
Abstract
This retrospective observational study aimed to evaluate the safety and efficacy of dexmedetomidine (DEX) for children with heart failure. The study was conducted in the cardiovascular intensive care unit (CVICU) of a single, tertiary care, academic children’s hospital. A retrospective review of the charts for all children (up to 18 years of age) with signs and symptoms consistent with congestive heart failure who received DEX in our CVICU between April 2006 and April 2011 was performed. The patients were divided into two groups for study purposes: the DEX group of 21 patients, who received a DEX infusion together with other conventional sedation agents, and the control group of 23 patients, who received conventional sedation agents without the use of DEX. To evaluate the safety of DEX, physiologic data were collected including heart rate, mean arterial pressure (MAP), and inotrope score. To assess the efficacy of DEX, the amount and duration of concomitant sedation and analgesic infusions in both the DEX and control groups were examined. The numbers of rescue boluses for each category before the initiation of sedative infusion and during the sedative infusion also were examined. The baseline characteristics of the patients in the two groups were similar. There was no effect of DEX infusion on heart rate, MAP, or inotrope score at the termination of infusion. The daily amount of midazolam administered was significantly less during the last 24 h of DEX infusion in the DEX group than in the control group (p = 0.04). The daily amount of morphine infusion did not differ between the DEX and control groups during any period. The numbers of sedation and analgesic rescue boluses were lower in DEX group throughout the infusion. No other significant side effects were noted. Two patients in the DEX group had a 50 % or greater drop in MAP compared with baseline in the first 3 h after initiation of DEX infusion, whereas one patient had a 50 % or greater drop in heart rate compared with baseline in the first 3 h after initiation of DEX infusion. Administration of DEX for children with heart failure appears to be safe but should be used cautiously. Furthermore, DEX use is associated with a decreased opiate and benzodiazepine requirement for children with heart failure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Children with heart failure represent 10–33 % of all cardiac admissions [14]. Providing adequate sedation for critically ill children with heart failure is a challenging clinical problem. These patients often have labile cardiovascular function and may require several days to weeks of sedation as a component of their intensive care unit (ICU) management. Although it has been proposed by some that dexmedetomidine (DEX) is a useful addition to the sedation armamentarium of pediatric cardiac intensive care physicians, the safety and efficacy of this agent for children with heart failure is unknown.
Elevations of the neurohormonal and inflammatory mediators such as renin, aldosterone, norepinephrine, brain natriuretic peptide (BNP), N-terminal prohormone BNP, and tumor necrosis factor-α receptor have been reported in children with heart failure [6, 7]. Infusion of DEX is associated with sympatholysis and a decrease in epinephrine and norepinephrine levels [19, 23]. In addition, DEX has been shown to diminish myocardial blood flow in parallel with reductions in plasma catecholamines, heart rate, and blood pressure.
Because heart failure is a proinflammatory state and DEX infusion is associated with complex direct and indirect cardiovascular responses that adversely affect coronary and myocardial perfusion, it becomes prudent to study the safety of this drug in children with heart failure [21].
In both adult and pediatric populations, DEX has been administered in several clinical scenarios. These have included its use intraoperatively as part of balanced anesthetic care, postoperatively to provide sedation and analgesia after surgery or during mechanical ventilation, and as a means of providing sedation for nonpainful and invasive procedures [2–4].
As an α2-adrenergic agonist, DEX (Precedex; Hospira Worldwide Inc., Lake Forest, IL, USA) has sedative, anxiolytic, and analgesic properties. It contains an imidazole ring and has a structure similar to that of clonidine but with an α2:α1 specificity of nearly 1,600–1 [24]. The shorter half-life of DEX (2–3 vs 12–24 h for clonidine) allows titration by a continuous infusion [24]. The enthusiasm for this newer agent stems from several factors, including the lack of an ideal agent for sedation during mechanical ventilation and adverse effects associated with existing agents.
