
Abstract
Our aim was to identify prognostic factors for an arrhythmic event (AE) in children with hypertrophic cardiomyopathy (HCM) without a previous AE. One hundred thirty-one nonconsecutive patients (≤20 years) with HCM but no previous AE were evaluated at the NIH Clinical Center from 1980 to 2001. At a median follow-up of 6.4 years, 22 patients experienced an AE [sudden death (SD) (n = 12), resuscitated cardiac arrest (n = 3), clinical sustained ventricular tachycardia (VT) (n = 2), and implantable cardiac defibrillator discharge (n = 5)], resulting in a 2% annual AE rate. Baseline factors that were most predictive in univariate risk analysis included ventricular septal thickness (ST) (P = 0.01), VT induction by programmed ventricular stimulation (PVS) (P = 0.01), age (P = 0.05), and presyncope/syncope (P = 0.05). In multivariate analysis, ST, age, presyncope/syncope, and PVS were not independently predictive of risk for an AE. However, the 5-year event rates for AE was 15% (95% CI: 5–23%) if ST ≥ 20 mm, 19% (95% CI: 6–31%) when age ≥ 13 years and ST ≥ 20 mm were combined together, and 23% (95% CI: 3–39%) when PVS and ST ≥ 20 mm were combined together. Of the various risk factors that were considered in our pediatric HCM cohort, ST and inducible VT were the most significant univariate predictors of risk for an AE. More traditional risk factors identified in older patients (family history of SD, VT on Holter, and exercise-induced hypotension) were not predictive of an AE in patients age under 21 years.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypertrophic cardiomyopathy (HCM) is the commonest inherited cardiomyopathy, with a prevalence of 0.2% [11]. It is an important cause of arrhythmic events (AEs) in young people with an estimated annual mortality varying between 1 and 6% [5, 11, 14, 17, 29, 33]. Implantable cardiac defibrillator (ICD) therapy can be life-saving. On the other hand, ICD therapy can result in considerable morbidity and psychological burden if used indiscriminately [2, 5, 23, 29, 30]. Recent reports have shown ICD lead failure rates varying from 2 to 21% in children and young adults [2, 23, 30] and a very high rate of inappropriate shock rates in the HCM population (23–33% of patients) [28, 52], highlighting the importance of selecting the highest risk population of children most likely to benefit from defibrillator therapy.
Traditional risk factors for sudden death (SD) in adult HCM patients include a family history of SD, young age, nonsustained ventricular tachycardia (VT) on Holter, increased septal thickness (ST), abnormal blood pressure (BP) response to exercise, and syncope [17].
The purpose of this study was to determine (1) whether risk factors for AE in adults would have predictive value in children and (2) if not, what clinical parameters would identify subgroups of children and adolescents that might benefit the most from ICD implantation.
Methods
Patient Population
Between 1980 and 2001, 144 children (≤20 years of age) with HCM were evaluated in the Cardiology Branch, National Heart Lung and Blood Institute (NHLBI), National Institutes of Health (NIH), Bethesda, MD and underwent an electrophysiology study (EPS). Two patient subsets were of interest: (Group 1) 131 patients who did not have a prior (preclinical presentation at the NIH) AE and who had an EPS and (Group 2) 13 patients who had a prior AE and who had an EPS. These two patient groups were identified retrospectively from our larger clinical database of patients with HCM who had undergone clinical evaluation. Study inclusion criteria were (1) clinical evaluation at the NIH between 1980 and 2001, (2) age ≤ 20 years, (3) consent to participate in a research protocol, (4) consent for publication of clinical data, and (5) an EPS. The primary focus of this article is the first group of 131 patients. This research study was conducted under a separate NHLBI Institutional Review Board approved protocol.
Hypertrophic cardiomyopathy was diagnosed by echocardiographic demonstration of a hypertrophied nondilated left ventricle (LV) in the absence of another cause of LV hypertrophy.
Baseline evaluation included an electrocardiogram and a two-dimensional echocardiogram. Ventricular ST was measured in mm and also indexed to body surface area (BSA) (mm/m2). Patients undertook additional tests depending on patient age, clinical presentation, patient/family willingness to participate in protocols, and year of presentation. These studies included 24–48-h ambulatory electrocardiographic monitoring, exercise stress test, myocardial perfusion scan, cardiac catheterization and angiography, and EPS.
Electrophysiologic Study
EPS was performed under fasting conditions and conscious sedation. Cardioactive drugs were discontinued five half-lives prior to study. Details of the EP protocol have been described [5, 16]. Programmed ventricular stimulation (PVS) involved up to three premature extrastimuli delivered during drive cycle lengths (600, 500, and 400 ms) at the right ventricular (RV) apex and RV outflow tract (OT). The end point of the stimulation protocol was induction of a sustained ventricular arrhythmia or shortening of the coupling intervals down to ventricular refractoriness. PVS was only performed in the baseline state; isoproterenol was not administered. VT was defined as three or more consecutive ventricular beats at a rate of >100 beats/min. Nonsustained VT was defined as VT terminating spontaneously in less than 30 s. Sustained VT was defined as VT lasting greater than 30 s in duration or requiring termination because of hemodynamic compromise. VT with continuously changing QRS morphology was termed polymorphic and that with uniform QRS complexes was termed monomorphic.
