
Abstract
Both scanning electron microscopy and atomic force microscopy (AFM) have shown that calcium oxalate monohydrate kidney stones are made up from arrangements of sub micron crystals. The purpose of this investigation was to determine the morphology of these crystals which was obscured by the presence of organic matrix in our earlier study. Sections of stones were treated to remove the protein component of the matrix and then imaged using AFM. Images obtained after proteolysis show that the crystals are in the form of plates stacked on (100) surfaces. These results were confirmed by scanning electron microscopy observations from selected regions of calcium oxalate kidney stone surfaces. The observed crystal sizes are consistent with both the known matrix mass fraction and crystallite growth in the passage through the collecting duct.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
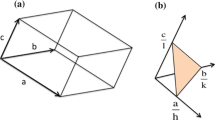
Kidney stone disease has become relatively common in developed countries with an incidence of about 12% [1]. Most stones have calcium oxalate as their main component, usually in its monohydrate form [2]. Calcium oxalate dihydrate (COD), mineral name weddellite, can also be found in mixed stones. It is easily recognizable as distinctive bipyramids corresponding to the (101) facets of the tetragonal structure, space group I 4/m. The COD bipyramids are usually found on the outside of a core of calcium oxalate monohydrate (COM) [3]. Calcium oxalate monohydrate, sometimes known by its mineral name whewellite, has a monoclinic cell described by space group P21/c. Crystal structures have been given by both Tazzoli and Domeneghetti [4] and Deganello [5] who used different conventions to describe the unit cell, the (100) plane of Tazzoli and Domeneghetti being the \( (10\ifmmode\expandafter\bar\else\expandafter\=\fi{1}) \) plane of Deganello. Tazzoli and Domeneghetti [4] also distinguish between a high temperature and a low temperature form of whewellite where the unit cell is doubled along [010] and there are slight differences in rotations of the oxalate group. When crystallized from solution COM crystals grow in a plate-like morphology whose faces are (100) in Tazzoli and Domeneghetti [4] notation, whose convention we follow. The sides are bounded by (010) and (021) and (121) facets, shown schematically in Fig. 1.