The Food and Drug Administration (FDA) has approved DEX to provide sedation up to 24 h for adults during mechanical ventilation and sedation for nonintubated patients before and during surgical and other procedures. In this study, we evaluated the safety and efficacy of DEX infusion for children with heart failure at a single center.
Patients and Methods
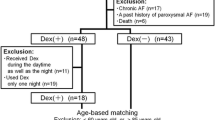
We retrospectively reviewed the charts of all children (up to 18 years of age) with signs and symptoms consistent with congestive heart failure who received DEX in our pediatric cardiovascular intensive care unit (CVICU) between April 2006 and April 2011. All critically ill children who received DEX within 18 days before orthotopic heart transplantation (OHT) were eligible for inclusion in the study. The patients were identified using records for our pharmacy and departmental surgical database.
The patients excluded from the study were those who had received any part of their DEX infusion outside the CVICU setting (i.e., operating room, acute care ward, or radiologic suite), those who had received DEX while receiving mechanical support in the form of either extracorporeal membrane oxygenation or ventricular assist device support, those with incomplete or missing medical records, those who did not have an arterial catheter for invasive blood pressure monitoring, and those with “do not resuscitate” orders. The Institutional Review Board at Arkansas Children’s Hospital approved the study protocol, and the need for informed consent or assent was waived.
According to routine clinical practice in our CVICU, DEX was initiated as a continuous infusion as a second or third sedative after the perceived failure of a conventional sedation strategy. The decision to initiate a DEX infusion was at the discretion of the medical team caring for the patient.
For study purposes, the DEX group included the patients who had received a DEX infusion together with other conventional agents (with DEX as the point drug for the purpose of collecting variables), whereas the control group included the patients who received conventional agents without the use of DEX (with midazolam as the point drug for the purpose of collecting variables).
The control patients were managed concurrently with those receiving DEX, and they were under the care of the same physicians. The conventional agents infused included midazolam, morphine, or both. We initiated DEX as a continuous infusion at a dose of 0.3–1.0 μg/kg/h without a bolus dose. The daily dose of DEX (μg/kg/day) was calculated by averaging the amount of drug each patient received over 24 h, expressed as μg/kg/day. Similarly, the midazolam and morphine doses were expressed by averaging the amount of each drug received by each patient during 24 h, expressed as mg/kg/day.
We collected demographic data including age, weight, sex, and diagnosis. We also collected detailed information on the dosages of DEX, midazolam, and morphine administered by infusion in the first and second 24-h periods after initiation of DEX and during the last 24 h before discontinuation of DEX, together with the total duration of infusion for each agent. The clinical outcomes evaluated in our study included days of mechanical ventilation, CVICU length of stay, hospital length of stay, and mortality.
To evaluate the safety of DEX, we collected physiologic data including heart rate, mean arterial pressure (MAP), respiratory rate, systemic oxygen saturation by pulse oximetry, and inotrope score [16]. Baseline data for these variables were collected for the following periods: 6 h before the initiation of sedation, the first hour after the initiation of sedation, 2–6 h after the initiation of sedation, 7–24 h after initiation of sedation, and the last 6 h before termination of sedation. When more than 1 h of data were recorded, the values were expressed as the mean value exceeding 1 h.
The potential side effects evaluated included nausea, vomiting, abdominal distension, dysrhythmias, neurologic abnormalities, and seizures. The other variables evaluated to assess the safety of DEX were blood pH, partial pressure of arterial oxygen (PaO2), partial pressure of arterial carbon dioxide (PaCO2), and base excess before the initiation of sedative infusion and during the infusion.
To assess the efficacy of DEX in terms of requirements for sedatives and analgesics, we examined the number of rescue boluses for each category before the initiation of sedative infusion and during the sedative infusion. We defined a rescue bolus as an extra sedative or analgesic bolus dose given in addition to the existing infusions required for maintaining adequate sedation or analgesia. Given the lack of an established protocol for sedation management in our unit, the rescue agents were multiple and included midazolam, lorazepam, ketamine, chloral hydrate, and diphenhydramine for sedation, and fentanyl and morphine for analgesia.