Patient Management
Therapy was based on the patient’s clinical presentation, cardiac catheterization, and EP findings. Treatment was initiated with the intent of relieving symptoms and treating identified mechanisms of syncope or AE. The study population dated back to 1980, when ICD therapy was very rarely used in children. Over time, with changes in technology and indications for device implantation, the frequency of ICD implantation increased. In the early era, only drug therapy was readily available. A total of 40 Group 1 subjects underwent ICD implantation. ICD implantation was also advised in 10 patients whose families refused. Beta-blockers were used in 70 of 125 (56%) patients for whom such data were available. However, only 46 of 70 (66%) patients continued beta-blocker therapy long term. Five patients underwent septal myomyectomy.
Follow-Up
Follow-up included annual visits to the NIH, reassessment by the community cardiologist, and telephone calls to the patient and local physicians. When possible, ICDs were interrogated to review electrograms, event markers, and counter data for VT or ventricular fibrillation (VF). Episodes of VT/VF associated with ICD discharge were considered to be an AE if coincident with a clinical event (e.g., lightheadedness, rapid palpitations, or loss of consciousness).
Definitions
A modified Hinkle–Thaler system was used to classify AEs [20]. AEs included sudden unwitnessed deaths, sustained VT, successful resuscitation from a cardiac arrest (CA), and ICD therapy for appropriately treated episodes of VT/VF. Exercise-induced hypotension was defined as a lack of increase in systolic BP by more than 10 mmHg during treadmill exercise. LV outflow tract obstruction was defined as a subvalvular gradient >30 mmHg at rest or ≥50 mmHg after provocation (isoproterenol or premature ventricular complex).
Predictors of Arrhythmic Events
Clinical features examined as potential predictors included: age at presentation to the NIH, family history of SD, presyncope or syncope, exercise-induced hypotension, ST, LV outflow obstruction, elevated LV end diastolic pressure, QRS duration, QT interval, VT on ambulatory electrocardiographic monitoring, myocardial ischemia on stress nuclear perfusion imaging, inducible VT, intracardiac conduction intervals, and ventricular refractory periods.
Statistical Analysis
The time of first NIH attendance was used as the baseline in the time-to-AE analyses. Statistical significance for the univariate time-to-event analyses was assessed using the log rank statistic for dichotomous variables and Cox’s score statistic for continuous variables. Statistical significance for the multivariate time-to-event analyses was assessed using Cox’s Wald statistic. Baseline differences between inducible VT and electrophysiologic findings were assessed using the t-test. For all analyses, a two-sided P-value ≤ 0.05 was deemed statistically significant. Unless otherwise indicated, data are reported as median ± standard deviation.
Results
Baseline Characteristics and Follow-Up
The baseline clinical characteristics and risk factors for AE are summarized in Table 1. The median follow-up was 6.4 years (interquartile range: 3.0–9.6; maximum: 21.3). Twenty-two patients experienced AEs that included SD (n = 12), resuscitated CA (n = 3), clinical sustained VT (n = 2), and ICD discharge (n = 5). Twenty of the 22 patients were ≥14 years of age at the time of their AE (mean age: 20.1 ± 6.2 years).
Forty patients had an ICD, 11 of whom had an AE. AEs occurred subsequent to ICD implantation in seven patients (ICD discharge = 5 and SD = 2).
Risk Factors for an Arrhythmic Event
Table 2 describes the univariate prognostic significance of baseline characteristics for a subsequent AE. Inducible VT and ST were most strongly associated with an AE. Other risk factors found significant in univariate analysis were older age at the time of initial NIH evaluation and presyncope/syncope. Multivariate analysis did not reveal ST (P = 0.13), age (P = 0.10), presyncope/syncope (P = 0.29), or inducible VT (P = 0.21) to be independently predictive of an AE. The traditional risk factors for AE in adult HCM patients of family history of SD, nonsustained VT on Holter, and abnormal BP response to exercise were not significantly related to the occurrence of AE.
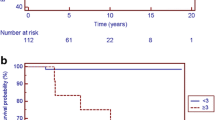
Patients with inducible VT had a significantly higher AE rate than those with noninducible VT (5-year rates of 22 vs. 6%, respectively; log rank P = 0.01; Fig. 1a).

