
Abstract
Historically, outcomes of patients with heterotaxy syndrome and congenital heart disease have been poor. Published series include patients treated over many decades or focus on specific patient/operative subgroups. This study was performed to evaluate midterm outcomes and determine anatomic risk factors for death in patients with all types of heterotaxy syndrome treated after 1985. We reviewed all infants with heterotaxy born between 1985 and 1997, presenting to one institution at <6 months age, followed for ≥5 years. Of 102 study patients (46 with asplenia phenotype, 56 with polysplenia phenotype), 48 (47%) died at a median age of 0.6 months, 12 without intervention. Survivors were followed for a median of 12.8 years (5–21.7 years). Independent predictors of mortality included obstructed totally anomalous pulmonary venous connection (TAPVC) (OR, 7.8; 95% CI, 1.9–32.9; p = 0.005), mild or greater atrioventricular (AV) valve regurgitation at presentation (OR, 3.5; 95% CI, 1.0–12.1; p = 0.03), and common AV canal (OR, 3.1; 95% CI, 1.1–8.5; p = 0.03). Sixteen patients developed pulmonary vein stenosis at a median age of 2 months, with 5 (31%) alive at follow-up. In patients with TAPVC, the mean indexed sum of pulmonary vein diameters was lower among patients who died than in survivors (42.3 ± 8.3 vs 49.5 ± 10.1 mm/m2; p = 0.02). Mortality remains high among patients with heterotaxy treated in the modern surgical era, particularly those with obstructed TAPVC. Pulmonary vein stenosis is common after repair of TAPVC in patients with heterotaxy, may be diagnosed beyond infancy, and is associated with poor outcomes.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Anomalies of thoracoabdominal visceral sidedness or laterality, a spectrum of congenital malformations commonly referred to as heterotaxy syndrome or visceroatrial heterotaxy, are frequently associated with cardiac defects that include abnormal segmental cardiac anatomy [10, 30]. Cardiac defects in individuals with heterotaxy syndrome are frequently among the most complex cardiac lesions and historically have been associated with particularly poor outcome [4, 9, 12, 18, 27]. Along with general progress in surgical and medical management of complex congenital heart disease, a number of investigators have described encouraging trends in recent outcomes of patients with heterotaxy-associated congenital heart disease [1, 16, 19, 20, 23, 29]. Many of these reports, however, have focused on specific procedures or stages in palliative management, and do not shed light on trends in outcomes of the broader population of newborns with heterotaxy syndrome. The goals of this study, therefore, were to describe the midterm outcomes of a large, unselected cohort of patients with all forms of heterotaxy syndrome, with or without congenital cardiovascular disease, managed at a single institution, and to determine independent anatomic predictors of outcome.
Methods
Patients
Patients with heterotaxy syndrome who were followed and/or treated at Children’s Hospital Boston were ascertained from the computerized database of the Cardiovascular Program. Records were reviewed and patients were included in this study if the following inclusion criteria were met: (1) structural congenital heart disease anatomically consistent with heterotaxy syndrome, as defined below; (2) date of birth between 1985 and 1997; (3) presentation to Children’s Hospital Boston by 6 months of age; (4) residence in the primary referral area for Children’s Hospital, which includes New England and upstate New York; (5) all surgery at Children’s Hospital, with the possible exception of early palliation with a pulmonary arterial band or systemic-to-pulmonary arterial shunt; and (6) patient status known at ≥5 years of follow-up or death prior to this time. The study was performed in accordance with a protocol approved by the Committee on Clinical Investigation at Children’s Hospital. Informed consent was waived for this deidentified retrospective assessment.
The definition of heterotaxy proposed by Van Praagh et al., “an abnormal symmetry of certain viscera and veins (lungs, liver, vena cavae) and situs discordance between the various organ systems, as well as between the various segments of the heart,” was used for inclusion in this study [30]. Patients with abnormalities of visceral situs and/or systemic venous return, coupled with a typical situs abnormality of at least one cardiac segment, were considered to have heterotaxy syndrome in this study. Characteristic anatomic features as set forth by Van Praagh et al. were used for reference, and patients were classified as having a phenotype most consistent with “asplenia syndrome” or “polysplenia syndrome,” based on commonly observed anatomic patterns [10, 30].