Calcium oxalate morphology showing prominent facets indexed according to Tazzoli and Domeneghetti [4]
Pure COM stones usually are spherulitic in nature, sometimes with multiple spherules joined together. Occasionally a small dimple is seen at the point of attachment to the renal papilla. Optical microscopy of stone sections reveals concentric rings and radial striations reminiscent of growth rings of a tree [6]. Sometimes dumbbell arrangements are also observed. Kidney stones are composites made up from the calcium oxalate mineral phase and an organic matrix phase. At some level the mineral phase must be organized as single crystals bounded by recognizable facets that are usually low index crystallographic planes under normal growth conditions. A characteristic of single crystals is that they give spot diffraction patterns and there is continuity of lattice planes from one side to the other. This does not exclude inclusions, point defects, line defects such as dislocations or planar defects such as stacking faults. The single crystals can clump together in a seemingly random arrangement, possibly with organic matrix between them, to form a polycrystalline aggregate. The size of these crystals [we sometimes refer to sub micron crystals whose presence is only detectable by scanning electron microscope (SEM), atomic force microscopy (AFM) or high angle resolution synchrotron X-ray diffraction (XRD) as crystallites] is significant for stone formation as it sets limits on where they could form, given a knowledge of crystal growth rates and urinary transit times through various parts of the nephron. High-resolution scanning electron microscopy and AFM showed that the fundamental single crystal sizes were less than 1 μm and they appeared to be stacked together as a compact aggregate [7]. Similar results were also found in TEM of demineralized stones induced in rats [8] and SEM studies of demineralized human stones [9]. However, the expected crystal facets for single crystals, as seen in crystals grown from solution, were not observed.
We hypothesized that remnants of organic matrix prevented the imaging of the true single crystal morphology. The matrix accounts for about 2–3% of the weight of the stone [10, 11] and is reported to be about 70% protein, 20% lipid and 10% other organic macromolecules [12]. However, later work suggested that lipid might account for a higher proportion of matrix in calcium oxalate stones [13]. If the stone consists of single crystals of COM surrounded by matrix macromolecules, then removing a major component of the matrix by proteolysis should help reveal the true morphology of the single crystals making up the mineral phase. The aim of this investigation is to identify single crystal mineral components and relate them to possible stone growth mechanisms.
Materials and methods
Seven COM stones no larger than 2 mm were selected based on XRD analysis. A small part of the stone was scraped away and crushed for powder diffractometry in a Rigaku D/MAX-IIB diffractometer. Stones that showed only COM peaks (a conservative estimate would put the presence of other constituents at less than 5%) were selected for further analysis. The stones were mounted in capsules in epoxy and polished using a succession of grinding powders to ensure a flat surface, following the procedure outlined in Shaapur et al. [14]. We need to polish the stones so that they are smooth enough for AFM examination. It is certainly a concern that the procedure alters the morphology of large single crystals. For this reason we have also examined exterior surfaces and, more recently, surfaces after fracture under liquid N2 by SEM. The fact that the general morphologies are the same gives us confidence that our procedure does not bring about gross morphological changes.
The proteolysis was performed following the procedure of Ryall et al. [15] described in their study of intracrystalline protein content of COM and COD crystals. A 12.5 mM Tris solution was prepared and HCl added to make the pH 6.0, typical of urine. Five units of Cathepsin D (Sigma) were added to 500 μl of distilled water. This was divided into five aliquots of 100 μl. To achieve a final dilution of 1 U per 200 μl, 100 μl of Tris buffer was added to each of these aliquots. The stones were immersed in the buffer and incubated for 3 days at 37°C. They were then removed from the solution, mounted on stubs and examined in a Digital Instruments Nanoscope II AFM. A 50× optical microscope was used to search for suitable flat regions and initial survey scans were taken as 256 × 256 pixel images of regions between 50 and 100 μm across. Higher resolution images were taken with 512 × 512 pixels from regions approximately 1 × 1 μm.
Two other COM stones whose identification was checked by XRD were mounted in the as-received state on aluminum stubs and the surfaces were examined using 3.0 kV beam in a Hitachi S 550 field emission SEM. Previous investigations [7] have shown that there is minimal charging of calcium oxalate at this accelerating voltage since the incident beam current is balanced by the combination of backscattered and secondary electron current. There was therefore no need to deposit a conductive coating on the specimens. Angles were measured from both AFM and SEM images using the ImageJ image analysis program [16].
Results
Figure 2 is a typical AFM image from a stone section surface. It shows particles ranging in size from 50 to 160 nm. After proteolysis the crystallite shapes became more visible and appeared to have a plate-like morphology as seen in Fig. 3. In these images it is still not possible to distinguish individual facets since the crystals are oriented almost perpendicular to the surface. In Fig. 4 there appears to be a stacked arrangement of flat crystals. The boundaries of the facets are not very distinct though those marked as A and B were measured as 103° and 106°, respectively, close to the angle between (021) and \( (0\ifmmode\expandafter\bar\else\expandafter\=\fi{2}1) \) faces, while C and D were measured as 125° and 121°, respectively, approximately the same as the 121° angle between (010) and \( (12\ifmmode\expandafter\bar\else\expandafter\=\fi{1}) \) planes. SEM images (Fig. 5) of exposed surfaces show rough plate-like morphology, with serrated edges very similar to those observed by Ryall et al. [17] after proteolysis of COM crystals grown in urine. In Fig. 6a, which is also an SEM image of a surface region, the crystals again appear to be stacked. The obtuse angle between the edges of the plates is approximately 120°. To measure the dihedral angles between planes more precisely the specimen was tilted as shown in Fig. 6b. The measurements are summarized in Table 1 and the mean is 119° ± 2°. This is close to 121°, the angle between (010) and \( (12\ifmmode\expandafter\bar\else\expandafter\=\fi{1}) \) type planes, though it is not that different from 116°, the angle between \( (12\ifmmode\expandafter\bar\else\expandafter\=\fi{1}) \) and \( (1\ifmmode\expandafter\bar\else\expandafter\=\fi{2}\ifmmode\expandafter\bar\else\expandafter\=\fi{1}) \) planes. A similar micrograph has been published by Holden [18] on the Herring web site. Although the size of the plates varied between about 0.5 and 1.5 μm for different specimens, the stacking was a common feature of all observations.

AFM image of the surface of a COM stone section, bar is 1 μm

AFM image of COM stone section surfaces after proteolysis, bar is 1 μm

AFM image showing COM crystal morphology in a stone section after proteolysis, bar is 1 μm. Angles A is 103° and B is 106° close to the angle between (021) and \( (0\ifmmode\expandafter\bar\else\expandafter\=\fi{2}1) \) faces, while C is 125° and D is 121°, approximately the same as the 121° between (010) and \( (12\ifmmode\expandafter\bar\else\expandafter\=\fi{1}) \) planes. The angle measurement error is approximately 3°

SEM image showing plate-like crystals on COM stone surface, bar is 0.5 μm

a SEM image showing COM crystal morphology on a stone surface, bar is 1.0 μm. Angles close to 120° are marked. b The same region tilted for more accurate measurement of angles a, b, c, d, and e. Values are given in Table 1. Bar is again 1.0 μm
Low resolution SEM and AFM images, given in Fig. 7, show that crystals are only stacked in the same orientation over relatively small regions, about 5 μm across. Small differences in crystal size or shape allow for changes in the relative orientation of arrays of plates. Any XRD experiment would therefore sample many random orientations which is why COM stones give polycrystalline diffraction patterns.