Cumulative probability, with 95% Greenwood confidence intervals, of an AE according to baseline inducibility of VT (a) and ventricular ST (b). a Log rank P-value = 0.01 for the difference between the EP+ and the EP− group for follow-up to 5 years. b Log rank P-value = 0.01 for the difference between the ≥20-mm group and the <20-mm group for follow-up to 5 years. Log rank P-value = 0.94 for the difference between the 30-mm group and the 20–29 mm group for follow-up to 5 years
Patients with ST < 20 mm were at significantly lower risk for an AE than were patients with ST ≥ 20 mm (Five-year event rates of 0 vs. 14%, respectively; P = 0.01; Fig. 1b). In particular, among the 51 patients with ST < 20 mm (median follow-up: 5.9 years), only 2 patients had an AE during follow-up (16 and 20 years after baseline evaluation). ST increased from 18 to 20 mm by year 15 of follow-up in the first patient and from 14 to 28 mm by year 14 of follow-up in the second patient. ST was ≥20 mm in 19 of 21 patients who developed an AE and had baseline ST data; ST data were missing in one patient who developed an AE.
When analyzed as a continuous variable, larger absolute ST (mm), as well as larger ST divided by BSA (mm/m2) were significantly predictive of an AE (Table 2). There was no statistical difference in age between patients with ST < 20 mm and patients with ST ≥ 20 mm (13.2 ± 5.1 vs. 13.9 ± 4.1 years, P = 0.37). ST ≥ 20 mm was independently predictive of age for an AE (P = 0.003).
When combined, children ≥ 13 years of age and a ST ≥ 20 mm had a 5-year event rate of 19% (95% CI: 6–31%). Children <13 years of age and ST ≥ 20 mm had a 5-year event rate of 6% (95% CI: 0–16%) (see Fig. 2).

Cumulative probability, with 95% Greenwood confidence intervals, of an AE according to baseline age and ventricular ST. Log rank P-value = 0.11 for the difference between the ≥20-mm and ≥13-year-old group and the ≥20-mm and <13-year-old group for follow-up to 5 years. Log rank P-value = 0.18 for the difference between the ≥20 mm and <13-year-old group and the <20-mm group for follow-up to 5 years
The probability of AE curves stratified according to VT inducibility and ST ≥ 20 mm are shown in Fig. 3. In particular, children without inducible VT but ST ≥ 20 mm had a 5-year event rate of 10% (95% CI: 4–25%). Children with inducible VT and ST ≥ 20 mm had a 5-year event rate of 23% (95% CI: 10–47%). Children with ST < 20 mm had a 5-year AE rate of 0%.

Cumulative probability, with 95% Greenwood confidence intervals, of an AE according to baseline inducibility of VT and ventricular ST. Log rank P-value = 0.12 for the difference between the ≥20-mm EP+ group and ≥20-mm EP− group for follow-up to 5 years. Log rank P-value = 0.04 for the difference between the ≥20-mm EP− group and the <20-mm group for follow-up to 5 years
By comparison, there were 13 children (Group 2) referred because of a prior AE during the same time period as the 131 patients who did not have a prior AE reported here. Six of those 13 patients had an AE during follow-up (5-year rate = 43%), making that group a higher AE risk group (P < 0.0001) than the 131 patients (5-year rate = 9%). In those 13 patients, VT was induced during EPS in 3 of the 6 patients (50%) who had a subsequent AE. Ventricular ST in the patient group with a prior AE did not differ significantly from the EPS patient group without a prior AE (24.6 ± 6.6 vs. 23.8 ± 9.4 mm; P = 0.76).
Electrophysiologic Findings and Inducible VT
Ventricular tachycardia was induced in 28/131 (21%) patients (nonsustained 11; sustained 17). The combination of sustained or nonsustained VT was significantly predictive of an AE (P = 0.01), as was sustained VT alone (P = 0.03). VT was induced using two extrastimuli in nine and three extrastimuli in 18 patients. The number of extrastimuli needed for VT induction was unknown in one patient studied outside of the NIH. There was no difference in AE outcome whether VT was induced using two extrastimuli or three extrastimuli (P = 0.39). Induced VT was polymorphic in 24 patients, monomorphic in 3 patients, and of unknown morphology in 1 patient. Ventricular extrastimulus testing was performed down to ventricular refractoriness. At a drive cycle length of 400 ms, the V1V2, V2V3, and V3V4 coupling intervals in the noninducible group were 268 ± 21, 228 ± 21, and 212 ± 21 ms, respectively; in the inducible group, they were 268 ± 19, 220 ± 20, and 209 ± 18 ms, respectively. Although statistically significant (P = 0.04), the 8-ms difference in the V2V3 coupling interval was not clinically significant.
