
Abstract
The exploration of spatial patterns of abundance and diversity patterns along precipitation gradients has focused for centuries on plants and animals; microbial profiles along such gradients are largely unknown. We studied the effects of soil pH, nutrient concentration, salinity, and water content on bacterial abundance and diversity in soils collected from Mediterranean, semi-arid, and arid sites receiving approximately 400, 300, and 100 mm annual precipitation, respectively. Bacterial diversity was evaluated by terminal restriction fragment length polymorphism and clone library analyses and the patterns obtained varied with the climatic regions. Over 75% of the sequenced clones were unique to their environment, while ∼2% were shared by all sites, yet, the Mediterranean and semi-arid sites had more common clones (∼9%) than either had with the arid site (4.7% and 6%, respectively). The microbial abundance, estimated by phospholipid fatty acids and real-time quantitative PCR assays, was significantly lower in the arid region. Our results indicate that although soil bacterial abundance decreases with precipitation, bacterial diversity is independent of precipitation gradient. Furthermore, community composition was found to be unique to each ecosystem.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Microbial biogeography offers an insight into the mechanisms generating and maintaining diversity, speciation, extinction, dispersal, and species interactions [1]. The spatial distribution of microbial diversity has been observed and documented at various scales and for different environmental gradients. It is well documented that profound differences exist between arid and temperate regions in vegetation coverage and animal abundance and diversity [2–5]. Yet, very little is known about the variation in microbial diversity along precipitation gradients. This presents a hindrance to our understanding of biodiversity, as bacteria, the most abundant and diverse group of organisms on earth, play a fundamental role in a vast array of ecological processes and ecosystem functioning [6].
Our perspective on spatial distribution of microbial diversity is still limited, in spite the advent of molecular tools during the last three decades. This might be because most studies have been site-specific, which has limited our understanding of the factors affecting the structure of soil bacterial communities to local scales [7, 8]. However, large-scale surveys conducted in recent years have shown that different ecosystems support unique microbial populations [9–13], indicating that microbial assemblages can exhibit small- (within site) and large-scale biogeographic distribution. It has been suggested that, along the climatic gradient from subtropical to tropical forests, bacterial diversity may be influenced by soil organic carbon and nutrient content levels but not by floristic richness [9, 14]. Another study [10] suggested that bacterial diversity is unrelated to site temperature, latitude, or other variables that typically predict plant and animal diversity; it was thus concluded that microbial biogeography is controlled by edaphic variables and differs fundamentally from the biogeography of macro-organisms. In contrast, Antarctic surveys across a range of terrestrial habitats revealed a negative relationship between bacterial diversity and latitude for bare soils, but no such pattern was observed for vegetated sites [15]. A meta-analysis of soil bacteria across seven of the major biomes on Earth revealed that all biomes are dominated by the same soil bacterial phyla (Acidobacteria, Actinobacteria, Proteobacteria, and Bacteroidetes). Yet, although distinct biomes harbored similar bacterial communities, the composition of the bacterial communities varied and could be explained mostly by soil pH [16].
In this study, we attempted to assess the role of natural water availability in microbial diversity patterns. We hypothesize that if precipitation affects biodiversity in the same fashion for micro- and macro-organisms, then diversity patterns will be similar in both groups, with gradual decrease in abundance and diversity along the precipitation gradient. Alternatively, if each group is shaped by different ecological processes, then they will have distinct diversity patterns. To test these hypotheses, we evaluated microbial diversity measures in three soils along a gradient ranging from 100 to 400 mm of annual precipitation. Bacterial community composition and abundance were examined in these arid, semi-arid, and Mediterranean ecosystems, together with abiotic factors that might shape their diversity patterns.
Materials and Methods
Site Description and Sampling
The study was conducted at three long-term ecological research (LTER) sites in Israel that represent three climatic regions: arid, semi-arid, and Mediterranean [17]. Details of these sites are presented in Table 1. The three 40 × 25 m plots sampled within each site had been undisturbed for at least 5 years, i.e., they were fenced to prevent livestock grazing or human activities.
