaugh J, Wendel D, et al. (2012) The biological observation matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome GigaScience 1:–7. doi:10.1186/2047-217X-1-7
Lin X, Kennedy D, Fredrickson J, Bjornstad B, Konopka A (2012) Vertical stratification of subsurface microbial community composition across geological formations at the Hanford site: Hanford site subsurface microbial communities Environ. Microbiol. 14:414–425. doi:10.1111/j1462-2920201102659x
Legendre P, Fortin MJ (2010) Comparison of the Mantel test and alternative approaches for detecting complex multivariate relationships in the spatial analysis of genetic data Mol. Ecol. Resour. 10:831–844. doi:10.1111/j1755-0998201002866x
Galtier N, Lobry JR (1997) Relationships between genomic G+C content, RNA secondary structures, and optimal growth temperature in prokaryotes J. Mol. Evol. 44:632–636
Kimura H, Ishibashi JI, Masuda H, Kato K, Hanada S (2007) Selective phylogenetic analysis (SePA) targeting 16S rRNA gene of hyperthermophilic archaea in the deep-subsurface hot biosphere Appl. Environ. Microbiol. 73:2110–2117. doi:10.1128/AEM02800-06
Agarwala R, Barrett T, Beck J, Benson DA (2016) Database resources of the National Center for biotechnology information Nucleic Acids Res. 44:D7-19. doi:10.1093/nar/gkv1290
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences Nat. Biotechnol.:8, 1–10. doi:10.1038/nbt2676
Kovácz J, Balázs I, Jákfalvi S (2013) Studiul forajelor geotermale. Program de cercetare în Euroregiunea Hajdú-Bihar – Bihor pentru cunoaşterea stării hidrogeologice a corpurilor de apă termală transfrontaliere. Proiect finanţat prin programul de Cooperare Transfrontalieră Ungaria - România 2007–2013
Itävaara M, Nyyssönen M, Kapanen A, Nousiainen A, Ahonen L, Kukkonen I (2011) Characterization of bacterial diversity to a depth of 1500 m in the Outokumpu Deep Borehole, Fennoscandian shield: deep terrestrial biodiversity FEMS Microbiol. Ecol. 77:295–309. doi:10.1111/j1574-6941201101111x
Purkamo L, Bomberg M, Kietäväinen R, Salavirta H, Nyyssönen M, Nuppunen-Puputti M, et al. (2016) Microbial co-occurrence patterns in deep Precambrian bedrock fracture fluids Biogeosciences 13:3091–3108. doi:10.5194/bg-13-3091-2016
Onstott TC, Lin LH, Davidson M, Mislowack B, Borcsik M, Hall J, et al. (2006) The origin and age of biogeochemical trends in deep fracture water of the Witwatersrand Basin, South Africa Geomicrobiol J. 23:369–414. doi:10.1080/01490450600875688
Moser DP, Onstott TC, Fredrickson JK, Brockman FJ, Balkwill DL, Drake GR, et al. (2003) Temporal shifts in the geochemistry and microbial community structure of an ultradeep mine borehole following isolation Geomicrobiol J. 20:517–548. doi:10.1080/713851170
Gihring TM, Moser DP, Lin LH, Davidson M, Onstott TC, Morgan L, et al. (2006) The distribution of microbial taxa in the subsurface water of the Kalahari shield, South Africa Geomicrobiol J. 23:415–430 doi:10.1080/01490450600875696
Oren A (2014) The Family Methanobacteriaceae in Rosenberg E, DeLong EF, Lory S, et al (eds) The Prokaryotes, Springer, Berlin, Heidelberg pp 165–193
Oren A (2014) The Family Rhodocyclaceae. In: Rosenberg E, EF DL, Lory S, et al. (eds) The Prokaryotes. Springer, Berlin, pp. 975–998
Hayashi NR, Ishida T, Yokota A, Kodama T, Igarashi Y (1999) Hydrogenophilus thermoluteolus gen nov, sp nov, a thermophilic, facultatively chemolithoautotrophic, hydrogen-oxidizing bacterium Int J Syst Evol Micr 49:783–786
Vesteinsdottir H, Reynisdottir DB, Orlygsson J (2011) Hydrogenophilus islandicus sp nov, a thermophilic hydrogen-oxidizing bacterium isolated from an Icelandic hot spring Intl J Syst Evol Micr 61:290–294. doi:10.1099/ijs0023572-0
Stöhr R, Waberski A, Liesack W, Völker H, Wehmeyer U, Thomm M (2001) Hydrogenophilus hirschii sp nov, a novel thermophilic hydrogen-oxidizing Beta-Proteobacterium isolated from Yellowstone national park Intl J Syst Evol Micr 51:481–488. doi:10.1099/00207713-51-2-481
Bhatnagar S, Badger JH, Madupu R, Khouri HM, O’Connor EM, Robb FT, et al. (2015) Genome sequence of a sulfate-reducing thermophilic bacterium, Thermodesulfobacterium commune DSM 2178T (phylum Thermodesulfobacteria) Genome Announc 3:e01490–e01414. doi:10.1128/genomeA01490-14