Patients with inducible VT were older (15.7 ± 2.3 vs. 12.9 ± 4.7 years, P = 0.003); nevertheless, inducible VT was nearly independent of age as an AE predictor (P = 0.06). Patients with inducible VT had a prolonged HV interval (55 ± 11 vs. 49 ± 10 ms, P = 0.01) and a marginally prolonged QTC interval (433 ± 27 vs. 419 ± 31 ms, P = 0.04). There were no significant relationships between inducible VT and AH interval, QRS interval, or ventricular refractory period in the RV apex or RVOT.
Discussion
In this study, the AE rate (SD, resuscitated CA, sustained VT, or defibrillator discharge) in children with HCM but no previous AE was 2% per year, quite comparable to the rate recently reported in a large pediatric cardiomyopathy registry (1.4% per year) [11]. Univariate analysis identified ST (either in absolute terms or indexed to BSA) and inducible VT as significantly associated with AE risk. Age, presyncope or syncope, QTC, and RVOT ERP were marginally predictive in univariate analysis for an AE; family history of SD, exercise-induced hypotension, nonsustained VT on Holter, and myocardial ischemia were not significant. Multivariate analysis did not reveal ST, inducible VT, age, or presyncope/syncope to be independently significant for an AE. This might be due to the relatively small number of AEs as well as the possible correlation among these risk factors. The combination of inducible VT and ST ≥ 20 mm or patient age ≥ 13 years and ST ≥ 20 mm had the highest 5-year AE rates. In contrast, patients with a ST < 20 mm had a 5-year event rate of 0%.
The 13 children in our database with a prior AE had a significantly higher subsequent AE rate than the 131 patients without a prior AE, despite similar ventricular ST at time of presentation. This identifies a previous AE as one of the highest risk factors for a subsequent AE. Maron et al. [31] found similar data for high risk of AE recurrence (53%) in HCM individuals who suffered a previous AE.
Left Ventricular Hypertrophy
Recent reports provide conflicting data on the association between the magnitude of LV hypertrophy and SD risk in adult HCM patients. Some authors have reported an increased SD risk when LV ST exceeds 15 mm [49], whereas others could not identify any relationship between LV ST and SD [15, 39]. The current data provide evidence for increased risk of AEs in children when ST is ≥20 mm. This risk was compounded in the presence of inducible VT and older age.
Östman-Smith et al. [40] reported that ST over 190% of the 95th percentile for age was a significant risk marker for AE in children with HCM (odds ratio = 6.2, 95% CI = 1.5–25.1). Östman-Smith et al. [40] used the following equation from their earlier work for the 95th percentile of ST in mm: 6.25 + (age in years × 0.0269). In the age group 14–21 years, a ST of 19–22 mm defines ST over 190% of the 95th percentile for age, agreeing with our finding of 20 mm as being associated with higher AE risk.
Basso et al. [4] studied morphologically the hearts from 19 patients with HCM in the age group ≤35 years who died suddenly. ST averaged 21.8 mm and measured ≥20 mm in 12 of 19 patients. Our results are concordant with these findings. In our study, ST measured at the time of clinical presentation was ≥20 mm in 19 of 21 patients who developed AE and had ST data (ST was unknown for one patient who had a subsequent AE).
Absence of LV hypertrophy on an electrocardiogram has been shown to have a lower SD rate in HCM [34]. ST averaged 17.3 mm in the group presenting with a normal ECG (CM). Similar to our study, patients with a ST < 20 mm had a 5-year event rate of 0%.
Induction of VT at EP Study and Polymorphic VT
In previous studies of children with congenital heart disease, inducible VT predicted a threefold increased risk of AEs, whereas noninducibility favored a good outcome (3% false negative rate) [1, 22]. In this study of children with HCM, inducible VT was an important univariate predictor of an AE (hazard ratio = 3.1, 95% CI = 1.2–8.1).
Previous studies have shown the value of EPS in adult patients with HCM and other forms of cardiomyopathy. Induction of VT during EPS was associated with increased risk for ICD firing in adult subjects with arrhythmogenic RV dysplasia (odds ratio = 11.2, 95% CI = 1.23–101.24) [43]. Although EPSs in adults with HCM have limited predictive value during short-term follow-up [3, 18, 21, 24, 26, 47, 54], a negative test predicted a better prognosis in studies with longer follow-up [54].
Anatomic studies have described myocardial fiber disarray and patchy fibrosis in HCM. Fragmentation of electrograms was detected at long premature ventricular coupling intervals in patients with previous VT/VF [46], which could provide the substrate for unstable conduction and reentry and, hence, polymorphic VT/VF.
Traditionally, inducible polymorphic VT was thought to have little predictive value. However, in the Multicenter UnSustained Tachycardia Trial, patients with coronary artery disease and inducible sustained VT/VF had higher mortality from CA than noninducible patients, irrespective of VT morphology [7]. The 2-year adverse AE rate was slightly higher in patients with sustained monomorphic or polymorphic VT/VF induced with two extra stimuli (18%) compared with sustained polymorphic VT induced using three extra stimuli (14%) [8]. More recently, Greenberg et al. [19] showed no difference in appropriate ICD discharge rate or mortality between patients dichotomized by type of arrhythmia induced during PVS (monomorphic VT = 20 vs. polymorphic VT/VF = 21%). Similarly, in congenital heart disease, sustained polymorphic VT trended toward the worse outcome [1].