For some patients, narcotics were used for both analgesia (primary effect) and sedation (sedation effect). Rescue agents were labeled as either “sedation bolus” or “analgesic bolus” depending on their primary effect.
Statistical Analysis
Continuous variables are presented as median and interquartile range (Q1, Q3), as mean ± standard deviation, or both, whereas categorical variables are presented as numbers and percentages. Calculations of p values were performed using the Chi-square test and/or Fisher’s exact test of independence for categorical variables and Wilcoxon rank-sum test for continuous variables. A p value of 0.05 was considered significant for study purposes. All analyses were performed using either SAS 6.12 (SAS Institute Inc., Cary, NC, USA) or Harrell’s RMS package in R (http://biostat.mc.vanderbilt.edu/wiki/Main/Rrms).
Results
During the study period, 21 patients received DEX, and we compared them with 23 control patients hospitalized during the same period. The baseline characteristics of the patients were similar in the two groups (Table 1). Both groups were similar in age, weight, and sex. Of the 44 patients in the combined cohort receiving DEX, 33 (77 %) had dilated cardiomyopathy before OHT. Most of the patients (43/44) were mechanically ventilated during the study period. Dexmedetomidine was administered as a continuous infusion for a median duration of 193 h (range, 102–605 h). The patients received 9.1 ± 4.9 μg/kg of DEX per day in the first 24 h, 11.1 ± 5.1 μg/kg/day in the 24- to 48-h period, and 13.5 ± 7.2 μg/kg/day in the last 24 h before infusion was terminated.
Table 2 compares the sedation effects between the two groups. The duration of concomitant continuous midazolam and morphine infusions after initiation of the study drug did not differ significantly between the DEX and control groups. The daily amount of midazolam administered was significantly less during the last 24 h of DEX infusion in the DEX group than in the control group (p = 0.04). However, the daily amount of morphine infusion did not differ between the DEX and control groups during any period.
Table 3 presents the additional rescue boluses of sedatives and analgesics during the DEX infusion. The number of sedation rescue boluses was lower in DEX group in the first 24 h and during the 24- to 48-h period of DEX infusion. The number of analgesic rescue boluses was lower in DEX group in the first 24 h, during the 24- to 48-h period, and in the last 24 h of DEX infusion.
Changes in blood pressure, inotrope score, and heart rate are depicted graphically in Figs. 1, 2, and 3, respectively. Comparison of the DEX and control groups showed that mean arterial blood pressures were lower in the 2-to 6-h period (p = 0.01) and during the 7- to 24-h period (p = 0.01) after initiation of DEX infusion. However, the mean arterial blood pressures were similar in two groups in last 6 h of DEX infusion (Fig. 1). The inotrope scores did not differ statistically at any time during DEX or after the termination of DEX infusion (Fig. 2).

Changes in mean arterial pressure stratified by treatment with dexmedetomidine (DEX). *Significant difference between the control and DEX groups

Changes in inotrope score stratified by treatment with dexmedetomidine (DEX)

Changes in heart rate stratified by treatment with dexmedetomidine (DEX).*Significant difference between the control and DEX groups
The baseline heart rate before initiation of DEX infusion was lower in the DEX group (p = 0.009). This trend continued in the first 6 h after the initiation of DEX infusion, with heart rate lower in the DEX group during the first hour (p = 0.01) and 2–6 h (p < 0.001) after initiation of DEX (Fig. 3). Heart rate did not differ between in the two groups in the 7- to 24-h period (p = 0.11) or the in last 24 h (p = 0.9).
Table 4 presents the patients with significant changes (defined as a change of ≥50 % compared with baseline) in either mean arterial blood pressure or heart rate in our cohort. Two patients in the DEX group had a 50 % or greater drop in MAP compared with baseline values in the first 3 h after initiation of DEX infusion, whereas one patient had a 50 % or greater drop in heart rate compared with baseline value in the first 3 h after initiation of DEX infusion. For the two patients with hypotension, DEX infusion was continued, whereas DEX infusion was terminated for the patient with significant bradycardia.
