
Abstract
Mounting evidence suggests that Archaea are widespread and abundant in aquatic and terrestrial habitats and play fundamental roles in global biogeochemical cycles, yet the pattern and its ecological drivers of biogeographic distribution of archaeal community in estuarine ecosystem are still not well understood. Here, we investigated planktonic and benthic archaeal communities in the human-impacted Jiulong River estuary (JRE), southern China by using real-time PCR (RT-PCR) and Illumina 16S ribosomal RNA (rRNA) amplicon sequencing. RT-PCR analysis indicated that Archaea accounted for an average of 0.79 and 5.31 % of prokaryotic biomass in water and sediment samples of the JRE, respectively. The diversity of planktonic archaeal community decreased gradually from the river runoff to seawater, whereas that of benthic community did not show the similar pattern. The results of taxonomic assignments indicated that Thaumarchaeota (Nitrosopumilus and Cenarchaeum), Methanocorpusculum, and Methanospirillum were significantly more abundant in planktonic than benthic communities, whereas the relative abundances of Miscellaneous Crenarchaeotic Group, Marine Benthic Group-B/-D, anaerobic methane-oxidizing Archaea -1/-2D, and South Africa Gold Mine Euryarchaeotic Group 1 were higher in sediments than in surface waters. Moreover, planktonic archaeal community composition varied significantly at broad and finer-scale taxonomic levels along the salinity gradient. Multivariate statistical analyses revealed that salinity is the main factor structuring the JRE planktonic but not benthic archaeal community at both total community and population level. SourceTrakcer analysis indicated that river might be a major source of archaea in the freshwater zone of the JRE. Overall, this study advances our understanding of the biogeographic patterns and its ecological drivers of estuarine archaeal communities.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The prokaryotes including Bacteria and Archaea, are the most abundant organisms with versatile ecological functions inhabiting the aquatic ecosystem and play the primary roles in recycling organic matters and nutrients as well as microbial food web [1]. Previous studies have investigated the diversity, composition, and structure of bacterial communities as well as their ecological function in estuarine environment as estuaries provide ideal environments for performing comprehensive studies in microbial ecology along sharp physicochemical gradients (e.g., salinity, pH, oxygen, nutrient, dissolved, and particulate organic matter) [2–5]. They primarily found that salinity is the major determinant of not only drastic changes in bacterial community composition but also of succession in metabolic characteristics of bacterial community in the estuarine area [2–4]. However, there is limited information on the abundance, diversity, and biogeographic distribution of Archaea in estuarine ecosystem despite their potential importance in biogeochemical cycling highlighted in recent years [6, 7].
The Archaea domain mainly consists of three phyla, Crenarchaeota, Euryarchaeota, and Thaumarchaeota, and the latter was previously known as “mesophilic Crenarchaeota” and was recently assigned as an independent archaeal phylum based on comparative genomic analysis [8]. Although Archaea were only found to be present in harsh environments during the early studies, subsequent molecular studies have provided mounting evidence that members of the Archaea are widespread in diverse natural environments, including ocean, soil, and freshwater ecosystems, and majority of them were distantly affiliated to the cultivated strains and resistant to culture [9]. The quantitative analyses have demonstrated that Archaea contribute up to 20 and 5 % of the total prokaryotic abundance in sea water and soils, respectively [10–12], and may represent 5–30 % of the total microbial biomass in marine surface sediments [13]. Meanwhile, there is growing evidence that some functional groups of Archaea, such as ammonia-oxidizing Archaea (AOA; phylum Thaumarchaeota) and methanogenic and anaerobic methane-oxidizing Archaea (ANME; phylum Euryarchaeota), play crucial roles in carbon and nitrogen biogeochemical cycles [7].
Given the considerable abundance of Archaea in natural environments and their significance to global biogeochemical cycles [6, 7], a detailed analysis of abundance, diversity, and composition of estuarine archaeal community is essential to gain an in-depth understanding on their ecological function. However, compared with extensive studies on marine and soil archaeal communities, there were only a few investigations concerning the assembly of archaeal community in several estuaries [14–18]. The available studies indicated that estuarine environments harbored multiple environmental sources of archaeal lineages or phylotypes which may be due to the combined influences of river and groundwater discharge, coastal erosion, seawater intrusions, and resuspension events in estuarine system [14, 16–18]. For example, marine group I Thaumarchaeota (MGI; also named as group 1.1a) and marine group II Euryarchaeota (MGII) dominated in planktonic archaeal communities of the marine-influenced region of the estuary, while estuarine freshwater zone often contained highly diverse archaeal assemblages associated with freshwater, soil, and sediment environments [16–18]. Besides, some specific benthic archaeal lineages that included MGI, Miscellaneous Crenarchaeotic Group (MCG), Marine Benthic Groups A, B, and D (MBGA, B, and D), methanogens, ANME, etc., thrived in the estuarine sediments [15, 19]. The sharp change in archaeal community composition and structure along the salinity gradient in different estuaries suggest that salinity also play an important role in shaping spatial distribution of estuarine archaeal community [14–18]. However, low-resolution molecular methods used by most previous studies confine our understanding on biogeographic pattern of estuarine archaeal community and its critical ecological drivers [14, 15]. Moreover, none of the studies have simultaneously investigated planktonic and benthic archaeal communities in an estuary.
In the current study, we aim to unravel the biogeographic distribution pattern of archaeal communities in water and sediments of the Jiulong River estuary (JRE), southern China, using an integrated approach including environmental parameter measurement, real-time PCR (RT-PCR), Illumina 16S ribosomal RNA (rRNA) amplicon sequencing, and multivariate statistical analysis. The JRE is profoundly influenced by its watershed—the Jiulong River Watershed which has a catchment area of 1.47 × 104 km2 [20–22]. A previous nutrient study of the JRE conducted by Yan et al. [20] estimated that there was 34.3 × 103 ton dissolved inorganic nitrogen (DIN) year−1, 0.63 × 103 ton soluble reactive phosphorus (SRP) year−1, and 72.7 × 103 ton dissolved silicate (DSi) year−1 discharged from watershed into the JRE, which lead to eutrophication and algal blooms in the JRE and the adjacent Xiamen Sea. The primary objectives of this study were (i) to investigate the abundance, diversity, and composition of both planktonic and benthic archaeal communities in the JRE and (ii) to identify the critical factor (local or regional factors) influencing biogeographic pattern of the JRE archaeal community at both community and population level. The possible environmental sources of planktonic archaeal communities in the JRE were also discussed based on SourceTracker analysis [23].