In our study, polymorphic VT was the predominant morphology of VT induced. Pablo Kaski et al. [42] reviewed the type of ventricular arrhythmias resulting in appropriate ICD shocks in 22 children with HCM. Arrhythmia morphology identified from 15 appropriate shocks were ventricular fibrillation (n = 11), polymorphic VT (n = 2) and monomorphic VT (n = 2), confirming the importance of ventricular fibrillation and polymorphic VT as a clinically significant arrhythmia in children with HCM [42]. These results differ from what has been reported in the adult HCM population in whom monomorphic VT and ventricular fibrillation predominate [10].
The safety of EPSs has been questioned following reports of HCM patients refractory to defibrillation [25, 51]. No complication related to failure of defibrillation was encountered in the current pediatric study or in an earlier adult study [16].
Other Risk Factors for Sudden Death
Risk factors for SD vary between different adult age groups with HCM [6, 9, 27, 32, 37, 38, 45, 50], but most studies include few patients aged less than 21 year of age. As in our study, Decker et al. [12] were unable to show that risks factors identified for SD in adults predicted SD in a pediatric HCM cohort of 96 patients. However, this might have been due to the limited statistical power of the two studies.
Syncope
Syncope has been regarded as an important predictor of SD in children given the low predictive value of other clinical/laboratory findings [32]. By comparison, syncope in adults had low predictive value except in combination with a family history of SD [14]. In this study, syncope/presyncope was of marginal statistical significance in univariate analysis.
Abnormal Blood Pressure Response to Exercise
In approximately one-third of HCM patients, systolic BP does not change or decreases during exercise, consistent with the current report. Lack of BP response to exercise has been attributed to exaggerated vasodilatory responses [27] and to abnormal reflex responses in venous capacitance [50], increasing LVOT obstruction, myocardial ischemia or abnormal diastolic relaxation. Abnormal BP response to exercise might be an independent predictor of CA in adults <40 years but not >40 years [17, 45]. Consistent with previous reports, the current study failed to show an association between exercise-induced hypotension and an AE [5, 38].
Family History of Sudden Death
A malignant family history of SD has been associated with increased adverse events [17], but detection of “malignant” genetic mutations did not always predict adverse outcomes [6]. In this study, a family history of SD was not predictive of an AE.
Nonsustained VT on Holter
Ventricular tachycardia on Holter occurred in 18% of children in this study and was not a risk marker. The incidence of VT on Holter is generally higher in adult HCM patients (14–50%) [9, 17, 21, 37] and has variable prognostic significance [9].
Myocardial Ischemia
Myocardial ischemia detected by thallium-201 scintigraphy was a frequent finding in this study. An earlier report suggested that myocardial ischemia was common in children with a history of CA or syncope [13]. It remains uncertain whether myocardial bridging contributes to ischemia and SD in both children and adults [36, 44, 48, 53].
Time-Limited Nature of Risk Stratification
Risk stratification for AEs should be considered a time-limited process in children. In medicine, risk is thought to evolve over time, usually increasing. Because of age-related changes in the myocardial substrate (changes in OT gradients, ST, and fibrosis), the substrate for AEs is likely to alter. The median age of our EP group was 14.4 ± 3.9 years, representing early adolescence. A 5-year time frame represents the transition to late adolescence and early adulthood, when myocardial septal hypertrophy might be nearing a plateau. We envisioned but do not have the data to support a reassessment at 18–21 years of age. Consequently, for smaller/younger patients for whom ICD implantation is technically unattractive, noninducibility, age < 13 years, and a ST < 20 mm might be viewed as good justification for delaying implant, but only until the patient is older and larger, when the patient should be reassessed. However, initial risk assessment should not be delayed to late adolescence because for the 13 patients who presented with an AE prior to NIH evaluation, their mean age at NIH evaluation was 13.8 ± 3.5 years.
Limitations
The effect of patient choice on enrollment, testing, and data collection cannot be quantified. Not all children evaluated at the NHLBI underwent PVS; technical limitations and younger patient age affected decisions as to whether to perform certain invasive procedures. Most patients were symptomatic at presentation and were treated with what was considered to be the best available therapy for hemodynamic and electrophysiologic abnormalities. The effect of such therapy on outcome is also unknown, although recent data have confirmed the absence of efficacy of pharmacologic treatment in the prevention of sudden death in HCM [35]. The small number of adverse events in our study group somewhat limited the predictive power of our analysis.