Soil samples were collected in May and June 2007. Sampling at each LTER site was based on a spatially stratified, random sampling approach: at each LTER site each of the three plots was divided into equal sectors of 1 m2, and eight sectors were randomly selected. In the field, we located the coordinates and sampled the nearest respective vegetation-free patch. The reason for sampling a vegetation-free patch was to reduce the effect of vegetation impact on soil microbial composition and abundance, and allow comparison between sites with different types of dominant vegetation. Each sample was collected aseptically by removing approximately 200 g of soil from the upper 5 cm of the profile (after carefully brushing aside any loose litter), using a sterile scoop. The samples were placed into individual sterile Whirl-Pak® sampling bags (Nasco, Fort Atkinson, WI, USA), and stored at 4°C until processing in the laboratory within 24 h. In the laboratory, the eight samples of each plot were pooled together and homogenized. A 50-g amount of the homogenate was stored at -80°C for subsequent extractions of DNA and phospholipid fatty acids (PLFA), and the remainder was used for chemical analysis. All subsequent analyses were carried out in all soil homogenates, except for the clone libraries that, as discussed below, were analyzed for one homogenate (i.e., one plot) per LTER.
Soil Chemical Analyses
Soil chemical analyses were according to standard protocols for soil analyses [31]: water content was determined by gravimetric method, organic matter content by dichromate oxidation method, pH and electrical conductivity (EC) in saturated soil extract (SSE), sodium, calcium, and magnesium in SSE by inductively coupled plasma spectroscopy and flame photometer, sodium adsorption ratio (SAR) by calculation from sodium, calcium and magnesium concentrations, phosphate by the “Olsen method” (in sodium bicarbonate extract), potassium in SSE, nitrogen as nitrate in aqueous extract, nitrogen as ammonium in KCl solution extract (including adsorbed nitrogen), and calcium carbonate content by HCl digestion.
DNA Extraction
The soil homogenates were extracted for DNA using the PowerSoil™ DNA Isolation Kit (MoBio, West Carlsbad, CA, USA) according to the manufacturer's recommendations. The extracts were measured spectrophotometrically, confirmed on an ethidium bromide-stained agarose gel and used for molecular analysis, as described below. On average, soil samples taken from Mediterranean, semi-arid, and arid sites yielded final concentrations of 10, 11, and 5 ng μL−1, respectively. All products were stored at −20°C until use.
Terminal Restriction Fragment Length Polymorphism Analyses
Soil DNA extracts were obtained as described above, and further subjected to Terminal Restriction Fragment Length Polymorphism (TRFLP) fingerprint analysis as previously described [17]. Briefly, community 16S rRNA-encoding gene fragments were amplified with FAM-labeled Eub-341F (5′-CCTACGGGAGGCAGCAGI-3′) and untagged Eub-907R (5′-CCGTCAATTCMTTTGAGTTI-3′) [17]. The resulting amplicons were treated with mung-bean nuclease (TaKara, Shiga, Japan) to eliminate single-strand DNA [18], and purified using a polymerase chain reaction (PCR) purification kit (Bioneer, Daejeon, South Korea). Bacterial amplicons were digested with restriction enzymes HhaI, TaqI (TaKara), and HpyCH 4 IV (New England Biolabs, Beverly, MAS, USA), selected in accordance with the RDPII [19] and MiCA [20] databases. The resultant DNA fragments were precipitated by 70% ethanol, and the pellets were dyed using Pellet Paint (Novagen, Darmstadt, Germany), washed, dried, suspended in purified water, and analyzed using ABI 3100 sequencer (Applied Biosystems, Foster City, CA, USA). The terminal restriction fragments (TRFs) were sorted according to size using Peak Scanner™ software v1.0 (Applied Biosystems), exported to MATLAB 7 (http://www.mathworks.com), and analyzed by considering the size in base pairs of each TRF as an operational taxonomic unit (OTU).
Statistical Analysis
The TRFLP patterns of each sample were standardized as described by Dunbar et al. [21]. Profiles were then aligned and a consensus profile was computed for each sample by eliminating non-reproducible peaks and averaging shared peaks. The Shannon index for the resultant peaks was then calculated for each sample.