Materials and Methods
Study Area
The JRE, situated in a subtropical monsoon zone, is a shallow estuary (3–16 m in depth) of the northern South China Sea, with a total length of approximately 21 km and a catchment area of 100 km2 [20, 22] (Fig. 1). The JRE is strongly influenced by semidiurnal tidal cycles with a tidal range of 2.7–4 m from the upper to the lower estuary. The estuarine water comes from the Jiulong River, which consists of two major tributaries, North River and West River, and flows into the sea mainly through the south bank [20, 22]. The flushing time of river water in the JRE varied from 2 to 3 days [24]. The total suspended matter concentration is highest in the freshwater area and decreases quickly in the low salinity area [25]. The coastlines of the lower salinity part of the JRE are boarded by mangroves. Due to rapid economic development in this area in the past 30 years, intensive human activities such as agricultural practices, livestock breeding, and urbanization have great impacts on both river and estuary nutrient biogeochemistry [21]. Recently, Yan et al. [20] have found the concentrations of NO3–N and SRP increased two to three times in the upper/middle areas of the JRE since the 1990s, which is the major cause resulting in eutrophication and algal blooms in the JRE and the adjacent Xiamen Bay.

Location of sampling stations in the JRE. Water samples were collected from all stations, and sediment samples were also collected from six of them (gray circles)
Sample Collection and Environmental Parameter Analysis
Sampling was carried out during early April, 2011 under normal hydrological condition (relative to wet or dry hydrological conditions, river discharge is under its average condition) [20]. As shown in Fig. 1, a total of 12 sampling sites were selected along the south bank from river to sea. Sites A3–A6 were strongly influenced by the freshwater plume from the Jiulong River, while sites JY0, JY1, JY3, and KM4 were subjected to the influence of coastal current. In addition, human activities of Xiamen area have an impact on the quality of water in site W. Surface water (∼0.5 m) samples were collected using 5-L Niskin bottles (model QCCC-5, National Ocean Technology Center, China). For molecular analysis, about 500 to 1000 mL water samples were pre-filtered through 20 μm mesh (Millipore, Billerica, MA, USA) and subsequently filtered onto 0.22-μm pore-size polycarbonate filters (47 mm, Millipore, USA), while sediments were collected from six sites (Fig. 1). All samples were stored at −80 °C until analysis.
Surface water temperature and salinity were determined in situ using a YSI-6600 probe (YSI, Yellow Springs, Ohio, USA). The concentration of dissolved oxygen (DO) in the water was measured using the standard Winkler titration method [26]. pH was determined in situ using a precision pH analyzer (Orion Star™, Thermo Electron Co., USA). Water samples for nutrient analysis (NH4–N, NO2–N, NO3–N, SRP, and DSi) were filtered through 0.45-μm pore-size cellulose acetate filters (Millipore, USA) and analyzed using an auto-analyzer. The detailed information can be found in a previous study [20].
Sediment samples were dried in a freeze-drier (Labconco, Kansas City, MO, USA). Total organic carbon (OrgC), organic nitrogen (OrgN), and organic sulfur (OrgS) were analyzed using a Vario Max CNS analyzer (Elementar, Hanau, Germany). pH was measured at a 1:2.5 sediment-to-water ratio (w/vol). Inorganic nitrogen was extracted from 2 g dried sample with 2 M KCl, and the concentrations of NH4–N, NO2–N, and NO3–N were determined using Lachat QC8500 Flow Injection Auto-analyzer (Lachat Instruments, Loveland, CO, USA) as described elsewhere [21].
DNA Extraction and Real-Time PCR Assay
DNA from environmental samples was extracted in duplicate following protocols established in our lab [21]. For quantification of bacterial and archaeal 16S rRNA genes, RT-PCR reactions were performed in triplicates using an Applied Biosystems 7500 Real-Time PCR system (Life Technologies, Forster City, CA, USA) with SYBR green method [27]. The abundance of bacterial and archaeal 16S rRNA genes were determined using Bacteria-specific primers 341F/518R [28] and Archaea-specific primers A364aF/A934b [29], respectively. Melting curve analysis and gel electrophoresis were performed after each run to confirm the specificity of RT-PCR.
Illumina 16S rRNA Amplicon Sequencing
The hypervariable V6 region of the archaeal 16S rRNA gene was amplified using the primer pair: Illumina sequencing adaptor-linker (AG)-8 bp barcode-958F (5′-AAT TGG ANT CAA CGC CGG-3′) and an equal mixture of 1048R major (5′-CGR CGG CCA TGC ACC WC-3′) and 1048R minor (5′-CGR CRG CCA TGY ACC WC-3′) [30]. Each sample was amplified in triplicate, pooled, and purified using the MinElute Gel Extraction Kit (Qiagen, Valencia, CA, USA). The concentration of amplicon was measured in duplicate using Quant-iT dsDNA HS assay kit (Life Technologies, Carlsbad, USA). All amplicons were mixed in equimolar concentration and sent to the Beijing Genomics Institute (Shenzhen, China). Paired-end sequencing (2 × 91 bp) was performed on a HiSeq 2000 device (Illumina Inc., San Diego, CA, USA). The raw sequence data were deposited into the NCBI short reads archive (SRA) database under accession number SRP029700.
Processing of Illumina Sequencing Data
The raw paired-end reads were assembled following the barcoded Illumina PE sequencing (BIPES) pipeline to improve the sequencing accuracy of the reads [31], and then the clean data were analyzed using Mothur v1.31 [32] and QIIME v1.7.0 [33]. Briefly, the reads shorter than 50 bp or longer than 90 bp were discarded, and any reads containing ambiguous bases (N), incorrect barcode, or primer sequences were excluded from further analysis. The potential sequencing errors were corrected using Mothur command “Pre.cluster” allowing 1 bp difference between sequences. The chimeras were removed using the UCHIME algorithm [34]. The remaining high-quality reads were clustered into operational taxonomic units (OTUs) at 98 % identity using a UCLUST-based [35] open-reference OTU picking pipeline using Greengenes v13_08 as reference database. Singletons were discarded in order to improve the quality of sequence analysis as recommended by a previous study [36]. Finally, the high-quality sequences were classified to taxonomic rank using RDP classifier with a 50 % bootstrap confidence threshold. Taxonomy was also assigned to each representative sequence using the Global Alignment for Sequence Taxonomy (GAST) algorithm [30] retrained on the Greengenes v13_08 database. Indicator taxa were identified within each sample category using LEfSe software [37].
To identify putative AOA occurrence in the JRE, 16S rRNA sequences of 20 thaumarchaeal isolates belonging to Nitrosopumilus, Nitrosotalea, Nitrososphaera, and Nitrosocaldus lineages were retrieved from GenBank (as at 30th April 2014) (Fig. S1) [38–47]. The sequences were aligned with the CLUSTALW, and a neighbor-joining tree was constructed using MEGA 6.05 [48]. Tree topology was evaluated by 1000 bootstrap resamplings. The representative sequence of each OTU was assigned to thaumarchaeal lineages by comparing it with sequences of thaumarchaeal isolates using the BLASTN algorithm with an e value of 1 × 10−10, at least 98 % identity and 100 % coverage.
The newly developed SourceTracker software, which employs a Bayesian approach to identify the potential sources and proportions of microorganism to a sink community, has recently been applied in microbial surveys [23]. In the present study, potential sources we examined included global soil archaeal communities from 146 soils obtained from North and South America and Antarctica [49] and the planktonic and benthic archaeal communities sampled from the Jiulong River during 16th–19th October 2011 [21] (Fig. S2) (our unpublished data, SRA accession number SRX527142). A OTU table combined with source communities (soil, river planktonic, and benthic Archaea) and sink communities (JRE Archaea) was used as input for analysis by the SourceTracker algorithm.