Pham VD, Hnatow LL, Zhang S, Fallon RD, Jackson SC, Tomb JF, et al. (2009) Characterizing microbial diversity in production water from an Alaskan mesothermic petroleum reservoir with two independent molecular methods Environ. Microbiol. 11:176–187. doi:10.1111/j1462-2920200801751x
Dong Y, Kumar CG, Chia N, Kim PJ, Miller PA, Price ND, et al. (2014) Halomonas sulfidaeris - dominated microbial community inhabits a 18 km-deep subsurface Cambrian sandstone reservoir Environ. Microbiol. 16:1695–1708. doi:10.1111/1462–292012325
Davidson MM, Silver BJ, Onstott TC, Moser DP, Gihring TH, Pratt LM, et al. (2011) Capture of planktonic microbial diversity in fractures by long-term monitoring of flowing boreholes, Evander Basin, South Africa Geomicrobiol J. 28:275–300. doi:10.1080/014904512010499928
Aüllo T, Ranchou-Peyruse A, Ollivier B, Magot M (2013) Desulfotomaculum spp and related gram-positive sulfate-reducing bacteria in deep subsurface environments Front. Microbiol. 4:362. doi:10.3389/fmicb201300362
Mladenovska Z, Mathrani IM (1995) Ahring, BK Isolation and characterization of Caldicellulosiruptor lactoaceticus sp nov, an extremely thermophilic, cellulolytic, anaerobic bacterium. Arch. Microbiol. 163: 223–230. doi:10.1007/BF00305357
Huang CY, Patel BK, Mah RA, Baresi L (1998) Caldicellulosiruptor owensensis sp nov, an anaerobic, extremely thermophilic, xylanolytic bacterium Intl J Syst Evol Micr 48:91–97
Miroshnichenko ML, Kublanov IV, Kostrikina NA, Tourova TP, Kolganova TV, Birkeland NK, et al. (2008) Caldicellulosiruptor kronotskyensis sp. nov. and Caldicellulosiruptor hydrothermalis sp. nov., two extremely thermophilic, cellulolytic, anaerobic bacteria from Kamchatka thermal springs Intl J Syst Evol Micr 58:1492–1496. doi:10.1099/ijs065236-0
Widdel F (2006) The Genus Desulfotomaculum. In: Dworkin M, Falkow S, Rosenberg E (eds) The Prokaryotes. Springer, New York, pp. 787–794
Nazina TN, Ivanova AE, Kanchaveli LP, Rozanova EP (1989) A new sporeforming thermophilic methylotrophic sulfate-reducing bacterium, Desulfotomaculum kutznetsovii sp. nov Mikrobiologiya (Translated) 57:823–827
O’Sullivan LA, Roussel EG, Weightman AJ, Webster G, Hubert CRJ, Bell E (2015) Survival of Desulfotomaculum spores form estuarine sediments after serial autoclaving and hith-temperature exposure ISME J 9:922–933
Zillig W, Holz I, Janekovic D, Schifer W, Reiter WD (1983) The archaebacterium Thermococcus celer represents, a novel genus within the thermophilic branch of the archaebacteria System Appl Microbiol 4:88–94
Huber H, Huber R, Steter KO (2006) Thermoproteales. In: Dworkin M, Falkow S, Rosenberg E (eds) The Prokaryotes. Springer, New York, pp. 10–22
Hartzell P, Reed DW (2006) The Genus Archaeoglobus. In: Dworkin M, Falkow S, Rosenberg E (eds) The Prokaryotes. Springer, New York, pp. 82–100
Palleroni NJ, Bradbury JF (1993) Stenotrophomonas, a new bacterial genus for Xanthomonas maltophilia (Hugh 1980) Swings et al 1983 Intl J Syst Evol Micr 43:606–609. doi:10.1099/00207713–43–3-606
Borsodi AK, Szirányi B, Krett G, Márialigeti K, Janurik E, Pekár F (2016) Changes in the water quality and bacterial community composition of an alkaline and saline oxbow lake used for temporary reservoir of geothermal waters Environ. Sci. Pollut. Res. 23:17676. doi:10.1007/s11356-016-6923-7
Cheng L, Shi S, Li Q, Chen J, Zhang H, Lu Y (2014) Progressive degradation of crude oil n-Alkanes coupled to methane production under mesophilic and thermophilic conditions PLoS ONE 9:e113253. doi:10.1371/journalpone0113253
Fang HHP, Liang DW, Zhang T, Liu Y (2006) Anaerobic treatment of phenol in wastewater under thermophilic condition Water Res. 40:427–434. doi:10.1016/jwatres200511025
Lladser ME, Gouet R, Reeder J (2011) Extrapolation of Urn models via poissonization: accurate measurements of the microbial unknown PLoS One 6:e21105. doi:10.1371/journalpone0021105
Inskeep WP, Rusch DB, Jay ZJ, Herrgard MJ, Kozubal MA, Richardson TH, et al. (2010) Metagenomes from high-temperature chemotrophic systems reveal geochemical controls on microbial community structure and function PLoS One 5:e9773. doi:10.1371/journalpone0009773