Clone Library Construction and Analysis
DNA was extracted from one soil homogenate of each LTER site as described above and further subjected to the following procedures:
PCR targeting of bacterial 16S rRNA genes
All amplifications were carried out in a MyCycler thermocycler (Bio-Rad, Richmond, CA, USA) using the universal bacterial 16S rRNA-targeting primers Eub-341F and Eub-1387R (5′-CCCGGGAACGTATTCACCGCI-3′) [22]. The outcome fragment of ca. 1 kb would allow sufficient information to identify the organism of origin in BLAST (see below). Each 50 μL reaction contained 10–20 ng of genomic DNA template, 4 mM of each deoxynucleoside triphosphate (TaKara), 1× PCR reaction buffer, 4 mM MgCl2, 1 μM of each primer and 0.5 U Taq DNA polymerase (Invitrogen Corp., Carlsbad, CA, USA). The mixtures were heated for 4 min at 94°C followed by 30 three-phase cycles of 30 s at 94°C, 30 s at 56°C, and 90 s at 72°C. A final elongation step was performed at 72°C for 10 min. Products were checked on a 1% agarose (Sigma) gel; successful amplifications were extracted from the gel using a gel purification kit (Bioneer).
Cloning and sequence analysis
Cloning of cleaned PCR products was performed at the Genome Sequencing Center (Washington University, St. Louis, MO, USA) and 384 fragments from each reaction were sequenced. All sequences were hand-picked using the freeware MEGA version 4 [23] when more than 450 bp per sequence of high-quality chromatograms were selected. The resulting sequences were aligned using CLUSTALW2 [24], trimmed from the Eub-341F primer sequence to remove the plasmid sequence, and checked for chimeras at the RDPII website using the CHIMERA_CHECK program 2.7 [25] and the BELLEROPHON program [26], which check for chimerical sequences from 16S rRNA gene-clone libraries. Non-chimerical, analyzed sequences were manually aligned for better positioning and processed for taxonomic assignment with the RDPII classifier tool [27] using 80% bootstrap value baseline, to test the composition of the main phyla at each library.
Statistical analyses
Each sequence was considered an OTU, and OTUs with 97% or more sequence similarity were regarded as identical. Using the RDPII pyrosequencing package [19], representative sequences were randomly picked by the Dereplicate tool, and the Shannon and Chao1 Index tool was used to generate these estimators. Rarefaction curves were calculated via 10,000 resampling trials (bootstrap) using the Resampling-Rarefaction tool downloaded from http://www.uga.edu/strata/software/index.html.
The relationships between the number of OTUs from the different groups (i.e., phylogenetic levels) and the chemical properties of the soil were examined, and significant correlations (P < 0.05) were recorded.
Accession Numbers
Sequence data generated in this study were deposited in GenBank under accession numbers GQ425232–GQ426030.
Quantitative (real-time) PCR
To estimate the number of 16S rRNA gene copies in the soil samples, a calibration curve of a known number of rRNA genes was performed as follows. (1) Environmental genomic fragments were amplified as described above, using the universal primers Eub-341F and Eub-519R (5′-GWATTACCGCGGCKGCTG-3′) [28]. The products were eluted from an agarose (Sigma) gel and concentrations were determined spectrophotometrically (NanoDrop Technology, Rockland, DE, USA). (2) A large number of available (4,142) 16S rRNA sequences of bacterial isolates were imported from the RDPII database, aligned, and trimmed in silico using the Eub-341F/519R primers. The molecular weight of all fragments was calculated using the “DNA molecular weight” tool of the sequence manipulation suite [29] and the average molecular weight of a fragment was calculated. The average fragment was found to be 61328 ± 68 g mol−1 (the statistical analysis of the 4,142 sequences is shown in Electronic Supplementary Material, Table S1). (3) Using this average value, we calculated the number of gene copies in a PCR amplification given its concentration, i.e., the amount of PCR product in weight divided by the average bacterial fragment weight equals the number of fragments in solution (see Electronic Supplementary Material, Table S1 regarding data and calculation). Stock solutions containing known fragment concentrations were used to calibrate the environmental genomic samples.