Definition and Assessment of Pulmonary Venous Anomalies
Native pulmonary venous anatomy was characterized as normal, totally anomalous pulmonary venous connection (TAPVC), partially anomalous pulmonary venous connection (PAPVC), or anomalous pulmonary venous drainage. TAPVC and PAPVC were defined as connection of all or some pulmonary veins, respectively, to a site other than the atria, and were distinguished from “anomalous pulmonary venous drainage,” which was defined as pulmonary venous drainage to an anatomic right atrium or with ipsilateral drainage of the left and right veins to a common atrium. “Obstructed” pulmonary venous drainage refers to native TAPVC or PAPVC, while “pulmonary vein stenosis” is used to describe pulmonary venous obstruction that develops after repair of TAPVC/PAPVC (including anastomotic obstruction), or in normally or anomalously draining veins (with or without surgery).
In patients with TAPVC, preoperative diameters of the individual pulmonary veins entering the confluence, or just proximal to the site of drainage if separate from a confluence, were measured from cross-sectional echocardiographic images according to previously reported methods, as was the diameter of the pulmonary venous confluence [14]. The sum of diameters of the individual pulmonary veins, and the diameter of the confluence, were indexed to body surface area.
Data Analysis
The primary outcome was survival. A secondary outcome was functional clinical status; surviving patients were classified in the following manner, based on review of medical records: (1) well—routine follow-up only; (2) intermediate morbidity—requiring frequent medical or surgical care, having mild to moderate developmental delay or a systemic oxygen saturation <90%; (3) severe morbidity—severe mental or physical impairment, listing for transplant; and (4) heart transplant. Other secondary outcomes included development of pulmonary vein stenosis, atrioventricular (AV) valve replacement, and pacemaker placement.
Survival probabilities from birth were estimated using the Kaplan-Meier method, and the log-rank test was used to compare survival between patients with and patients without TAPVC. However, because all surviving patients were followed for at least 5 years, and there were variable exposures to mortality-associated procedures, primary analyses of factors associated with mortality were performed using logistic regression rather than survival analysis techniques. Independent variables significant at the 0.10 level in univariate analysis were considered for inclusion in a multivariate logistic regression model. Analyses were performed for the entire cohort of patients and separately for the “treatment cohort” to determine if the latter group had a different outcome as compared with the entire cohort. Anatomic characteristics were compared between patients in the treatment group and those who died without surgery using Fisher’s exact test. For patients with TAPVC, the mean sum of pulmonary vein diameters indexed to body surface area and the mean diameter of the pulmonary confluence were compared between patients who survived and those who died using the two-sample t-test. Odds ratios (OR) are presented with 95% confidence intervals (CI).
Results
Patients
Of the 114 patients who met inclusion criteria for the study, 12 were excluded due to insufficient follow-up information at 5 years. Thus, the study cohort consisted of 102 patients. Table 1 summarizes the demographic features and cardiovascular interventions of the study patients and Table 2 summarizes their anatomic features.
Eighty-seven of the 102 patients (85%) underwent some form of cardiac surgical intervention (Table 1). Of the 15 patients who did not undergo surgery, 3 had heterotaxy syndrome with cardiovascular anatomy that did not require surgery, and 12 had congenital heart defects that would have required intervention. The “treatment cohort” included 90 patients, including the 87 patients who underwent intervention and the 3 patients with cardiovascular anatomy that did not require surgery.
Outcomes
Survival
A total of 48 patients (47%) died during the study period (Fig. 1). The proportion of patients with cardiovascular intervention did not differ between the 3 birth-year cohorts (p = 0.43). Of the 58 patients who underwent univentricular palliation, 25 (43%) died, as did 9 of the 27 (33%) who had biventricular repair and the 2 patients who had pacemaker placement only.