No patients in the control group experienced this significant drop in either MAP or heart rate. None of the patients in our cohort were noted for significant arrhythmias except for sinus bradycardia.
No adverse respiratory effects were associated with DEX infusion. The DEX and control groups did not differ in near-infrared spectroscopy (NIRS), oxygen saturation, blood pH, PaO2, PaCO2, or base excess. We found no difference in days of mechanical ventilation (p = 0.44) or hospital length of stay (p = 0.34) between the two groups. The two groups did not differ significantly in terms of hospital mortality (10 % in the DEX group vs 4 % in the control group; p = 0.5).
No significant abdominal signs or symptoms were described. None of the patients had increased vomiting or development of a distended abdomen. No other adverse effects were reported in the limited medical records associated with initiation or continuation of DEX. No neurologic abnormalities were noted during or after the termination of DEX infusion.
Discussion
We report the use of DEX infusion for critically ill children with heart failure at doses of 0.1–1.0 μg/kg/h for a median duration of 193 h. We found that DEX infusion appears to be safe from a hemodynamic standpoint but should be used cautiously for children with heart failure. There was no effect of DEX infusion on heart rate, MAP, or inotrope score at the time of infusion termination. Furthermore, we demonstrated that DEX infusion could reduce the concomitant dosing of opiate and benzodiazepine agents in these patients.
In a recent study, our group reported the safe use of DEX in neonates and infants with heart disease. However, this particular study included only children 12 months old or younger receiving DEX for a median period of 78 h. A control group with another agent was missing in this particular study, and the hemodynamic effects were compared with the baseline values used as controls [18].
In another study from our group, we reported that prolonged DEX administration (≥96 h) for children with heart disease appeared to be safe and associated with decreased opioid and benzodiazepine requirement as well as decreased inotropic support [13]. In this same study, we reported that the inotrope score was significantly lower in the DEX group than in the control group during the last 6 h before termination of DEX infusion (p < 0.001) and then 1 h (p < 0.001) and 6 h (p < 0.001) after termination of DEX infusion [13]. Chrysostomou et al. [9] were the first to describe the use of DEX as a sedative agent after cardiac surgery in children and to show no significant adverse cardiovascular, respiratory, or gastrointestinal effects.
Findings have shown DEX to have a significant sympatholytic effect and the ability to blunt endogenous catecholamine release in response to various stimuli, including surgical trauma [23]. A similar sympatholytic effect has been demonstrated with DEX in pediatric patients undergoing cardiopulmonary bypass and surgery for congenital heart disease [19]. Dexmedetomidine has the potential, especially in larger doses or in specific clinical circumstances, to induce direct coronary vasoconstriction resulting in ischemia [10, 15]. Plasma levels of epinephrine and norepinephrine decrease to ~70 % with DEX infusion [22]. In addition, DEX has been shown to diminish myocardial blood flow in parallel with reductions in plasma catecholamines, heart rate, and blood pressure [22].
The most frequently reported adverse effects associated with DEX infusion in published reports are dose-related hypotension and bradycardia [5, 9]. In our cohort, the baseline heart rate in the DEX group was lower before initiation of DEX infusion. Although not statistically significant, this difference could have been due to the fact that the patients in the DEX group were older and larger than the subjects in the control group. This trend in the heart rate continued for 6 h after initiation of DEX infusion. After this period, the two groups showed no difference in heart rate until the termination of DEX infusion. There was a difference in MAP, with lower blood pressures noted in the control group between 2 and 24 h after initiation of DEX infusion. After this period, the two groups did not differ in terms of MAP until termination of DEX infusion.
In this study, we did not notice any difference in inotropic scores in any period during DEX infusion. In one of our prior studies, we observed an improvement in inotrope score with increasing duration of DEX infusion without any significant change in hemodynamics.