DNA was extracted as described above. Primer set (Eub-341F/519R) and sample concentrations were optimized to 200 nM and 10–20 ng μL−1, respectively. The qPCR measurements were performed using the DyNAmo Flash SYBR Green qPCR Kit core mix (Finnzymes, Espoo, Finland) and run in a real-time thermocycler (Corbett Research, Sidney, Australia) in accordance with the manufacturer's recommendations.
Analysis of Phospholipid Fatty Acids
Analyses of PLFA were conducted on 4 g of soil homogenate by the procedure described by White and Ringelberg [30]. Briefly, lipids were extracted and fractionated by stepwise elution using Bond Elut JR-S1 of a 500 mg Sep-Pak cartridge (Waters, Milford, MA, USA) into neutral lipids, glycolipids and polar lipids. Phospholipids (the polar fraction) were converted to fatty acid methyl esters (FAMEs), then extracted using 4:1 (v/v) hexane/chloroform, evaporated under a stream of nitrogen gas, and stored at −20°C. Prior to gas chromatography (GC) analyses, FAMEs were reconstituted in 20 μL hexane, and 10 μL hexane containing FAME 17:0 (heptadecanoic acid, 0.5 mg mL−1) was added as an internal standard. Aliquots of 2 μL were analyzed using a GC system 5890II (GMI) (Hewlett Packard, Wilmington, DE, USA) consisting of flame ionization detection, a 30 m × 0.32 mm × 0.25 μm Cowax-10 column (Supelco, Inc., Bellefonte, PA. USA) and helium as the carrier gas (flow rate 3.5 mL min−1). The injector and detector were maintained at 250°C, and the column temperature was programmed from 120°C for 2.5 min to 195°C for 1.5 min at the ramping rate of 2.5°C min−1, each run lasting approximately 36 min. Peaks were identified using a bacterial acid methyl esters mix (catalog number 47080-U; Sigma-Aldrich, St. Louis, MO) and ClarityTM 2.3 software (DataApex, Czech Republic).
Results
The main chemical and physical properties of the soils sampled at the different sites are summarized in Table 2. Water and organic matter content increased, while the percentage of calcium carbonate, SAR, and the concentrations of sodium and potassium decreased along the precipitation gradient. In the semi-arid site nitrate concentration was lower, and that of ammonium was higher, compared to the other two sites. The pH values were similar (averaging 7.3 ± 0.3) in all sites.
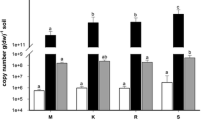
The bacterial abundance at each site was assessed by both PLFA and qPCR analyses demonstrating a statistically significant reduction in bacterial biomass in the arid versus semi-arid and Mediterranean soils (Fig. 1); no significant differences in microbial abundance were detected between the latter.

Bacterial abundance indicated by PLFA (empty bars) and quantitative analysis of bacterial 16S rDNA (gray bars) at the Mediterranean, arid, and semi-arid sites. The measures presented are the sum of the peaks corresponding to bacterial PLFA calculated in accordance to the standard
In our attempt to study the bacterial community composition within each of the three LTER sites we relied on a previous study [17] in which we tested bacterial diversity, using a scheme that enabled us to examine triplicate composite soil samples in each plot within each LTER site. Statistical analysis of bacterial fingerprints taken by TRFLP revealed that the differences among plots within each LTER site are not statistically significant (P = 0.33) [17], indicating that the samples taken from the three plots within a site are indeed replicates. Thus, we decided to study the soil bacterial community composition at a single representative plot within each site.
The clone libraries amplified from the arid, semi-arid, and Mediterranean soils yielded 276, 247, and 276 high-quality clones, respectively. Rarefaction curves generated for the three soils examined produced similar and overlapping curves (see Electronic Supplementary Material Fig. S1). The rarefaction curves indicated that 250 to 280 clones per sample is not enough to reach saturation and that over 400 clones need to be analyzed to yield the minimal bacterial estimation in the soils used in this study; thus, we regard our cloning results as minimal values. Diversity appears to be very high among the three sites, as deduced from the Chao1 and Shannon indices calculated in accordance to the cloning analysis (Table 3).