Flow diagram depicting outcomes among 102 patients with heterotaxy and congenital heart disease. *Median duration of follow-up, 12.8 years (5–21.7 years). †Median age at death, 6 months (0–10.3 years)
The cause of death was determined to be cardiac in 27 treatment cohort patients, 9 of whom were known to have pulmonary vein stenosis, 8 after prior repair of TAPVC. The most common noncardiac cause of death was infection (n = 3). Another patient died of S. aureus sepsis following cardiac surgery and was assigned a cardiac cause of death. The cause of death was undetermined in four patients, with three dying suddenly while in hospital and one dying at home.
Among surviving patients, the median age at follow-up was 12.8 years (5 to 21.7 years). Estimated 10-year survival by the Kaplan-Meier method was 55% for the overall study population and 62% for patients in the treatment cohort (Fig. 2a). For both groups, mortality was high in the first month of life; after this, there was a fairly constant risk of death up to age 4 years, and few deaths beyond age 4 years. Factors associated with increased odds of death by univariate and multivariate analyses are summarized in Table 3. Patients with an asplenia syndrome phenotype had worse survival over time than those with a polysplenia phenotype (Fig. 2b), although this variable was not independently associated with worse survival by multivariate analysis. By multiple logistic regression analysis, independent predictors of death among all patients and among patients receiving surgical intervention included obstructed TAPVC, AV canal defect, and ≥mild AV valve regurgitation at the time of presentation. There was a strong correlation between infradiaphragmatic/mixed TAPVC and obstructed TAPVC, and the logistic regression results were similar using either variable. Therefore, only obstructed TAPVC was included in the final model.

Kaplan-Meier survival curves for (a) all patients and patients who underwent surgery (treatment cohort), (b) patients with asplenia and polysplenia syndrome phenotypes, and (c) all patients, stratified for the presence of TAPVC
Functional status and other secondary outcomes
Intermediate and severe morbidity were present in 15 (28%) and 4 (7%) of the 54 surviving patients (Table 4). Two other patients underwent orthotopic heart transplantation, one at 21 months of age for severe ventricular dysfunction after staged univentricular palliation, and the other at 14 years of age for severe cyanosis and ventricular dysfunction, as an initial surgical intervention. The first patient died with transplant coronary arteriopathy, and the other is alive and well. Other secondary outcomes are summarized in Table 4.
Pulmonary venous anomalies
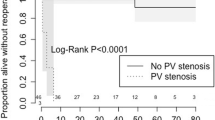
TAPVC was present in 36 of 102 patients (35%), 24 of whom (66%) died, 6 without intervention and 18 following surgical procedures. TAPVC was found predominantly, but not exclusively, in patients with an asplenia syndrome phenotype (33 of 46 [72%] vs 3 of 56 [5%]; p < 0.001). As depicted in Fig. 2c, survival over time was significantly better for patients without TAPVC than for those with TAPVC (p = 0.001); estimated 10-year survival was 39% in patients with TAPVC and 64% in those without TAPVC. For the 30 surgical patients with TAPVC, mortality was higher in those with infracardiac or mixed TAPVC (10 of 11) than those with supracardiac TAPVC (8 of 19; p = 0.001) (Fig. 3).

Breakdown of outcomes according to type of TAPVC and presence of native TAPVC obstruction
Among patients with TAPVC, the prevalence of obstructed TAPVC did not differ based on the presence of pulmonary atresia or severe pulmonary stenosis (p > 0.25). These findings applied to both overall and treatment cohorts.
Of the 30 patients with TAPVC in the treatment cohort, 14 (47%) developed postoperative pulmonary vein stenosis, 5 with obstruction exclusively at the pulmonary vein-to-atrium anastomosis. The remainder developed intrinsic pulmonary vein stenosis, either in isolation or in addition to anastomotic obstruction. In addition, two patients without TAPVC developed pulmonary vein stenosis: one of these had severe congenital pulmonary venous hypoplasia and died as a newborn without intervention; the other died after multiple surgical and transcatheter interventions. The median age at diagnosis of pulmonary vein stenosis was 2 months (0.1 month to 7 years). Among the 16 patients with pulmonary vein stenosis, only 5 were alive at most recent follow-up.