It has been proposed that α2 agonists such as DEX aid in restoring vascular reactivity to exogenously administered catecholamines in patients with catecholamine-refractory shock [20]. Sympathetic inhibitors such as α2 agonists lower intrasynaptic catecholamine concentrations, leading to reduction in α1 desensitization. This eventually leads to gradual resensitization of α1 receptors and a better response to exogenously administered catecholamines [11, 20]. These results should be interpreted with caution and investigated in future studies because our findings appear counterintuitive regarding sympatho-inhibition in the setting of catecholamine-refractory shock.
We did not observe any clinical respiratory depression or significant changes in arterial blood gases or oxygen saturation. These results are consistent with outcomes described in other studies with DEX [2, 25]. Other investigators have demonstrated that DEX use is associated with mild decreases in PaO2 levels, oxygen saturation, and mild hypercapnia. These changes, however, appear to be clinically insignificant [12, 17].
We found that DEX administration significantly reduced the overall concomitant daily dosing of opiates and benzodiazepines, which is consistent with other pediatric studies performed in cardiovascular intensive care unit settings [1, 8]. We found that the dose of continuous midazolam was significantly less in the DEX group than in the control group during the last 24 h of DEX infusion. However, the two groups did not differ in the requirement of continuous morphine dose at any time during DEX infusion. The numbers of analgesic and sedation rescue boluses were significantly lower in the DEX group than in the control group.
This was a single-center study, and the results may not be generalizable to other CVICUs with different sedation and analgesia practices. Furthermore, the study included a small number of patients, which may have limited the effect of DEX infusion on sedation, analgesia, and hemodynamics. The retrospective design of the study renders it susceptible to design flaws and bias. We may have introduced selection bias in our study because DEX was used for patients who had failed conventional sedation.
The retrospective nature of the study does not allow us to affirm that the hemodynamic and sedation effects associated with the use of DEX were due to the drug itself and not due to any other reason. We may have potentially missed adverse effects associated with DEX due the limited data availability and the presence of several confounding variables in this population of pediatric cardiac patients that may account for some of the changes in hemodynamic variables. Although the clinical management of the two study groups was similar except for the type of sedation regimen, we cannot exclude the possibility of unrecognized differences or differing treatment practices during the course of the study.
Conclusion
We have evaluated the use of DEX for children with heart failure. Based on our findings, we conclude that DEX administration for children with heart failure appears to be safe and should be used cautiously. Furthermore, DEX use is associated with decreased opiate and benzodiazepine requirements in children with heart failure.
References
Bejian S, Valasek C, Nigro JJ, Cleveland DC, Willis BC (2009) Prolonged use of dexmedetomidine in the paediatric cardiothoracic intensive care unit. Cardiol Young 19:98–104
Belleville JP, Ward DS, Bloor BC, Maze M (1992) Effects of intravenous dexmedetomidine in humans: I. Sedation, ventilation, and metabolic rate. Anesthesiology 77:1125–1133
Berkenbosch JW, Wankum P, Tobias JD (2005) Prospective evaluation of dexmedetomidine for noninvasive procedural sedation in children. Pediatr Crit Care Med 6:435–439
Biccard BM, Goga S, de Beurs J (2008) Dexmedetomidine and cardiac protection for noncardiac surgery: a meta-analysis of randomised controlled trials. Anaesthesia 63:4–14