Over 73% of the 799 sequenced clones were found to be unique to their climatic region. Figure 2 depicts a Venn diagram representing the unique and overlapping OTUs at the three sites, indicating that most of the members comprising each of the soil communities were unique to their environment. Only a small percentage (11 out of 587) of the OTUs was represented at all sites. Clone sequences were attributed to 11 phyla that were shared by the three sites. Figure 3 presents the different bacterial phyla as percentage of all group members detected in each sampling site; observed significant (P < 0.05) correlations with soil parameters are shown in Table 4.

Venn diagram depicting individual sequenced clones from soil samples collected from Mediterranean, semi-arid, and arid sites

Phylum comparison of the bacterial community composition in soil samples collected from Mediterranean, semi-arid, and arid sites, as revealed by partial cloning of the 16S rRNA gene. Fractions are shown as relative percentage of the total number of clones. Asterisk indicates significant difference (P < 0.05)
Gemmatimonadetes and Planctomycetes were not significantly abundant in any of the sites, although an increasing trend along the precipitation gradient was detected for the latter. Similarly, the numbers of Bacteroidetes were not significantly different among sites, yet the Adhaeribacter genera (family Flexibacteraceae) composed 3.3% of the clones in the Mediterranean site, while none were detected in the arid soil. Both Bacteroidetes and Gemmatimonadetes correlated significantly only with organic matter percentage in the soil.
Acidobacteria (family Acidobacteriaceae) were significantly more abundant in the semi-arid site. Their abundance was negatively correlated to nitrate and magnesium, but positively correlated to ammonium and phosphorus. In contrast, Chloroflexi were significantly less abundant in the semi-arid site compared to the other two environments, and the phylum's abundance was positively correlated with nitrate and magnesium, but negatively correlated with phosphorus.
The relative proportion of Cyanobacteria, Thermomicrobia, and Verrucomicrobia increased with aridity, although not statistically so. Accordingly, these phyla were positively correlated with SAR and with the soil concentrations of potassium, sodium and calcium carbonate. Likewise, Firmicutes were significantly more abundant in the arid site than in the other two sites. In our samples, the second most abundant clone found was a member of the family Bacillaceae, and its abundance was correlated to pH, electrical conductivity, sodium, SAR and potassium. In contrast to Firmicutes, Proteobacteria were significantly less abundant in the arid soil than at the other two sites. A negative correlation was found between community abundance of Proteobacteria and EC, SAR, sodium, and potassium. Two families within the α-Proteobacteria class (the Sphingomonadaceae and the Rhizobiaceae) were significantly less prominent in the arid site.
The most abundant phylum in the soil samples collected from the three climatic regions was Actinobacteria. However, families within this phylum varied among sites: members of the Micromonosporaceae were barely detected in clones amplified from the arid soil, while being significantly more abundant in the semi-arid soil. Likewise, members of the Geodermatophilaceae were significantly more abundant in clones from the Mediterranean soil sample compared to those obtained from the other soils. More than 11% of the clones amplified from the semi-arid site belonged to the family Rubrobacteraceae, while no members of this family were detected in the other regions. On the other hand, significantly more clones were affiliated with the family Geodermatophilaceae in the Mediterranean soil than in the semi-arid soil. Only calcium showed a significant correlation with Actinobacteria abundance.
Overall measures of soil microbial diversity were estimated using the clone library estimated with the Chao1 and Shannon indices (Table 3). The Shannon measures (H′) and Chao1 estimates, calculated in accordance with the unique OTUs detected by the sequenced clones, were nearly equal among sites, both indicating high and similar diversity along the precipitation gradient.
Discussion
For many years, the ecological paradigm has been that water availability is positively correlated with richness, diversity and abundance of communities [32]. Yet, in recent years, it has been reported that the rules that apply to macro-organisms do not necessarily hold for micro-organisms, and that water availability is not directly correlated with the diversity of all organisms [10].