Pulmonary vein and TAPVC confluence diameter
In patients with TAPVC, the mean sum of pulmonary vein diameters indexed to body surface area was significantly lower among patients who died than for survivors (42.3 ± 8.3 vs. 49.5 ± 10.1 mm/m2; p = 0.02), as was the indexed diameter of the pulmonary venous confluence (16.2 ± 6.3 vs. 22.0 ± 7.1 mm/m2; p = 0.01). There was no association between these measurements and obstructed TAPVC or the development of pulmonary venous stenosis. Notably, summed pulmonary vein diameters in 20 of the 30 patients (67%) were in the lowest quartile (<50 mm/mm2) of patients reported in the study by Jenkins et al. [14].
Discussion
Outcomes
This study examined outcomes of an unselected population of patients with heterotaxy syndrome, defined using relatively strict criteria, who presented at <6 months of age during a 13-year time period in the modern surgical era, with at least 5 years of follow-up in survivors. Consistent with previous studies of patients with various subtypes of heterotaxy [9, 12, 27, 32], outcomes in our series were discouraging, with overall 10-year survival of 55% and 10-year survival in the treatment cohort of 62%. Patients with obstructed TAPVC, AV canal defect, and AV valve regurgitation at the time of presentation were at increased risk of death. Notably, several previously proposed predictors of mortality, including asplenia syndrome, univentricular physiology, pulmonary atresia/stenosis, and congenital AV block [3, 9, 25, 27], were not independently associated with increased mortality in our series.
Importance of Pulmonary Venous Anomalies
In this cohort, pulmonary venous anomalies—both TAPVC and pulmonary vein stenosis—were key factors in the poor survival of patients with heterotaxy syndrome. As found in prior studies [3, 9, 25], obstructed TAPVC was a dominant independent predictor of death in our series: 10-year survival among all patients with TAPVC was 39%, and only 3 of 21 patients (9%) with obstructed TAPVC were alive at follow-up. Pulmonary vein stenosis, which developed in almost one-half of patients with repaired TAPVC and up to 7 years after repair, was also critically important.
In several recent series of TAPVC in patients without heterotaxy syndrome, including a report from our center, mortality ranged from 5% to 15% and the reported frequency of pulmonary vein stenosis was 2% to 13% [2, 11, 17, 21]. Although comparison of survival between patients with TAPVC with and those without associated heterotaxy syndrome is confounded by the frequent presence of other cardiovascular anomalies in the latter group, the difference in the incidence of pulmonary vein stenosis following repair, 47% in our series compared with 2%–13%, is notable.
It has been suggested that low native pulmonary blood flow, as is frequently present in the form of pulmonary atresia or stenosis in patients with heterotaxy syndrome, may predispose to obstruction of native TAPVC [15, 23]. We did not find an association between pulmonary stenosis or atresia and obstructed TAPVC in the present cohort. Caldarone et al. noted a decreased survival in patients with TAPVC requiring pulmonary arterial bands or fixed systemic-arterial shunts [2]. They suggested that the fixed resistance imposed by these procedures, in concert with abnormal pulmonary vasculature, may result in the inability to regulate pulmonary blood flow in the dynamic postoperative period, potentially leading to early postoperative death. Alternatively, it may be that increasing pulmonary blood flow by placement of a systemic-to-pulmonary artery shunt simply unmasks existing pulmonary venous obstruction [7, 23]. While some investigators have reported increased mortality in heterotaxy patients with TAPVC who require placement of a systemic-to-pulmonary artery shunt, we did not.
Regardless of whether decreased pulmonary blood flow impairs pulmonary venous growth and predisposes to obstruction, previous studies have shown that smaller pulmonary vein size in patients with TAPVC is associated with increased probability of death, independent of the presence of heterotaxy syndrome [14]. We also found that the indexed sum of individual pulmonary vein diameters and the indexed diameter of the pulmonary vein confluence were significantly lower in patients who died than in survivors, but there was no correlation between either measurement and obstructed TAPVC or development of pulmonary vein stenosis.