Bloor BC, Ward DS, Belleville JP, Maze M (1992) Effects of intravenous dexmedetomidine in humans: II. Hemodynamic changes. Anesthesiology 77:1134–1142
Buchhorn R, Ross RD, Bartmus D, Wessel A, Hulpke-Wette M, Bursch J (2001) Activity of the renin-angiotensin-aldosterone and sympathetic nervous system and their relation to hemodynamic and clinical abnormalities in infants with left-to-right shunts. Int J Cardiol 78:225–230
Buchhorn R, Wessel A, Hulpke-Wette M, Bursch J, Werdan K, Loppnow H (2001) Endogenous nitric oxide and soluble tumor necrosis factor receptor levels are enhanced in infants with congenital heart disease. Crit Care Med 29:2208–2210
Buck ML, Willson DF (2008) Use of dexmedetomidine in the pediatric intensive care unit. Pharmacotherapy 28:1–57
Chrysostomou C, Di Filippo S, Manrique AM et al (2006) Use of dexmedetomidine in children after cardiac and thoracic surgery. Pediatr Crit Care Med 7:126–131
Coughlan MG, Lee JG, Bosnjak ZJ, Schmeling WT, Kampine JP, Warltier DC (1992) Direct coronary and cerebral vascular responses to dexmedetomidine: significance of endogenous nitric oxide synthesis. Anesthesiology 77:998–1006
Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R (2008) Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med 34:17–60
Eisenach JC (1994) Alpha2-agonists and analgesia. Expert Opin Investig Drugs 3:1005–1010
Gupta P, Whiteside W, Sabati A, Tesoro T, Gossett JM, Tobias JD, Roth SJ (2012) Safety and efficacy of prolonged dexmedetomidine use in critically ill children with heart disease. Pediatr Crit Care Med. doi:10.1097/PCC.0b013e318253c7f1
Hsu DT, Pearson GD (2009) Heart failure in children: part I: history, etiology, and pathophysiology. Circ Heart Fail 2:63–70
Jalonen J, Halkola L, Kuttila K, Perttilä J, Rajalin A, Savunen T, Scheinin M, Valtonen M (1995) Effects of dexmedetomidine on coronary hemodynamics and myocardial oxygen balance. J Cardiothorac Vasc Anesth 9:519–524
Ko WJ, Lin CY, Chen RJ, Wang SS, Lin FY, Chen YS (2002) Extracorporeal membrane oxygenation support for adult postcardiotomy cardiogenic shock. Ann Thorac Surg 73:538–545
Koroglu A, Demirbilek S, Teksan H (2005) Sedative, haemodynamic, and respiratory effects of dexmedetomidine in children undergoing magnetic resonance imaging examination: preliminary results. Br J Anaesth 94:821–824
Lam F, Bhutta AT, Tobias JD, Gossett JM, Morales L, Gupta P (2012) Hemodynamic effects of dexmedetomidine in critically ill neonates and infants with heart disease. Pediatr Cardiol. Epub ahead of publication
Mukhtar AM, Obayah EM, Hassona AM (2006) The use of dexmedetomidine in pediatric cardiac surgery. Anesth Analg 103:52–56
Pichot C, Géloën A, Ghignone M, Quintin L (2010) Alpha-2 agonists to reduce vasopressor requirements in septic shock? Med Hypotheses 75:652–656
Roekaerts PMH, Prinzen FW, De Lange S (1996) Beneficial effects of dexmedetomidine on ischaemic myocardium of anaesthetized dogs. Br J Anaesth 77:427–429
Snapir A, Posti J, Kentala E, Koskenvuo J, Sundell J, Tuunanen H, Hakala K, Scheinin H, Knuuti J, Scheinin M (2006) Effects of low and high plasma concentrations of dexmedetomidine on myocardial perfusion and cardiac function in healthy male subjects. Anesthesiology 105:902–910
Talke P, Richardson CA, Scheinin M, Fisher DM (1997) Postoperative pharmacokinetics and sympatholytic effects of dexmedetomidine. Anesth Analg 85:1136–1142
Tobias JD (2007) Dexmedetomidine: applications in pediatric critical care and pediatric anesthesiology. Pediatr Crit Care Med 8:115–131
Zornow MH (1991) Ventilatory, hemodynamic, and sedative effects of the alpha 2-adrenergic agonist, dexmedetomidine. Neuropharmacology 30:1065–1071
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lam, F., Ransom, C., Gossett, J.M. et al. Safety and Efficacy of Dexmedetomidine in Children With Heart Failure. Pediatr Cardiol 34, 835–841 (2013). https://doi.org/10.1007/s00246-012-0546-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-012-0546-7