A broad scale survey aiming to find robust patterns in the structure of soil microbial and faunal communities showed that unlike plant communities, which typically harbor distinct ecosystems, bacterial communities are rather similar in composition (harboring the same dominant phyla), yet they differ in structure and biomass [16]. However, explaining these differences is challenging: among the physico-chemical soil parameters evaluated only soil pH and available carbon could explain the spatial variance of some bacterial phyla (Acidobacteria, β-Proteobacteria, and Bacteroidetes). Yet, the variance of Actinobacteria, α-Proteobacteria and Firmicutes could not be assigned to any of the soil characteristics [14].
The current study, focused on three relatively dry environments, suggests that although bacterial abundance follows the water-availability paradigm, bacterial richness and diversity may not. The richness index, obtained using clone library analysis, suggests that soil microbial richness is not significantly affected by precipitation (Table 3). These findings are consistent with those of Meier and his colleagues [36] who found that bacterial richness fluctuates very little among seasons with different water availability. Similarly, it was shown that low-carbon surface soils exhibited a uniform diversity pattern distinct from saturated subsurface soil [13]. The surface, low-carbon soils tested in our study (Table 2) exhibited similarly high bacterial diversity (Table 3). However, each site supported a unique array of OTUs, which had little in common with the others (Fig. 2). Such low share of OTUs at the level of 97% similarity was demonstrated among soils from different, albeit more distant, sites in South and North America [33]. These observations support the hypothesis suggested by Treves and his colleagues [34] that spatial isolation of microbial populations in soils is key determinant of microbial community structure.
Inherent PCR-associated biases [35] accompany any molecular method used to explore bacterial diversity. Nevertheless, we draw attention to the similar pattern of bacterial abundance generated by the qPCR and PLFA analyses, and to the equal diversity patterns obtained from the TRFLP [17] and the clone library analyses. All these independent approaches support the claim that, unlike bacterial abundance, bacterial richness and diversity are not affected by precipitation in low-carbon-input soils [34]. These findings are in contrast to richness indices for annual plants and beetles collected at the same LTER sites during the spring of 2007, showing positive correlation with precipitation (Y. Lubin, unpublished data), and in agreement with biogeographical patterns reported for macro-organisms all around the world, from Australia to Africa and South America [32].
Community composition can be correlated to an array of environmental factors (Table 4). Cyanobacteria, a prominent phylum that has been found in the harshest of deserts, such as the Atacama [36] and Sonoran [37], was significantly more abundant in the desert region than in the northern, wetter regions. Likewise, the phylum Firmicutes, which correlated negatively with soil moisture [14], and was abundant in the deserts of Namib [38] and Tataouine [39] was identified, mainly, in the arid soil of the Negev desert. It is conceivable that we, and maybe others studying desert soils, amplified DNA of dormant Firmicutes spores [40], and thus we cannot determine with certainty whether the observed higher abundance of this phylum (Fig. 3) truly represents its in situ activity or ecological importance at the time of sampling. In contrast, the phylum Proteobacteria dominated by its most abundant and diverse group, the α-Proteobacteria, was positively correlated with precipitation (Fig. 3).
The mechanism that maintains high microbial richness is not clear but water availability may well be one of the important factors affecting microbial biomass and community composition in soils. Although we detected high correlations between specific taxonomic groups and environmental factors, such as calcium or magnesium concentrations (Table 4), we do not know, at this point, whether these correlations indicate true biochemical dependence. This stems mainly from the fact that at the phylum level such correlations are obscure, as no true biochemical pathways and/or chemical dependence function can be accredited to any one phylum. Therefore, we are currently focusing our attention on expanding the phylogenetic resolution, to better observe the differences in specific selected phyla along the precipitation gradient. One promising lead for such studies could be members of the class α-Proteobacteria and in particular, the Rhizobiaceae and Sphingomonadaceae, as their abundance increased with precipitation. Another interesting question is the lack of correlation between water availability and the high abundance of Actinobacteria (Fig. 3), although the abundance of families within this phylum differed according to site. It has been suggested that different chemical components alter microbial richness in soils [13], yet it remains to be seen which of the detected correlations will prove to be significant to phylum diversity and composition and which will prove redundant.