There are likely intrinsic abnormalities of the pulmonary veins that predispose to obstruction, and patients with heterotaxy syndrome may differ from those with nonheterotaxy TAPVC in this respect. Recent data suggest that pulmonary vein stenosis may be related to myofibroblastic proliferation, possibly in response to increased activity receptor tyrosine kinases such as platelet-derived growth factor receptors and vascular endothelial growth factor receptors [26, 28]. Several clinical and experimental studies have demonstrated increased medial thickness in association with pulmonary venous obstruction. Yamaki et al. found that preoperative pulmonary vein obstruction in patients with TAPVC was associated with increased medial thickness of the pulmonary arteries and veins compared to patients with ventricular septal defects and normal patients [33]. Others have confirmed these findings in patients with obstructed TAPVC, both with and without heterotaxy, but there was no comparison of the findings between the two groups [8]. Similarly, it has been suggested that a greater degree of medial thickness may increase a patient’s predisposition to developing postoperative intrinsic pulmonary vein stenosis [22]. However, evidence from a porcine model of unilaterally induced pulmonary vein stenosis, which resulted in the development of medial thickening proximal to the obstruction, suggests that that medial thickening is a consequence rather than a cause of pulmonary venous obstruction [6]. Differences in pulmonary venous pathology between patients with and patients without heterotaxy may be important in understanding the biology of these conditions.
Another potential etiologic factor in pulmonary vein stenosis that develops after repair of TAPVC in patients with heterotaxy syndrome is obstruction related to surgery. This may be due to anastomotic torsion or compression in the setting of complex intracardiac anatomy and a difficult repair, complex baffling procedures to separate the pulmonary and systemic venous returns, or impingement of intracardiac Fontan connections on the abnormal or repaired pulmonary veins. Although this is an important consideration in general, and indeed, a number of studies report surgical approaches specifically tailored to patients with heterotaxy syndrome and abnormal systemic and pulmonary venous anatomy [13, 15, 20, 22, 24, 31], the majority of the patients in our cohort developed intrinsic pulmonary vein stenosis, suggesting an abnormality of the pulmonary venous vasculature in patients with heterotaxy.
Limitations
As with all reviews of medical records, there are inherent limitations in the retrospective nature of this study. Although every effort was made to reduce selection bias through conservative entry criteria, there are several other surgical centers in the region and we may still have a more complex patient group than the general population. In addition, referring centers may have chosen to not pursue treatment in some of the more severely affected patients. As is inherent to any chart review, there is likely to be some degree of ascertainment bias, in particular, underascertainment of subclinical noncardiac anomalies and intermediate morbidities. By limiting our study to patients with a minimum of 5 years’ follow-up, we did not include those from our most recent experience, and as a result our findings may not reflect changes in outcome over the past 5–10 years.
Conclusions
Patients with heterotaxy syndrome continue to have a high rate of mortality, especially in the presence of TAPVC. Pulmonary vein obstruction is relatively common in this group of patients, particularly after repair of infracardiac or mixed TAPVC. Although a lower index of summed pulmonary vein diameters to body surface area was associated with death, this measure was not associated with obstructed TAPVC or pulmonary vein stenosis. However, because of competing risks (i.e., the high mortality in this group), detection of an association between preintervention pulmonary vein size and pulmonary vein stenosis may have been confounded. Additional research on the pathophysiology of and potential treatments for pulmonary vein stenosis are critical to improving outcomes in patients with heterotaxy syndrome.