Whereas the children evaluated at the NHLBI might be considered a referral population, our study group is not unlike other reported pediatric HCM series. The incidence of an AE in the study reported by Östman-Smith et al. [41] from a multicenter European HCM study group was 39/150 (26%), higher than what was observed in our study population 22/131(17%). Olivotto et al. [39] found the incidence of severe ST (≥30 mm) in patients ≤15 years to be 24%, which was lower than in our study 33%.
Conclusions
One hundred thirty-one children and adolescents with HCM but no prior AE had an annualized AE rate of 2% per year. Patients with an AE prior to evaluation continued to have a high incidence of subsequent AEs (11% per year). The current study found in univariate analyses that ST and VT induced at EPS were significant predictors of AEs in children with HCM. Older age and presyncope or syncope were of marginal statistical significance. However, perhaps due to the limited statistical power of our study, ST, age, presyncope/syncope, and inducible VT were not independently predictive of risk for an AE.
Our data suggest that ICD implantation has stronger justification when both ST is ≥20 mm and there is a positive EP test (5-year event rate for AE = 23%) than when either factor occurs alone. Nevertheless, the ST ≥ 20 mm and EP negative group had a 5-year event rate of 10% and was at significantly greater risk (P = 0.04) than the ST < 20 mm group. Thus, newer technologies using smaller ICD leads and devices with improved algorithms for VT detection might suggest that ST ≥ 20 mm alone is sufficient to justify ICD implantation.
References
Alexander ME, Walsh EP, Saul JP, Epsein MR, Triedman JK (1999) Value of programmed ventricular stimulation in patients with congenital heart disease. J Cardiovasc Electrophysiol 10:1033–1044
Alexander ME, Cecchin F, Walsh EP, Triedman JK, Bevilacqua, Berul CI (2004) Implications of implantable cardioverter defibrillator therapy in congenital heart disease and pediatrics. J Cardiovasc Electrophysiol 15:72–76
Anderson KP, Stinson EB, Derby GC, Oyer PE, Mason JW (1983) Vulnerability of patients with obstructive hypertrophic cardiomyopathy to ventricular arrhythmia induction in the operating room. Analysis of 17 patients. Am J Cardiol 51:811–816
Basso C, Thiene G, Corrado D, Buja G, Melacini P, Nava A (2000) Hypertrophic cardiomyopathy and sudden death in the young: pathologic evidence of myocardial ischemia. Hum Pathol 31:988–998
Begley DA, Mohiddin SA, Tripodi D, Winkler JB, Fananapazir L (2003) Efficacy of implantable cardioverter defibrillator therapy for primary and secondary prevention of sudden cardiac death in hypertrophic cardiomyopathy. Pacing Clin Electrophysiol 26:1887–1896
Brito D, Richard P, Isnard R, Pipa J, Komajda M, Madeira H (2003) Familial hypertrophic cardiomyopathy: the same mutation, different prognosis. Comparison of two families with long follow-up. Rev Port Cardiol 22:1445–1461
Buxton AE, Lee KL, Fisher JD, Josephson ME, Prystowsky EN, Hafley G (1999) A randomized study of the prevention of sudden death in patients with coronary artery disease. N Engl J Med 341:1882–1890
Buxton AE, Lee KL, DiCarlo L, Gold MR, Greer GS, Prystowsky EN, O’Toole MF, Tang A, Fisher JD, Coromilas J, Talajic M, Hafley G (2000) Electrophysiologic testing to identify patients with coronary artery disease who are at risk for sudden death. N Engl J Med 342:1937–1945
Cecchi F, Olivotto I, Montereggi A, Squillatini G, Dolara A, Maron BJ (1998) Prognostic value of non-sustained ventricular tachycardia and the potential role of amiodarone treatment in hypertrophic cardiomyopathy: assessment in an unselected non-referral based patient population. Heart 79:331–336
Cha YM, Gersh BJ, Maron BJ, Boriani G, Spirito P, Hodge DO, Weivoda PL, Trusty JM, Friedman PA, Hammill SC, Rea RF, Shen WK (2007) Electrophysiologic manifestations of ventricular tachyarrhythmias provoking appropriate defibrillator interventions in high-risk patients with hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol 18:483–487
Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF, Lurie PR, Orav EJ, Towbin JA (2007) Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children. Findings from the pediatric cardiomyopathy registry. Circulation 15:773–781
Decker JA, Rossano JW, Smith EO, Cannon B, Clunie SK, Gates C, Jefferies J, Kim J, Price JF, Dreyer WJ, Towbin JA, Denfield SW (2009) Risk factors and mode of death in isolated hypertrophic cardiomyopathy in children. J Am Coll Cardiol 54:250–254