We conclude that bacterial biomass follows a pattern similar to that of macro-organisms, being more abundant in Mediterranean ecosystems compared with arid sites. However, unlike macro-organisms, bacterial diversity is not constrained by precipitation. Moreover, the composition of the bacterial community was found to be distinct to each site along the rainfall gradient.
References
Lomolino MV (2005) Body size evolution in insular vertebrates: generality of the island rule. J Biogeogr 32:1683–1699
Gentry AH (1982) Patterns of neotropical plant-species diversity. Evol Biol 15:1–85
Heaney LR (2001) Small mammal diversity along elevational gradients in the Philippines: an assessment of patterns and hypotheses. Glob Ecol Biogeogr 10:15–39
McCoy ED (1990) The distribution of insect along elevational gradients. Oikos 58:313–322
Terborgh J (1977) Bird species-diversity on an Andean elevational gradient. Ecology 58:1007–1019
Usher MB, Sier ARJ, Hornung M, Millard P (2006) Understanding biological diversity in soil: the UK's Soil Biodiversity Research Programme. Appl Soil Ecol 33:101–113
Navarro-Gonzalez R, Rainey FA, Molina P, Bagaley DR, Hollen BJ, de la Rosa J, Small AM, Quinn RC, Grunthaner FJ, Caceres L, Gomez-Silva B, McKay CP (2003) Mars-like soils in the Atacama Desert, Chile, and the dry limit of microbial life. Science 302:1018–1021
Zhou JZ, Xia BC, Huang H, Palumbo AV, Tiedje JM (2004) Microbial diversity and heterogeneity in sandy subsurface soils. Appl Environ Microbiol 70:1723–1734
Chan OC, Yang XD, Fu Y, Feng ZL, Sha LQ, Casper P, Zou XM (2006) 16S rRNA gene analyses of bacterial community structures in the soils of evergreen broad-leaved forests in south-west China. FEMS Microbiol Ecol 58:247–259
Fierer N, Jackson RB (2006) The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci U S A 103:626–631
Green J, Bohannan BJM (2006) Spatial scaling of microbial biodiversity. Trends Ecol Evol 21:501–507
Vishniac HS (2006) A multivariate analysis of soil yeasts isolated from a latitudinal gradient. Microb Ecol 52:90–103
Zhou J, Xia B, Treves DS, Wu LY, Marsh TL, O'Neill RV, Palumbo AV, Tiedje JM (2002) Spatial and resource factors influencing high microbial diversity in soil. Appl Environ Microbiol 68:326–334
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88:1354–1364
Yergeau E, Newsham KK, Pearce DA, Kowalchuk GA (2007) Patterns of bacterial diversity across a range of Antarctic terrestrial habitats. Environ Microbiol 9:2670–2682
Fierer N, Strickland MS, Liptzin D, Bradford MA, Cleveland CC (2009) Global patterns in belowground communities. Ecol Lett 12:1238–1249
Angel R, Soares MIM, Ungar ED, Gillor O (2009) Biogeography of soil archaea and bacteria along a steep precipitation gradient. ISME J 4:553–563
Egert M, Friedrich MW (2003) Formation of pseudo-terminal restriction fragments, a PCR-related bias affecting terminal restriction fragment length polymorphism analysis of microbial community structure. Appl Environ Microbiol 69:2555–2562
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145
Shyu C, Soule T, Bent SJ, Foster JA, Forney LJ (2007) MiCA: a web-based tool for the analysis of microbial communities based on terminal-restriction fragment length polymorphisms of 16S and 18S rRNA genes. Microb Ecol 53:562–570
Dunbar J, Ticknor LO, Kuske CR (2001) Phylogenetic specificity and reproducibility and new method for analysis of terminal restriction fragment profiles of 16S rRNA genes from bacterial communities. Appl Environ Microbiol 67:190–197
Iwamoto T, Tani K, Nakamura K, Suzuki Y, Kitagawa M, Eguchi M, Nasu M (2000) Monitoring impact of in situ biostimulation treatment on groundwater bacterial community by DGGE. FEMS Microbiol Ecol 32:129–141