Statistical Analysis
Principal components analysis (PCA) was used to display main gradients in the physicochemical variables. The pattern of archaeal community structures was analyzed by principal coordinate analysis (PCoA) based on unweighted and weighted UniFrac distance matrices using QIIME v1.7.0 [33]. The permutational multivariate analysis of variance (PERMANOVA) analysis was used to test the significant difference between bacterial community compositions from different environmental categories of samples [50]. The Mantel test was performed to determine the relationship between environmental variables (e.g., geographic distance and physicochemical parameters) and archaeal community structures, whereas a partial Mantel test examines the correlation between local environmental variables and archaeal community structures controlling for the effects of geographic distance or salinity. Prior to performing multivariate statistical analyses, highly correlated environmental variables were removed if the variance inflation factor for independent variables is more than 10 [21]. The redundancy analysis (RDA) and partial RDA were used to distinguish the effects of space (dispersal) and environmental factors (environmental filtering) on archaeal community variation in the JRE. For RDA analysis, the spatial structure was represented by a set of spatial variables generated using the principal coordinates of the neighbor matrices algorithm (PCNM) approach [51, 52]. A forward selection with a double stopping criterion was performed to select significant environmental and spatial variables [53]. The significance of RDA models was determined with 999 Monte Carlo permutations, and the adjusted R 2 value of RDA models was calculated using “RsquareAdj” in the package vegan [54]. Multivariate statistical analyses were performed using R v2.14 with the packages vegan [54], PCNM [52], and packfor [53].
Results
Characterization of Environmental Variables
The main physicochemical parameters of water and sediment samples collected from the JRE are summarized in Tables S1 and S2 in the Supplemental Materials, respectively. For surface water samples, temperature, salinity, DO, and pH ranged from 14.41 to 18.44 °C, 0.14 to 30.98 ‰, 6.12 to 8.83 mg L−1, and 7.16–8.02, respectively (Dr. Weidong Zhai, unpublished data). The nutrient concentrations including NH4–N, NO3–N, NO2–N, SRP, and DSi were highest at the freshwater site and decreased along the salinity gradient (Table S1), indicating anthropogenic input from the Jiulong River. PCA analysis indicated that salinity was highly correlated with other physicochemical variables, and the axis 1 explained 95.5 % of the total variation among the environmental variables (Fig. S3). The surface water samples of the JRE could be divided into three groups, freshwater-oligosaline (salinity, 0.14–1.53 ‰), mesosaline (8.38–15.53 ‰), and polysaline groups (22.7–30.98 ‰), on the basis of salinity (Fig. S3). For sediment samples, sediment pH ranges from 6.92 to 8.05. The sediment from freshwater zone (A4s and A5s) contained the highest concentration of NH4–N and dissolved inorganic nitrogen (DIN), also demonstrating serious nutrient loading to the JRE. The proportions of OrgC, OrgN, and OrgS ranged from 0.35 to 1.28 %, 0.04 to 0.14 %, and 0.12 to 0.93 % respectively, while the sediment OrgC/OrgN ratios were constant and ranged from 10.46 to 12.65.
Abundance of Bacterial and Archaeal 16S rRNA Genes
RT-PCR results demonstrated that the abundance of bacterial and archaeal 16S rRNA genes in surface waters of the JRE ranged from 1.16 × 107 to 2.70 × 108 and 5.38 × 104 to 8.29 × 105 copies ng DNA−1, respectively (Fig. 2). In the sediment samples, the abundance of bacterial 16S rRNA genes compared with those occurring in waters of the corresponding stations, ranged from 6.12 × 108 to 1.44 × 108 copies ng DNA−1, whereas archaeal 16S rRNA gene abundances were significantly higher than those in water samples (Wilcoxon test, P < 0.05) (Fig. 2). Statistical analysis indicated that the abundance of archaeal 16S rRNA genes were significantly lower than those of bacteria (Wilcoxon test, P < 0.001), and relative abundances of archaeal 16S rRNA gene to total archaeal and bacterial 16S rRNA genes were averaged at 0.79 and 5.31 % in JRE water and sediment samples, respectively.

Abundance of bacterial and archaeal 16S rRNA genes in the water and sediment samples obtained from the JRE. The numbers in parentheses after the site name indicated salinity of surface water of the JRE
α-Diversity of the JRE Archaeal Communities
To identify the diversity and composition of archaeal communities in the JRE, 12 water and 6 sediment samples were collected from the JRE along the salinity gradient for Illumina 16S rRNA amplicon sequencing (Fig. 1), and a total of 8,522,645 high-quality reads were obtained. After randomly resampling 300,000 reads per sample, 23,803 OTUs were clustered at the 98 % identity level using open-reference picking OTU approach (Table S3). The majority of OTUs (96.9 %) did not match representative sequences of the Greengene v13_08 database (OTUs fail to match the reference if they exhibit <98 % sequence identity with representative sequences), although singletons were discarded to insure analysis quality. α-diversity indices were also calculated on the basis of 300,000 sequences randomly resampled from each sample (Table S3). The Shannon index of planktonic archaeal communities ranged from 2.45 to 5.25 and decreased gradually from the river runoff to seawater (Fig. 3a), whereas benthic archaeal community did not show a similar pattern (Fig. 3b). This pattern was also shown by other α-diversity indices (Table S3). Statistical analysis indicated that there was no significant difference between diversity, richness, and evenness of planktonic and benthic archaeal communities (Wilcoxon test, P > 0.05).

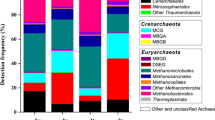
Shannon index of planktonic (a) and benthic (b) archaeal community and the taxonomic composition of planktonic (c) and benthic (d) archaeal community in the JRE. Minor phyla accounting for <0.5 % of total sequences and unclassified Archaea are summarized in the group “Other and unclassified Archaea.” The arrows under the site names represent an increase in surface water salinity
Composition of the JRE Archaeal Communities
In order to guarantee the accuracy of taxonomic identification, the RDP and GAST classification approaches, which were based on the Bayesian and “nearest-neighbor” algorithm respectively, were applied in this study. Generally, the taxonomic results at all classification levels based on RDP Classifier were generally similar to those generated by GAST algorithm (Fig. S4) (Spearman correlation analysis, r > 0.68, P < 0.05). Hence, only the results of RDP taxonomic assignment were demonstrated in the following text. In general, the majority of high-quality reads obtained from the JRE could be assigned into three archaeal phyla, i.e., Thaumarchaeota, Crenarchaeota, and Euryarchaeota (Fig. 3c, d). The planktonic archaeal community composition shifted along the salinity gradient. For example, MCG and methanogenic Archaea (Methanomicrobiales, Methanosarcinales, Methanocellales, Methanobacteriales, etc.) were dominant phylotypes or lineages in freshwater-oligosaline sites, whereas the relative abundances of Cenarchaeales (most belong to MGI) and Thermoplasmata (most belong to MGII) were high in polysaline waters (Fig. 3c). The benthic archaeal community composition was significantly different from that of archaeaplankton communities (PERMANOVA, pseudo-F = 3.001, P = 0.009). Several typical benthic archaeal lineages, such as MCG (55.19 % of all sequences from sediments), MBGB (also known as Deep Sea Archaeal Group (DSAG); 5.00 %), MBGD (also named as Deep Sea Hydrothermal Vent Euryarchaeal Group 1 (DHVEG-1); 7.91 %), and ANME-1 (1.39 %) were found abundant in sediment samples (Fig. 3d). Moreover, a concordant taxonomic composition pattern was obtained based on Silva 111 rRNA database [55] (Fig. S5).