Dilsizian V, Bonow R, Epstein SE, Fananapazir L (1993) Myocardial ischemia detected by thallium scintigraphy is frequently related to cardiac arrest and syncope in young patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 22:796–804
Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, Mahon NG, McKenna WJ (2000) Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 36:2212–2218
Elliott PM, Gimeno Blanes JR, Mahon NJ, Poloniecki JD, McKenna WJ (2001) Relation between severity of left ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet 357:420–424
Fananapazir L, Tracy CM, Leon MB, Winkler JB, Cannon RO 3rd, Bonow RO, Maron BJ, Epstein SE (1989) Electropysiologic abnormalities in patients with hypertrophic cardiomyopathy. A consecutive analysis in 155 patients. Circulation 80:1259–1268
Frenneaux MP (2004) Assessing the risk of sudden cardiac death in a patient with hypertrophic cardiomyopathy. Heart 90:570–575
Geibel A, Brugada P, Zehender M, Stevenson W, Waldecker B, Wellens HJJ (1987) Value of programmed electrical stimulation using a standardized ventricular stimulation protocol in hypertrophic cardiomyopathy. Am J Cardiol 60:738–739
Greenberg SL, Mauricio Sánchez J, Cooper JA, Cain ME, Chen J, Gleva MJ, Lindsay BD, Smith TW, Faddis MN (2007) Sustained polymorphic arrhythmias induced by programmed ventricular stimulation have prognostic value in patients receiving defibrillators. Pacing Clin Electrophysiol 30:1067–1075
Hinkle LE, Thaler HT, Merke DP, Renier-Berg D, Morton NE (1988) The risk factors for arrhythmic death in a sample of men followed for 20 years. Am J Epidemiol 127:500–515
Jansson K, Dahlstrom U, Karlsson E, Nylander E, Walfridsson H, Sonnhag C (1990) The value of exercise test, Holter monitoring, and programmed electrical stimulation in detection of ventricular arrhythmias in patients with hypertrophic cardiomyopathy. Pacing Clin Electrophysiol 13:1261–1267
Khairy P, Landzberg MJ, Gatzoulis MA, Lucron H, Lambert J, Marçon F, Alexander ME, Walsh EP (2004) Value of programmed ventricular stimulation after tetralogy of Fallot repair: a multicenter study. Circulation 109:1994–2000
Kleemann T, Becker T, Doenges K, Vater M, Senges J, Schneider S, Saggau W, Weisse U, Seidl K (2007) Annual rate of transvenous defibrillation lead defects in implantable cardioverter-defibrillators over a period of >10 years. Circulation 115:2474–2480
Kowey PR, Eisenberg R, Engel TR (1984) Sustained arrhythmias in hypertrophic cardiomyopathy. N Engl J Med 310:1566–1569
Krikler DM, Davies MJ, Fowland E, Goodwin JF, Evans RC, Shaw DB (1980) Sudden death in hypertrophic cardiomyopathy: associated accessory atrioventricular pathways. Br Heart J 43:245–251
Kuck KH, Kunze KP, Schluter M, Nienaber CA, Costard A (1988) Programmed electrical stimulation in hypertrophic cardiomyopathy. Results in patients with and without cardiac arrest or syncope. Eur Heart J 9:177–185
Lim PO, Morris-Thurgood JA, Frenneaux MP (2002) Vascular mechanisms of sudden death in hypertrophic cardiomyopathy, including blood pressure responses to exercise. Cardiol Rev 10:15–23
Lin G, Nishimura RA, Gersh GJ, Phil D, Ommen SR, Ackerman MJ, Brady PA (2009) Device complications and inappropriate implantable cardioverter defibrillator shocks in patients with hypertrophic cardiomyopathy. Heart 95:709–714
Maron BJ, Shen WK, Link MS, Epstein AE, Almquist AK, Daubert JP, Bardy GH, Favale S, Rea RF, Boriani G, Estes NA 3rd, Spirito P (2000) Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med 342:365–373
Maron BJ, Spirito P, Shen WK, Haas TS, Formisano F, Link MS, Epstein AE, Almquist AK, Daubert JP, Lawrenz T, Boriani G, Estes NA III, Favale S, Piccininno M, Winters SL, Santini M, Betocchi S, Arribas F, Sherrid MV, Buja G, Semsarian C, Bruzzi P (2007) Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA 298:405–412
Maron BJ, Haas TS, Shannon KM, Almquist AK, Hodges JS (2009) Long term survival after cardiac arrest in hypertrophic cardiomyopathy. Heart Rhythm 6:993–997
McKenna WJ, Deanfield JE (1984) Hypertrophic cardiomyopathy: an important cause of sudden death. Arch Dis Child 59:971–975
McKenna WJ, Franklin RC, Nihoyannopoulos P, Robinson KC, Deanfield JE (1988) Arrhythmia and prognosis in infants, children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiolol 11:147–153
McLeod CJ, Ackerman MJ, Nishimura RA, Tajik AJ, Gersh BJ, Ommen SR (2009) Outcome of patients with hypertrophic cardiomyopathy and a normal electrocardiogram. J Am Coll Cardiolol 54:229–233