References
Azakie A, Merklinger SL, Williams WG, Van Arsdell GS, Coles JG, Adatia I (2001) Improving outcomes of the Fontan operation in children with atrial isomerism and heterotaxy syndromes. Ann Thorac Surg 72:1636–1640
Caldarone CA, Najm HK, Kadletz M, Smallhorn JF, Freedom RM, Williams, Cole JG (1998) Surgical management of total anomalous pulmonary venous drainage: impact of coexisting cardiac anomalies. Ann Thorac Surg 66:1521–1526
Cheung YF, Cheng VY, Chau AK, Chiu CS, Yung TC, Leung MP (2002) Outcome of infants with right atrial isomerism: Is prognosis better with normal pulmonary venous drainage? Heart 87:146–152
Cohen MS, Schultz AH, Tian ZY, Donaghue DD, Weinberg PM, Gaynor JW, Rychik J (2006) Heterotaxy syndrome with functional single ventricle: Does prenatal diagnosis improve survival? Ann Thorac Surg 82:1629–1636
DeLeon SY, Gidding SS, Ilbawi MN, Idriss FS, Muster AJ, Cole RB, Paul MH (1987) Surgical management of infants with complex cardiac anomalies associated with reduced pulmonary blood flow and total anomalous pulmonary venous drainage. Ann Thorac Surg 43:207–211
Endo M, Yamaki S, Hata M, Saiki Y, Tabayashi K (2002) Pulmonary vascular changes induced by unilateral pulmonary venous obstruction. Pediatr Cardiol 23:420–425
Freedom RM, Olley PM, Coceani F, Rowe RD (1978) The prostaglandin challenge. Test to unmask obstructed total anomalous pulmonary venous connections in asplenia syndrome. Br Heart J 40:91–94
Gaynor JW, Collins MH, Rychik J, Gaughan JP, Spray TL (1999) Long-term outcome of infants with single ventricle and total anomalous pulmonary venous connection. J Thorac Cardiovasc Surg 117:506–513
Gilljam T, McCrindle BW, Smallhorn JF, Williams WG, Freedom RM (2000) Outcomes of left atrial isomerism over a 28-year period at a single institution. J Am Coll Cardiol 36:908–916
Gutgesell H (1992) Cardiac malpositions and heterotaxy. In: Garson A (ed) The science and practice of pediatric cardiology, 1st ed. Williams & Wilkins, Baltimore, pp 1547–1561
Hancock Friesen CL, Zurakowski D, Thiagarajan RR, Forbess JM, del Nido PJ, Mayer JE, Jonas RA (2005) Total anomalous pulmonary venous connection: an analysis of current management strategies in a single institution. Ann Thorac Surg 79:596–606
Hashmi A, Abu-Sulaiman R, McCrindle BW, Smallhorm JF, Williams WG, Freedom RM (1998) Management and outcomes of right atrial isomerism: a 26-year experience. J Am Coll Cardiol 31:1120–1126
Humes RA, Feldt RH, Porter CJ, Julsrud PR, Puga FJ, Danielson GK (1988) The modified Fontan operation for asplenia and polysplenia syndromes. J Thorac Cardiovasc Surg 96:212–218
Jenkins KJ, Sanders SP, Orav EJ, Coleman EA, Mayer JE Jr, Colan SD (1993) Individual pulmonary vein size and survival in infants with totally anomalous pulmonary venous connection. J Am Coll Cardiol 22:201–206
Kawashima Y, Kitamura S, Matsuda H, Shimazaki Y, Nakano S, Hirose H (1984) Total cavopulmonary shunt operation in complex cardiac anomalies. A new operation. J Thorac Cardiovasc Surg 87:74–81
Kim SJ, Kim WH, Lim HG, Lee CH, Lee JY (2006) Improving results of the Fontan procedure in patients with heterotaxy syndrome. Ann Thorac Surg 82:1245–1251
Kirshbom PM, Myung RJ, Gaynor JW, Ittenbach RF, Paridon SM, DeCampli WM, Karl TR, Spray TL (2002) Preoperative pulmonary venous obstruction affects long-term outcome for survivors of total anomalous pulmonary venous connection repair. Ann Thor Surg 74:1616–1620
Lim JS, McCrindle BW, Smallhorn JF, Golding F, Caldarone CA, Taketazu M, Jaeggi ET (2005) Clinical features, management, and outcome of children with fetal and postnatal diagnoses of isomerism syndromes. Circulation 112:2454–2461