Archaeal Community Structure and Indicator Taxa
UniFrac-based PCoA analysis was used to reveal the distribution pattern of the JRE archaeal community structure. Consistent with the results of taxonomic composition analysis, PCoA analysis demonstrated that, generally, the JRE archaeal communities formed two groups based on habitat types, water and sediment (Fig. 4a), although 40.5 % of OTUs were shared between them (Fig. S6), suggesting that water and sediments of the JRE harbored unique archaeal taxa. Moreover, while the PC1 axis separated archaeaplankton communities of freshwater-oligosaline and mesosaline sites from planktonic polysaline communities (Fig. 4a), the sediment samples obtained from freshwater-oligosaline and mesosaline-polysaline sites were separated along the third principal coordinate axis (Fig. 4b). In addition, PCoA analyses of three dominant archaeal lineages (MCG, Thermoplamata and Thaumarchaeota) revealed that their population structures also shifted along the salinity gradient (Fig. 4c–e), implying again that salinity might be the most important factor influencing the distribution of the JRE archaeal communities.

PCoA analysis of community structures of total Archaea (a), MCG (Crenarchaeota) (b), Thermoplasmata (Euryarchaeota) (c), and Thaumarchaeota (d) in the JRE using unweighted UniFrac distances. Upper triangles and squares indicated planktonic and benthic communities, respectively. Figures colored with green indicate the freshwater-oligosaline sites, orange indicate the mesosaline sites, and blue indicate the polysaline sites. All sediment samples were removed from analysis of thaumarchaeal population structure because few sequences were obtained from those samples
To identify indicator taxa associated with different habitat types, i.e., (1) freshwater-oligosaline vs polysaline environments (sediment samples were excluded since sediment sample sizes were not large enough to perform comparative analysis.) and (2) water vs sediment, we used LEFSe software to determine taxa which significantly enriched in each habitat type. Across all taxonomic levels, 43 and 23 indicators were identified for freshwater-oligosaline and polysaline groups, respectively (Fig. 5). Some lineages belonged to Crenarchaeota such as MCG, B10 (MCG), pGrfC26 (MCG), and Terrestrial Hot Springs Crenarchaeotic Group (THSCG) and Euryarchaeota, Class Methanobacteria, Methanococci, and Methanomicrobia, and MBGD, were significantly higher in freshwater-oligosaline waters, while marine Thaumarchaeota and Euryarchaeota groups, Cenarchaeum (Thaumarchaeota), Nitrosopumilus (Thaumarchaeota), and MGII were identified as potential biomarkers for polysaline waters. Meanwhile, we observed that terrestrial Thaumarchaeota lineages including AK31 and Nitrososphaera were enriched in the freshwater-oligosaline group.

Indicator archaeal taxa were identified as significantly different abundance between freshwater-oligosaline and polysaline communities using LEfSe analysis
In comparison with water and sediment samples, 7 and 16 archaeal lineages were determined as potential biomarkers for water and sediment groups, respectively (Fig. S7). Typical benthic or terrestrial archaeal lineages such as MCG, MBGB, MBGD, ANME-1, ANME-2D, and South Africa gold mine euryarchaeotic group (SAGMEG)-1 were significantly more abundant in benthic than planktonic communities, whereas the relative abundances of Thaumarchaeota, Nitrosopumilus, Cenarchaeum, Methanocorpusculum, and Methanospirillum were higher in surface waters than in sediments.
Distribution of Putative AOA in the JRE
To explore the distribution of putative AOA in the JRE, we retrieved 20 nearly full-length 16S rRNA sequence of AOA isolates from GenBank. A phylogenetic tree of AOA isolates was constructed with the sequences of the V6 region of 16S rRNA gene. The result demonstrated that V6 region of archaeal 16S rRNA gene distinguish between different AOA lineages (Nitrosopumilus (MGI), Nitrosotalea (SAGMGC-1), Nitrososphaera (Soil Thaumarchaeota group 1.1b), and Nitrosocaldus lineages) very well (Fig. S1). The pairwise sequence comparison indicated that different AOA lineages shared <98 % identity of V6 region sequence except for Nitrosotenuis uzonensis and Nitrosoterreus chungbukensis MY2 (Table S4). Although these two species formed a unique cluster close to Nitrosopumilus lineage, they shared 98.4 % identity with Nitrosopumilus species. Hence, the reads that within a 98 % identity of the sequences of known AOA isolates were considered as putative AOA lineages. Then, a pairwise sequence comparison was performed to ensure whether the sequences belong to Nitrosopumilus lineage. Overall, 25 of 1402 thaumarchaeal OTUs, which represented about 74.77 % of total thaumarchaeal population, were identified as putative AOA. Of them, 13 OTUs were classified into Nitrosopumilus lineage, while three and nine OTUs had ≥98 % identity with the sequences of Nitrosotalea and Nitrososphaera lineages, respectively. The distribution of putative AOA OTUs in water and sediment samples of the JRE was shown in Fig. 6. It is clear that, with a few exceptions, the relative abundance of Nitrosopumilus-like OTUs in water samples decreased with the increased salinity, whereas Nitrosotalea- and Nitrososphaera-like OTUs show an opposite trend. In sediment samples, moreover, Nitrosotalea- and Nitrososphaera-like OTUs had higher relative abundance than that of Nitrosopumilus-like OTUs, but all of them did not have a salinity-related distribution pattern.

A heatmap of the distribution of putative AOA in water and sediments of the JRE. The sequence number of each OTU was transformed into Z-score. The arrows represent an increase in surface water salinity
Linking Environmental and Spatial Factors to Archaeal Community Structure
Mantel test analysis generally supported the results of PCoA analyses, indicating that the community structures of planktonic Archaea, MCG, Thermoplasmata, and Thaumarchaeota significantly correlated with salinity (r ≥ 0.618, P < 0.001) (Table 1). However, sediment NH4–N but not salinity was found to significantly correlated with benthic archaeal and MCG community structures (r ≥ 0.731, P < 0.05), and no environmental factors have shown a correlation with MBGB and MBGD population structures (Table 1). Although Mantel test analyses demonstrated that there were significant correlation between geographic distance and planktonic community structures (r ≥ 0.506, P < 0.01), the correlation disappeared when controlling for the effect of salinity except for planktonic Archaea, as shown by partial Mantel tests (Table 1). Moreover, geographic distance did not have correlation with benthic archaeal community structures (Table 1). Furthermore, consistent with the results of partial Mantel test analyses, RDA analyses indicated that the selected environmental factors explained significant variance (adjust R 2 ranged from 0.262 to 0.762) of both planktonic and benthic archaeal community structures except for benthic Archaea and MBGB, while none of PCNM variables were significant (P > 0.05) to include in the RDA model (Table 1).