Sharp CE, Brady AL, Sharp GH, Grasby SE, Stott MB, Dunfield PF (2014) Humboldt’s spa: microbial diversity is controlled by temperature in geothermal environments ISME J 8:1166–1174
Lau CY, Jing HM, Aitchison JC, Pointing SB (2006) Highly diverse community structure in a remote central Tibetan geothermal spring does not display monotonic variation to thermal stress FEMS Microbiol. Ecol. 57(1):80–91. doi:10.1111/j1574-6941200600104x
Purcell D, Sompong U, Lau CY, Barraclough TG, Peerapornpisal Y, Pointing SB (2007) The effects of temperature, pH and sulphide on the community structure of Hyperthermophilic streamers in Hot Springs of northern Thailand FEMS Microbiol. Ecol. 60(3):456–466. doi:10.1111/j1574-6941200700302x
Wang S, Weiguo H, Hailiang D, Hongchen J, Liuqin H, Geng W, Chuanlun Z, et al. (2013) Control of temperature on microbial community structure in Hot Springs of the Tibetan plateau PLoS One 8(5):e62901. doi:10.1371/journalpone0062901
Meyer-Dombard DR, Amend JP (2014) Geochemistry and microbial ecology in alkaline hot springs of Ambitle Island, Papua New Guinea Extremophiles 18:763. doi:10.1007/s00792-014-0657-6
Headd B, Engel AS (2014) Biogeographic congruency among bacterial communities from terrestrial sulfidic springs Front. Microbiol. 5:473. doi:10.3389/fmicb201400473
Lozupone CA, Hamady M, Kelley ST, Knight R (2007) Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities Appl. Environ. Microbiol. 73:1576–1585. doi:10.1128/AEM01996-06
Bomberg M, Lamminmäki T, Itävaara M (2016) Microbial communities and their predicted metabolic characteristics in deep fracture groundwaters of the crystalline bedrock of Olkiluoto, Finland Biogeosciences 13:6031–6047. doi:10.5194/bg-13-6031-2016
Kamagata Y, Kawasaki H, Oyaizu H, Nakamura K, Mikami E, Endo G, Koga Y, Yamasato K (1992) Characterization of three thermophilic strains of Methanothrix (“Methanosaeta”) thermophila sp nov and rejection of Methanothrix (“Methanosaeta”) thermoacetophila Int. J. Syst. Bacteriol. 42:463–468
Wasserfallen A, Nölling J, Pfister P, Reeve J, Conway de Macario E (2000) Phylogenetic analysis of 18 thermophilic Methanobacterium isolates supports the proposals to create a new genus, Methanothermobacter gen. nov., and to reclassify several isolates in three species, Methanothermobacter thermautotrophicus comb. nov., Methanothermobacter wolfeii comb. nov., and Methanothermobacter marburgensis sp. nov Intl J Syst Evol Mic 50:43–53
Oremland RS, Polcin S (1982) Methanogenesis and sulfate reduction: competitive and noncompetitive substrates in estuarine sediments Appl. Environ. Microbiol. 44:1270–1276
Berg IA (2011) Ecological aspects of the distribution of different autotrophic CO2 fixation pathways Appl. Environ. Microbiol. 77:1925–1936
Campbell BJ, Cary C (2004) Abundance of reverse tricarboxylic acid cycle genes in free-living microorganisms at deep-sea hydrothermal vents Appl. Environ. Microbiol. 70:6282–6289. doi:10.1128/AEM70106282-62892014
Magnabosco C, Ryan K, Lau MC, Kuloyo O, Lollar BS, Kieft TL, van Heerden E, Onstott TC (2016) A metagenomic window into carbon metabolism at 3 km depth in Precambrian continental crust ISME J 10:730–741
Acknowledgements
This work was partially supported by the EnviroAMR project (grant no. 3499/20.05.2015, financed through the EEA 2009-2014 Financial Mechanism) through the EnviroBioInfo bioinformatic center. CMC and ES were also supported by the POSDRU/187/1.5/S/155383 research scholarship programme. All the samples were collected with the approval of S.C. TRANSGEX S.A. company. The authors wish to thank Dr. Codruta Bendea (University of Oradea) and Sanda Sferle (TRANSGEX) for their valuable support towards obtaining the water samples.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
ESM 1
(Zip 564 kb)
Rights and permissions
About this article

Cite this article
Chiriac, C.M., Baricz, A., Szekeres, E. et al. Microbial Composition and Diversity Patterns in Deep Hyperthermal Aquifers from the Western Plain of Romania. Microb Ecol 75, 38–51 (2018). https://doi.org/10.1007/s00248-017-1031-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-017-1031-x