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM (2005) The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res 33:D294–D296
Huber T, Faulkner G, Hugenholtz P (2004) Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20:2317–2319
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, Chichester, pp 115–147
Stothard P (2000) The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28:1102
White DC, Ringelberg DB (1998) Signature lipid biomarker analysis. In: Burlage RS, Atlas R, Stahl D, Geesey G, Sayler G (eds) Techniques in Microbial Ecology. Oxford University Press, New York, pp 255–272
SSSA (1996) Methods of soil analysis, Part 3—chemical methods and processes. Soil Science Society of America, Madison
Hawkins BA, Field R, Cornell HV, Currie DJ, Guegan JF, Kaufman DM, Kerr JT, Mittelbach GG, Oberdorff T, O'Brien EM, Porter EE, Turner JRG (2003) Energy, water, and broad-scale geographic patterns of species richness. Ecology 84:3105–3117
Fulthorpe RR, Roesch LFW, Riva A, Triplett EW (2008) Distantly sampled soils carry few species in common. ISME J 2:901–910
Treves DS, Xia B, Zhou J, Tiedje JM (2003) A two-species test of the hypothesis that spatial isolation influences microbial diversity in soil. Microb Ecol 45:20–28
Suzuki MT, Giovannoni SJ (1996) Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62:625–630
Warren-Rhodes KA, Rhodes KL, Pointing SB, Ewing SA, Lacap DC, Gomez-Silva B, Amundson R, Friedmann EI, McKay CP (2006) Hypolithic cyanobacteria, dry limit of photosynthesis, and microbial ecology in the hyperarid Atacama Desert. Microb Ecol 52:389–398
Nagy ML, Perez A, Garcia-Pichel F (2005) The prokaryotic diversity of biological soil crusts in the Sonoran Desert (Organ Pipe Cactus National Monument, AZ). FEMS Microbiol Ecol 54:233–245
Prestel E, Salamitou S, Dubow MS (2008) An examination of the bacteriophages and bacteria of the Namib desert. J Microbiol 46:364–372
Chanal A, Chapon V, Benzerara K, Barakat M, Christen R, Achouak W, Barras F, Heulin T (2006) The desert of Tataouine: an extreme environment that hosts a wide diversity of microorganisms and radiotolerant bacteria. Environ Microbiol 8:514–525
Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (2007) The prokaryotes: an evolving electronic resource for the microbiology communitybacteria, firmicutes, vol. 3b. Springer, New York
Evenari M, Shanan L, Tadmor N (1982) The Negev: the challenge of a desert. Harvard University Press, Cambridge
Yair A, Danin A (1980) Spatial variations in vegetation as related to the soil moisture regime over an arid limestone hillside, northern Negev, Israel. Oecologia 47:83–88
Danin A, Orshan G (1990) The distribution of raunkiaer life forms in Israel in relation to the environment. J Veg Sci 1:41–48
Danin A, Orshan G, Zohary M (1975) The vegetation of the northern Negev and the Judean desert of Israel. Isr J Bot 24:118–172
Acknowledgments
The authors would like to acknowledge the contribution of the Michigan State University (Ribosomal Database Project), and the technical assistance of Rina Myaskovsky and Reuma Arusi. We thank Yael Lubin for allowing the use of unpublished data and for fruitful discussions, and the Israeli LTER (http://lter.bgu.ac.il) for allowing sampling on their sites.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Table S1
(DOC 33 kb)
Supplementary Fig. S1
(DOC 80 kb)
Rights and permissions
About this article
Cite this article
Bachar, A., Al-Ashhab, A., Soares, M.I.M. et al. Soil Microbial Abundance and Diversity Along a Low Precipitation Gradient. Microb Ecol 60, 453–461 (2010). https://doi.org/10.1007/s00248-010-9727-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-010-9727-1