Discussion
It is well known that Archaea are a persistent component in marine and soil environments [10, 49], but limited information is available regarding the archaeal diversity and distribution in freshwater and estuarine habitats. Well et al. [56] observed an elevated archaeal abundance in waters of the Makenzie River, which was correlated with particle concentration, suggesting that a portion of archaeal cells in coastal waters of Beaufort Sea could be from the river. A large number of particle matters are also discharged from the Jiulong River to the JRE, and the concentration decreases rapidly along the estuarine salinity profile [25]. However, the abundance of archaeal 16S rRNA gene in JRE waters did not vary significantly across the estuary, while we found that the archaeal 16S rRNA gene abundance in sediments slightly decreases from freshwater to marine zone (Fig. 2). The latter result is in accordance with the investigation of Xie et al. [15], who observed that the highest abundance of archaeal 16S rRNA gene in the sediment of Pearl River was affected by anthropogenic pollution (high concentration of NH4–N in pore water). Nevertheless, our results are within the range of the quantitative estimations published elsewhere, which generally showed that Archaea are usually a minor component of a microbial assemblage in surface water or surface sediments [10, 13, 19, 56]. However, because different primers targeting different region of 16S rRNA gene were used for determining the abundance of bacterial (341F/518R) and archaeal (A364aF/A934b) 16S rRNA genes, differences in primer efficiency and amplicon length for these two primer pairs may cause some bias in estimating the relative abundance of Archaea in prokaryotic community. Thus, further study combined with multiple methods such as rRNA slot blot hybridization, catalyzed reporter deposition FISH, and RT-PCR is needed to provide more accurate results.
Despite that the abundance of planktonic archaeal 16S rRNA gene was constant across all sites we surveyed, α-diversity analysis indicated that the diversity (H′) and richness (Chao1) of planktonic archaeal communities decreased from freshwater zone to marine zone (Fig. 3a; Table S3), which well supports the earlier findings of the investigations on estuarine archaeaplankton [17, 18, 57]. For instance, Galand et al. have reported the highest archaeaplankton diversity in the Mackenzie river when comparing with archaeaplankton communities from the Beaufort Sea [17, 18], while a study of the spatial-temporal dynamics of archaeal communities in the Tomoe River Mouth and Suruga Bay found that the Shannon diversity of river archaeaplankton was threefold higher than that in coastal waters [57]. This finding may be due to the input of allochthonous archaeal populations from multiple sources including terrigenous-fluvial supply, mangrove input, and estuarine sediment suspension, which may lead to high diverse planktonic archaeal communities assembling in the freshwater zone of the JRE [17]. In fact, the various environmental origins of chromophoric dissolved organic matter in the low-salinity region of the JRE were validated by a previous study [22], thereby supporting our hypothesis. Another explanation for the observed diversity trend could be that a phylogenetically rich diversity of archaeal assemblage tended to occur in the heterogeneous freshwater habitats [58]; however, most of which were not reported for archaeal ecology. In contrast, α-diversity pattern of benthic archaeal communities of the JRE was clearly different from that of planktonic communities (Fig. 3b; Table S3), reflecting the influence of the complex properties of sediments [21]. However, we did not observe a relationship between α-diversity index of benthic communities and sediment environmental parameters, which may be owing to the limited number of sediment samples. More studies are required to reveal the real diversity pattern of the JRE benthic archaeal communities.
Taxonomic analysis indicated that the overall composition of planktonic archaeal communities changed significantly over the salinity gradient, as previously demonstrated for estuarine bacterioplankton [2–5, 59] and archaeaplankton communities [14, 16–18]. The prevalence of MGI and MGII in the marine zone of the JRE is concordant with previous observations, where it was found that MGI and MGII are ubiquitous and abundant in the water column of the world ocean [10, 27, 30, 60]. MGII are often discovered to be more abundant than MGI in oligotrophic oceanic surface waters [10], yet some studies surveying estuarine archaeal communities reported that MGI could be in the ascendant in coastal waters of estuaries [57, 61]. Strikingly, MCG and diverse methanogenic Archaea, which were frequently recovered from anaerobic habitats including marine and estuarine sediments, anaerobic wastewater reactors, landfill leachate, and a certain type of soils [62, 63], predominated in the freshwater zone of the JRE (Fig. 3c). High concentrations of particles suspended in freshwaters of the JRE may provide suitable anaerobic microniches for these populations [64]. However, owing to the short hydraulic residence time in the JRE (2–3 days), it seems not possible to develop an autochthonous planktonic archaeal communities in the freshwater region of this estuary, and therefore may represent allochthonous inputs of river populations [5]. Although this finding is unexpected, it is in line with the results of previous works which observed MCG and methanogens in oxygen-rich water of lakes [11, 64–66] and rivers [57, 67–69], suggesting that these two archaeal populations may have a more cosmopolitan distribution than conventional observations. Indeed, we also observed that MCG and some groups of methanogens are consistent components of archaeal communities in water and sediments of the Jiulong River (our unpublished data).
In order to evaluate the contribution of allochthonous archaeal communities to JRE planktonic archaeal communities, a SourceTracker analysis was performed (The detailed information is shown in the “Materials and Methods”) [23]. The results indicated that 4.83 to 58.53 % and 0 to 20.71 % of the sequences in JRE water samples may derive from water and sediments of the Jiulong River, respectively, with the relative contribution to JRE planktonic archaeal communities decreasing along the estuarine salinity gradient (Fig. 7), supporting our speculation (above) that Jiulong River might be a major source of archaea in freshwaters of the JRE. The little contribution of the soil archaeal communities might be ascribed to the fact that the relatively low sequencing effort (<1500 reads per sample) in the work of Bates et al. [49] may not unravel the full archaeal diversity in global soils. Previous studies have demonstrated that some typical soil archaeal lineages such as group 1.1b, which was also retrieved from JRE planktonic communities (Fig. 3c), have a patchy distribution in freshwater plankton [11, 14, 16, 67]. In addition, it is very likely that JRE sediments and surrounding mangroves are important sources of archaeaplankton in the freshwater zone of the JRE, because a high proportion of OTUs was found to share between planktonic and benthic archaeal communities (Fig. S6), and the indicator taxa for freshwater-oligosaline group were widely recovered from estuarine sediments and mangroves [15, 19, 70].