Melacini P, Maron BJ, Bobbo F, Basso C, Tokajuk B, Thiene G, Iliceto S (2007) Evidence that pharmacologic strategies lack efficacy for the prevention of sudden death in hypertrophic cardiomyopathy. Heart 93:708–710
Mohiddin SA, Begley D, Shih J, Fananapazir L (2000) Myocardial bridging does not predict sudden death in children with hypertrophic cardiomyopathy but is associated with more severe cardiac disease. J Am Coll Cardiol 36:2270–2278
Monserrat L, Elliott PM, Gimeno JR, Sharma S, Penas-Lado M, McKenna WJ (2003) Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. J Am Coll Cardiol 42:873–879
Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F (1999) Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 33:2044–2051
Olivotto I, Gistri R, Petrone P, Pedemonte E, Vargiu D, Cecchi F (2003) Maximum left ventricular thickness and risk of sudden death in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 41:315–321
Östman-Smith I, Wettrell G, Keeton B, Riesenfeld T, Holmgren D, Ergander U (2005) Echocardiographic and electrocardiographic identification of those children with hypertrophic cardiomyopathy who should be considered at high-risk of dying suddenly. Cardiol Young 15:632–642
Östman-Smith I, Wettrell G, Keeton B, Holmgren D, Ergander U, Gould S, Bowker C, Verdicchio M (2008) Age and gender-specific mortality rates in childhood hypertrophic cardiomyopathy. Eur Heart J 29:1160–1167
Pablo Kaski JP, Tome Esteban MT, Lowe M, Sporton S, Rees P, Deanfield JE, McKenna WJ, Elliott PM (2007) Outcomes after implantable cardioverter-defibrillator treatment in children with hypertrophic cardiomyopathy. Heart 93:372–374
Roguin A, Bomma CS, Nasir K, Tandri H, Tichnell C, James C, Rutberg J, Crosson J, Spevak PJ, Berger RD, Halperin HR, Calkins H (2004) Implantable cardioverter-defibrillators in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol 43:1843–1852
Romero-Farina G, Candell-Riera J, Pereztol-Valdés O, Castell J, Aguadé S, Galve E, Palet J, Oller-Martínez G, Armadans L, Reina D, Soler-Soler J (2001) Myocardial perfusion SPECT and isotopic ventriculography in obstructive and non-obstructive hypertrophic myocardiopathy. Rev Esp Med Nucl 20:530–536
Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ (1997) Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 96:2987–2991
Saumarez RC, Slade AK, Grace AA, Sadoul N, Camm AJ, McKenna WJ (1995) The significance of paced electrogram fractionation in hypertrophic cardiomyopathy: a prospective study. Circulation 91:2762–2768
Schiavone WA, Maliney JD, Lever HM, Castle LW, Sterba R, Morant V (1986) Electrophysiologic studies of patients with hypertrophic cardiomyopathy presenting with syncope of undetermined etiology. Pacing Clin Electrophysiol 9:476–481
Sorajja P, Ommen SR, Nishimura RA, Gersh BJ, Tajik AJ, Holmes DR (2003) Myocardial bridging in adult patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 42:889–894
Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ (2000) Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med 342:1778–1785
Thomas HL, Morris-Thurgood J, Atherton J, McKenna WJ, Frenneaux MP (1998) Reflex responses of venous capacitance vessels in patients with hypertrophic cardiomyopathy. Clin Sci 94:339–346
Wellens JHH, Bar FW, Vanagt EJ (1980) Death after ajmaline administration. Am J Cardiol 45:905
Woo A, Monakier D, Harris L, Hill A, Shah P, Douglas Wigle E, Rakowski H, Rozenblyum E, Cameron DA (2007) Determinants of implantable defibrillator discharges in high risk patients with hypertrophic cardiomyopathy. Heart 93:1044–1045
Yetman AT, McCrindle BW, MacDonald C, Freedom RM, Gow R (1998) Myocardial bridging in children with hypertrophic cardiomyopathy: a risk factor for sudden death. N Engl J Med 339:1201–1209
Zhu DWX, Sun H, Hill R, Roberts R (1998) The value of electrophysiologic study and prophylactic implantation of cardioverter defibrillator in patients with hypertrophic cardiomyopathy. Pacing Clin Electrophysiol 21:299–302
Acknowledgment
This work was supported by intramural research funds from the National Heart, Lung and Blood Institute, NIH, Bethesda, MD.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moak, J.P., Leifer, E.S., Tripodi, D. et al. Long-Term Follow-Up of Children and Adolescents Diagnosed with Hypertrophic Cardiomyopathy: Risk Factors for Adverse Arrhythmic Events. Pediatr Cardiol 32, 1096–1105 (2011). https://doi.org/10.1007/s00246-011-9967-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-011-9967-y