Lodge AJ, Rychik J, Nicolson SC, Ittenbach RF, Spray TL, Gaynor JW (2004) Improving outcomes in functional single ventricle and total anomalous pulmonary venous connection. Ann Thorac Surg 78:1688–1695
McElhinney DB, Reddy VM, Moore P, Hanley FL (1997) Bidirectional cavopulmonary shunt in patients with anomalies of systemic and pulmonary venous drainage. Ann Thorac Surg 63:1676–1684
Michielon G, DiDonato RM, Pasquini L, Giannico S, Brancaccio G, Mazzera E, Squitieri C, Catena G (2002) Total anomalous pulmonary venous connection: long-term appraisal with evolving technical solutions. Eur J Cardiothor Surg 22:184–191
Michielon G, Gharagozloo F, Julsrud PR, Danielson GK, Puga FJ (1993) Modified Fontan operation in the presence of anomalies of systemic and pulmonary venous connection. Circulation 88:II141–148
Morales DLS, Braud BE, Booth JH, Graves DE, Heinle JS, McKenzie ED, Fraser CD (2006) Heterotaxy patients with total anomalous pulmonary venous return: improving surgical results. Ann Thorac Surg 82:1621–1628
Najm HK, Caldarone CA, Smallhorn J, Coles JG (1998) A sutureless technique for the relief of pulmonary vein stenosis with the use of in situ pericardium. J Thor Cardiovasc Surg 115:468–470
Phoon CK, Neill CA (1994) Asplenia syndrome—risk factors for early unfavorable outcome. Am J Cardiol 73:1235–1237
Riedlinger WF, Juraszek AL, Jenkins KJ, Nugent AW, Balasubramanian S, Calicchio ML, Kieran MW, Collins T (2006) Pulmonary vein stenosis: expression of receptor tyrosine kinases by lesional cells. Cardiovasc Pathol 15:91–99
Sadiq M, Stumper O, De Giovanni JV, Wright JG, Sethia B, Brawn WJ, Silove ED (1996) Management and outcome of infants and children with right atrial isomerism. Heart 75:314–319
Sadr IM, Tan PE, Kieran MW, Jenkins KJ (2000) Mechanism of pulmonary vein stenosis in infants with normally connected veins. Am J Cardiol 86:577–579
Stamm C, Friehs I, Duebener LF, Zurakowski D, Mayer JE Jr, Jonas RA, del Nido PJ (2002) Improving results of the modified Fontan operation in patients with heterotaxy syndrome. Ann Thorac Surg 74:1967–1977
Van Praagh S, Santini F, Sanders SP (1992) Cardiac malpositions with special emphasis on visceral heterotaxy (asplenia and polysplenia syndromes). In: Fyler DC (ed) Nadas’ pediatric cardiology. Hanley & Belfus, Philadelphia, pp 589–608
Vargas FJ, Mayer JE Jr, Jonas RA, Castaneda AR (1987) Anomalous systemic and pulmonary venous connections in conjunction with atriopulmonary anastomosis (Fontan-Kreutzer). Technical considerations. J Thorac Cardiovasc Surg 93:523–532
Webber SA, Sandor GGS, Patterson MWH, Taylor GP, Wadsworth LD, LeBlanc JG (1992) Prognosis in asplenia syndrome—a population-based review. Cardiol Young 2:129–135
Yamaki S, Tsunemoto M, Shimada M, Ishizawa R, Endo M, Nakayama S, Hata M, Mohri H (1992) Quantitative analysis of pulmonary vascular disease in total anomalous pulmonary venous connection in sixty infants. J Thorac Cardiovasc Surg 104:728–735
Acknowledgments
This study was supported in part by the Higgins Family Noninvasive Cardiology Research Fund at Children’s Hospital Boston. We thank Julia Edwards, RDCS, RCIS for her assistance in the measurement of pulmonary vein diameters.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Foerster, S.R., Gauvreau, K., McElhinney, D.B. et al. Importance of Totally Anomalous Pulmonary Venous Connection and Postoperative Pulmonary Vein Stenosis in Outcomes of Heterotaxy Syndrome. Pediatr Cardiol 29, 536–544 (2008). https://doi.org/10.1007/s00246-007-9128-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-007-9128-5