Results of SourceTracker analysis showing average relative contributions of indicator taxa from different potential sources to JRE planktonic archaeal communities. The numbers in parentheses after the site name indicated salinity of surface water of the JRE
Previous surveys based on the amoA gene, encoding the key enzyme—ammonia monooxygenase responsible for ammonia oxidation, demonstrated that AOA community composition and structure in estuaries are highly variable in spatial and temporal scales due to the steep environmental gradients [71–72]. Although we investigated JRE archaeal communities by analysis of 16S rRNA gene rather than the functional amoA gene, our results may provide some clues on biogeographic distribution of AOA in this estuary because many sequences (22.18 % of total sequences) were assigned into Thaumarchaeota (Fig. 3c, d), which were tentatively considered to be the principal members of AOA [6]. In addition, an increasing amount of cultured AOA strains [38–47] provide an opportunity for us to study the diversity and distribution of AOA in the JRE at finer taxonomic level (Fig. 6). Consistent spatial distribution of Thaumarchaeota in the JRE was found either at broad or finer taxonomic levels. The relative abundance of Nitrososphaerales or the OTUs affiliated to Nitrosotalea (SAGMAGC-1) and Nitrososphaera (group 1.1b) lineages in surface water of the JRE decreased with increasing salinity and nearly disappeared in polysaline water, while that of MGI or Nitrosopumilus-like OTUs showed the opposite trend across the salinity gradient (Figs. 3c and 6). It is reasonable because numerous studies have shown that MGI and group 1.1b are restrictedly distributed in marine and terrestrial environments, respectively [12], but accumulating evidences suggest that many MGI phylotypes are also widespread in soil and freshwater environments [11, 14, 49, 67].
It has been suggested that salinity is the major determining factor in structuring the estuarine bacterial communities although other abiotic and biotic factors such as DO, pH, nutrients, concentration and quality of organic matter, and phytoplankton community may have certain impacts on spatial distribution of bacterial communities in estuaries [2–5]. Similar to these studies, two independent studies have recently shown that spatial biogeography of planktonic and sediment archaeal communities in the Pearl River Estuary was mainly influenced by the salinity gradient [14, 15]. Analogously, multivariate statistical analyses including PCoA analysis, partial Mantel test and RDA analysis showed that salinity is the most significant environmental variable in shaping planktonic archaeal community structure in the JRE. This finding, together with analysis of taxonomic classification and SourceTracker discussed above, provided convergent evidence that salinity is a primary barrier in preventing the spread of freshwater and marine microorganisms to each other’s habitat [73]. The ultra high-throughput sequencing data presented here provides insights into understanding the mechanism governing estuarine archaeal community assembly not only at total community level but also at population level (Table 1; Fig. 4). Especially, to the best of our knowledge, our result provided for the first time evidence that population structure of planktonic MCG was controlled by salinity, whereas sediment NH4–N may have an effect on spatial distribution pattern of benthic MCG population in the JRE (Table 1). Kubo and colleagues have conducted a comprehensive analysis of biogeographical distribution of MCG in various types of marine sediments, in which they found that MCG are the dominant archaeal population in anoxic, low-activity subsurface sediments [62]. However, the factor controlling the distribution of MCG in natural environments is still unclear. Although our work opens a window on that issue, further studies with the use of newly emerging molecular tools such as meta- and single-cell genomics are needed to shed light on biogeographic pattern and its ecological drivers of estuarine archaeal community and population.
References
Fuhrman JA (2009) Microbial community structure and its functional implications. Nature 459:193–199. doi:10.1038/nature08058
Dupont CL, Larsson J, Yooseph S, Ininbergs K, Goll J, Asplund-Samuelsson J, McCrow JP, Celepli N, Allen LZ, Ekman M, Lucas AJ, Hagstrom A, Thiagarajan M, Brindefalk B, Richter AR, Andersson AF, Tenney A, Lundin D, Tovchigrechko A, Nylander JAA, Brami D, Badger JH, Allen AE, Rusch DB, Hoffman J, Norrby E, Friedman R, Pinhassi J, Venter JC, Bergman B (2014) Functional tradeoffs underpin salinity-driven divergence in microbial community composition. PLoS One 9:e89549. doi:10.1371/journal.pone.0089549
Herlemann DPR, Labrenz M, Jurgens K, Bertilsson S, Waniek JJ, Andersson AF (2011) Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5:1571–1579. doi:10.1038/ismej.2011.41
Fortunato CS, Eiler A, Herfort L, Needoba JA, Peterson TD, Crump BC (2013) Determining indicator taxa across spatial and seasonal gradients in the Columbia River coastal margin. ISME J 7:1899–1911. doi:10.1038/ismej.2013.79
Crump BC, Hopkinson CS, Sogin ML, Hobbie JE (2004) Microbial biogeography along an estuarine salinity gradient: combined influences of bacterial growth and residence time. Appl Environ Microbiol 70:1494–1505. doi:10.1128/AEM. 70.3.1494-1505.2004
Stahl DA, de la Torre JR (2012) Physiology and diversity of ammonia-oxidizing archaea. Annu Rev Microbiol 66:83–101. doi:10.1146/annurev-micro-092611-150128
Offre P, Spang A, Schleper C (2013) Archaea in biogeochemical cycles. Annu Rev Microbiol 67:437–457. doi:10.1146/annurev-micro-092412-155614
Brochier-Armanet C, Boussau B, Gribaldo S, Forterre P (2008) Mesophilic Crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat Rev Microbiol 6:245–252. doi:10.1038/nrmicro1852
Schleper C, Jurgens G, Jonuscheit M (2005) Genomic studies of uncultivated archaea. Nat Rev Microbiol 3:479–488. doi:10.1038/nrmicro1159
Karner MB, DeLong EF, Karl DM (2001) Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 409:507–510. doi:10.1038/35054051
Ochsenreiter T, Selezi D, Quaiser A, Bonch-Osmolovskaya L, Schleper C (2003) Diversity and abundance of Crenarchaeota in terrestrial habitats studied by 16S RNA surveys and real time PCR. Environ Microbiol 5:787–797. doi:10.1046/j.1462-2920.2003.00476.x
Nicol GW, Schleper C (2006) Ammonia-oxidising Crenarchaeota: important players in the nitrogen cycle? Trends Microbiol 14:207–212. doi:10.1016/j.tim.2006.03.004
Molari M, Giovannelli D, d’Errico G, Manini E (2012) Factors influencing prokaryotic community structure composition in sub-surface coastal sediments. Estuar Coast Shelf Sci 97:141–148. doi:10.1016/j.ecss.2011.11.036
Liu J, Yu S, Zhao M, He B, Zhang XH (2014) Shifts in archaeaplankton community structure along ecological gradients of Pearl Estuary. FEMS Microbiol Ecol. doi:10.1111/1574-6941.12404
Xie W, Zhang C, Zhou X, Wang P (2014) Salinity-dominated change in community structure and ecological function of Archaea from the lower Pearl River to coastal South China Sea. Appl Microbiol Biotechnol 98:7971–7982. doi:10.1007/s00253-014-5838-9
Crump BC, Baross JA (2000) Archaeaplankton in the Columbia River, its estuary and the adjacent coastal ocean, USA. FEMS Microbiol Ecol 31:231–239. doi:10.1111/j.1574-6941.2000.tb00688.x
Galand PE, Lovejoy C, Vincent WF (2006) Remarkably diverse and contrasting archaeal communities in a large arctic river and the coastal Arctic Ocean. Aquat Microb Ecol 44:115–126. doi:10.3354/ame044115
Galand PE, Lovejoy C, Pouliot J, Vincent WF (2008) Heterogeneous archaeal communities in the particle-rich environment of an arctic shelf ecosystem. J Mar Syst 74:774–782. doi:10.1016/j.jmarsys.2007.12.001
Li Q, Wang F, Chen Z, Yin X, Xiao X (2012) Stratified active archaeal communities in the sediments of Jiulong River estuary, China. Front Microbiol 3:311. doi:10.3389/fmicb.2012.00311
Yan XL, Zhai WD, Hong HS, Li Y, Guo WD, Huang X (2012) Distribution, fluxes and decadal changes of nutrients in the Jiulong River Estuary, Southwest Taiwan Strait. Chin Sci Bull 57:2307–2318. doi:10.1007/s11434-012-5084-4
Hu A, Yang X, Chen N, Hou L, Ma Y, Yu CP (2014) Response of bacterial communities to environmental changes in a mesoscale subtropical watershed, Southeast China. Sci Total Environ 472:746–756. doi:10.1016/j.scitotenv.2013.11.097
Guo W, Yang L, Hong H, Stedmon CA, Wang F, Xu J, Xie Y (2011) Assessing the dynamics of chromophoric dissolved organic matter in a subtropical estuary using parallel factor analysis. Mar Chem 124:125–133. doi:10.1016/j.marchem.2011.01.003
Knights D, Kuczynski J, Charlson ES, Zaneveld J, Mozer MC, Collman RG, Bushman FD, Knight R, Kelley ST (2011) Bayesian community-wide culture-independent microbial source tracking. Nat Meth 8:761–763. doi:10.1038/nmeth.1650
Cao WZ, Hong HS, Yue SP (2005) Modelling agricultural nitrogen contributions to the Jiulong River estuary and coastal water. Glob Planet Chang 47:111–121. doi:10.1016/j.gloplacha.2004.10.006
Yang YP, Hu MH (1997) The geochemistry of Jiulong River Estuary. In: Zhang J (ed) The biogeochemistry of major Chinese estuaries-transport of chemical materials and environment. Ocean Press, Beijing, pp 54–67
Zhai WD, Zhao HD, Zheng N, Xu Y (2012) Coastal acidification in summer bottom oxygen-depleted waters in northwestern-northern Bohai Sea from June to August in 2011. Chin Sci Bull 57:1062–1068. doi:10.1007/s11434-011-4949-2
Hu A, Jiao N, Zhang CL (2011) Community structure and function of planktonic Crenarchaeota: changes with depth in the South China Sea. Microb Ecol 62:549–563. doi:10.1007/s00248-011-9866-z
Muyzer G, de Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Kemnitz D, Kolb S, Conrad R (2005) Phenotypic characterization of Rice Cluster III archaea without prior isolation by applying quantitative polymerase chain reaction to an enrichment culture. Environ Microbiol 7:553–565. doi:10.1111/j.1462-2920.2005.00723.x
Galand PE, Casamayor EO, Kirchman DL, Potvin M, Lovejoy C (2009) Unique archaeal assemblages in the Arctic Ocean unveiled by massively parallel tag sequencing. ISME J 3:860–869. doi:10.1038/ismej.2009.23
Zhou HW, Li DF, Tam NFY, Jiang XT, Zhang H, Sheng HF, Qin J, Liu X, Zou F (2011) BIPES, a cost-effective high-throughput method for assessing microbial diversity. ISME J 5:741–749. doi:10.1038/ismej.2010.160
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi:10.1128/AEM. 01541-09
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7:335–336. doi:10.1038/nmeth.f.303
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi:10.1093/bioinformatics/btr381
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi:10.1093/bioinformatics/btq461
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Meth 10:996–998. doi:10.1038/nmeth.2604
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12:R60. doi:10.1186/gb-2011-12-6-r60
Blainey PC, Mosier AC, Potanina A, Francis CA, Quake SR (2011) Genome of a low-salinity ammonia-oxidizing archaeon determined by single-cell and metagenomic analysis. PLoS ONE 6:e16626. doi:10.1371/journal.pone.0016626
de la Torre JR, Walker CB, Ingalls AE, Konneke M, Stahl DA (2008) Cultivation of a thermophilic ammonia oxidizing archaeon synthesizing crenarchaeol. Environ Microbiol 10:810–818. doi:10.1111/j.1462-2920.2007.01506.x
Hatzenpichler R, Lebedeva EV, Spieck E, Stoecker K, Richter A, Daims H, Wagner M (2008) A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc Natl Acad Sci U S A 105:2134–2139. doi:10.1073/pnas.0708857105
Jung MY, Well R, Min D, Giesemann A, Park SJ, Kim JG, Kim SJ, Rhee SK (2014) Isotopic signatures of N2O produced by ammonia-oxidizing archaea from soils. ISME J 8:1115–1125. doi:10.1038/ismej.2013.205
Könneke M, Bernhard AE, de la Torre JR, Walker CB, Waterbury JB, Stahl DA (2005) Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437:543–546. doi:10.1038/nature03911
Lebedeva EV, Hatzenpichler R, Pelletier E, Schuster N, Hauzmayer S, Bulaev A, Grigor’eva NV, Galushko A, Schmid M, Palatinszky M, Le Paslier D, Daims H, Wagner M (2013) Enrichment and genome sequence of the group I. 1a ammonia-oxidizing Archaeon “Ca. Nitrosotenuis uzonensis” representing a clade globally distributed in thermal habitats. PLoS One 8:e80835. doi:10.1371/journal.pone.0080835
Lehtovirta-Morley LE, Stoecker K, Vilcinskas A, Prosser JI, Nicol GW (2011) Cultivation of an obligate acidophilic ammonia oxidizer from a nitrifying acid soil. Proc Natl Acad Sci U S A 108:15892–15897. doi:10.1073/pnas.1107196108
Mosier AC, Allen EE, Kim M, Ferriera S, Francis CA (2012) Genome sequence of “Candidatus Nitrosoarchaeum limnia” BG20, a low-salinity ammonia-oxidizing archaeon from the San Francisco Bay Estuary. J Bacteriol 194:2119–2120. doi:10.1128/JB.00007-12
Mosier AC, Allen EE, Kim M, Ferriera S, Francis CA (2012) Genome sequence of “Candidatus Nitrosopumilus salaria” BD31, an ammonia-oxidizing archaeon from the San Francisco Bay Estuary. J Bacteriol 194:2121–2122. doi:10.1128/JB.00013-12
Tourna M, Stieglmeier M, Spang A, Konneke M, Schintlmeister A, Urich T, Engel M, Schloter M, Wagner M, Richter A, Schleper C (2011) Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc Natl Acad Sci U S A 108:8420–8425. doi:10.1073/pnas.1013488108
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi:10.1093/molbev/mst197
Bates ST, Berg-Lyons D, Caporaso JG, Walters WA, Knight R, Fierer N (2011) Examining the global distribution of dominant archaeal populations in soil. ISME J 5:908–917. doi:10.1038/ismej.2010.171
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46. doi:10.1111/j.1442-9993.2001.01070.pp.x
Borcard D, Legendre P (2002) All-scale spatial analysis of ecological data by means of principal coordinates of neighbour matrices. Ecol Model 153:51–68. doi:10.1016/S0304-3800(01)00501-4
Dray S, Legendre P, Peres-Neto PR (2006) Spatial modelling: a comprehensive framework for principal coordinate analysis of neighbour matrices (PCNM). Ecol Model 196:483–493. doi:10.1016/j.ecolmodel.2006.02.015
Blanchet FG, Legendre P, Borcard D (2008) Forward selection of explanatory variables. Ecology 89:2623–2632. doi:10.1890/07-0986.1
Oksanen J, Kindt R, Legendre P, O’Hara B, Simpson G, Solymos P, Stevens M, Wagner H (2009) Vegan: community ecology package. R package version 115-2
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi:10.1093/nar/gks1219
Wells LE, Cordray M, Bowerman S, Miller LA, Vincent WF, Deming JW (2006) Archaea in particle-rich waters of the Beaufort Shelf and Franklin Bay, Canadian Arctic: clues to an allochthonous origin? Limnol Oceanogr 51:47–59. doi:10.4319/lo.2006.51.1.0047
Hao DM, Tashiro T, Kato M, Sohrin R, Ishibashi T, Katsuyama C, Nagaosa K, Kimura H, Thanh TD, Kato K (2010) Population dynamics of Crenarchaeota and Euryarchaeota in the mixing front of river and marine waters. Microbes Environ 25:126–132
Auguet JC, Barberan A, Casamayor EO (2010) Global ecological patterns in uncultured Archaea. ISME J 4:182–190. doi:10.1038/ismej.2009.109
Campbell BJ, Kirchman DL (2013) Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J 7:210–220. doi:10.1038/ismej.2012.93
Massana R, DeLong EF, Pedros-Alio C (2000) A few cosmopolitan phylotypes dominate planktonic archaeal assemblages in widely different oceanic provinces. Appl Environ Microbiol 66:1777–1787. doi:10.1128/AEM. 66.5.1777-1787.2000
Zeng YH, Li HY, Jiao NZ (2007) Phylogenetic diversity of planktonic Archaea in the estuarine region of East China Sea. Microbiol Res 162:26–36. doi:10.1016/j.micres.2006.03.007
Kubo K, Lloyd KG, Biddle JF, Amann R, Teske A, Knittel K (2012) Archaea of the Miscellaneous Crenarchaeotal Group are abundant, diverse and widespread in marine sediments. ISME J 6:1949–1965. doi:10.1038/ismej.2012.37
Liu Y, Whitman WB (2008) Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci 1125:171–189. doi:10.1196/annals.1419.019
Grossart HP, Frindte K, Dziallas C, Eckert W, Tang KW (2011) Microbial methane production in oxygenated water column of an oligotrophic lake. Proc Natl Acad Sci U S A 108:19657–19661. doi:10.1073/pnas.1110716108
Jurgens G, Glockner FO, Amann R, Saano A, Montonen L, Likolammi M, Munster U (2000) Identification of novel Archaea in bacterioplankton of a boreal forest lake by phylogenetic analysis and fluorescent in situ hybridization. FEMS Microbiol Ecol 34:45–56. doi:10.1111/j.1574-6941.2000.tb00753.x
Auguet JC, Casamayor EO (2008) A hotspot for cold crenarchaeota in the neuston of high mountain lakes. Environ Microbiol 10:1080–1086. doi:10.1111/j.1462-2920.2007.01498.x
Herfort L, Kim JH, Coolen MJL, Abbas B, Schouten S, Herndl GJ, Damste JSS (2009) Diversity of Archaea and detection of crenarchaeotal amoA genes in the rivers Rhine and Têt. Aquat Microb Ecol 55:189–201. doi:10.3354/ame01294
Wang S, Dong RM, Dong CZ, Huang L, Jiang H, Wei Y, Feng L, Liu D, Yang G, Zhang C, Dong H (2012) Diversity of microbial plankton across the Three Gorges Dam of the Yangtze River, China. Geosci Front 3:335–349. doi:10.1016/j.gsf.2011.11.013
Vieira RP, Clementino MM, Cardoso AM, Oliveira DN, Albano RM, Gonzalez AM, Paranhos R, Martins OB (2007) Archaeal communities in a tropical estuarine ecosystem: Guanabara Bay, Brazil. Microb Ecol 54:460–468. doi:10.1007/s00248-007-9261-y
Pires ACC, Cleary DFR, Almeida A, Cunha A, Dealtry S, Mendonca-Hagler LCS, Smalla K, Gomes NCM (2012) Denaturing gradient Gel electrophoresis and barcoded pyrosequencing reveal unprecedented archaeal diversity in mangrove sediment and rhizosphere samples. Appl Environ Microbiol 78:5520–5528. doi:10.1128/AEM. 00386-12
Mosier AC, Francis CA (2008) Relative abundance and diversity of ammonia-oxidizing archaea and bacteria in the San Francisco Bay estuary. Environ Microbiol 10:3002–3016. doi:10.1111/j.1462-2920.2008.01764.x
Beman JM, Francis CA (2006) Diversity of ammonia-oxidizing archaea and bacteria in the sediments of a hypernutrified subtropical estuary: Bahia del Tobari, Mexico. Appl Environ Microbiol 72:7767–7777. doi:10.1128/AEM. 00946-06
Logares R, Brate J, Bertilsson S, Clasen JL, Shalchian-Tabrizi K, Rengefors K (2009) Infrequent marine-freshwater transitions in the microbial world. Trends Microbiol 17:414–422. doi:10.1016/j.tim.2009.05.010
Acknowledgments
We thank Dr. Weidong Zhai and Ms. Xiuli Yan for providing physicochemical parameters of surface waters of the Jiulong River estuary and Mr. Jibing He and Mr. Jiajun Hong for assistance in collecting the samples. This work was supported by the National Natural Science Foundation of China (41106096), the Knowledge Innovation Program of the Chinese Academy of Sciences (IUEQN201307), Science and Technology Planning Project of Xiamen, China (3502Z20142021), and Natural Science Foundation of Ningbo, China (2013A610174, 2012C5011).
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Anyi Hu got his Ph.D degree from Xiamen University.
Chang-Ping Yu got his Ph.D degree from National Taiwan University.
Electronic Supplementary Material
Rights and permissions
About this article

Cite this article
Hu, A., Hou, L. & Yu, CP. Biogeography of Planktonic and Benthic Archaeal Communities in a Subtropical Eutrophic Estuary of China. Microb Ecol 70, 322–335 (2015). https://doi.org/10.1007/s00248-015-0597-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-015-0597-4