a AFM image from section after proteolysis, bar is 2 μm, and b SEM image from surface, bar is 5 μm, showing stacking
Discussion
Both the AFM images after proteolysis and the SEM images of selected surface region suggest that COM stones are formed from the aggregation of stacked plate-like single crystals. If growth in the kidney is similar to crystal growth from solution or nucleation of crystals on Langmuir Blodgett films [19–21] the face on which stacking occurs is (100), as this is the largest face and the general morphology supports this conclusion. The crystals appear to be about 1 μm across, which is larger than the objects identified as possible crystals in our earlier study [7]. It is possible, especially for the surface observations, that there is a bias towards larger sizes as these would be more prominent. Alternatively the earlier observations could have mistakenly identified some of the serrations observed at the plate edges (see Fig. 5) as separate crystals. The crystallite sizes are the same as those derived from the peak widths in synchrotron XRD studies of COM crystals grown in ultrafiltered urine and various solutions of aspartic and glutamic acids and their dimers [22].
The stacking on (100) planes is consistent with the observations of Sheng et al. [23] who investigated the forces between various molecular groups and COM surfaces by linking molecules to AFM tips with the relevant group exposed. They showed greater adhesion of carboxylate and amidinium groups on COM (100) which would imply that macromolecules have a positive role in enhancing adhesion.
The crystal size can be estimated for various cases where a monolayer of an organic macromolecule surrounds the crystal, as proposed by Leal and Finalyson [24]. For simplicity consider a cuboid crystal, dimensions a, b and d, where d is the shortest dimension. The volume of organic macromolecule is
where l is the monolayer thickness. The volume fraction f vol of organic material is
and the mass fraction f m is
where ρ prot and ρ ox are the densities of protein (1.3 gm/cc) and COM (2.2 gm/cc), respectively. The mass fraction can be set equal to 0.03, the observed average macroscopic weight fraction of matrix in COM stones. There are two limiting cases, the first when the crystallite is almost cubic and b = a = d. The mass fraction is, which is proportional to the ratio of surface area to volume, is
and the crystal size, d, is
This is the size estimate given in the third column of Table 2 and is shown schematically as Fig. 8a. It is interesting to note that the expression for the mass fraction would be the same if it were assumed that the crystallites were spheres coated with one monolayer, since the ratio of surface area to volume is identical. In this case the mass fraction of organic matrix is
As it is likely that the interstices between the spheres will be filled with organic material the mass fraction of organic material in practice would be \( 0.363{\left( {\frac{{\rho _{{{\text{prot}}}} }} {{\rho _{{{\text{ox}}}} }}} \right)} \) for identical close packed spheres, independent of the size of spheres when their diameters are much larger than a monolayer. The other limiting case, which is probably closer to the observations, is shown in Fig. 8b and assumes that the crystallites are thin plates such that a or b are much greater than the thickness d. The organic matrix mass fraction becomes
However, this derivation assumes a stacking of one monolayer associated with each side of the plate, so that there are two macromolecule layers between each crystallite. If there were only one macromolecule layer between continuous plates, the mass fraction would be
which leads to a plate thickness d
If the plates were not continuous and there were regions of organic matter between them, total area a, as shown schematically in Fig. 8a then Eq. 8 would be modified as
where A is the plate area. From micrographs such as Fig. 6 the area between plates is small, probably less than 10% of the crystallite area, so f m is only marginally bigger than the value given by Eq. 8. Furthermore Eq. 8 still defines the limiting case.

Schematic illustration showing a plan view showing macromolecule surrounding crystals similar to those observed in crystal growth experiments. b Macromolecules between crystal slabs in a sandwich arrangement. It is still possible that there are macromolecules between the crystals in a slab, though as explained in the text this will make only small changes to the organic mass fraction
Estimates for various macromolecules between the plates in this case are given in the fourth column of Table 2. The third and fourth column of Table 2 can be viewed as size bounds for a crystallite in the limiting case of the organic matrix being present as a single molecule layer. The observations of plate thickness are between 0.2 and 0.5 μm (see Fig. 7) which implies that there is more than one monolayer even for the large macromolecules.
A possible mechanism for stone formation would be heterogeneous nucleation of the crystals in the nephron, with subsequent aggregation/agglomeration on a favored site such as the renal papilla. This is in some ways similar to a model suggested by Vermeulen [25] who postulated that growth occurs by agglomeration around a crystal blocking a duct of Bellini on the renal papilla. The crystal sizes would then also be consistent with what is known about crystal growth rates. Kok and Khan [26] and Finlayson and Reid [27] give growth rates for COM as 1–2 μm/min while Werness [28] gives a lower estimate of 0.14 μm/min. According to Kok and Khan [26] the transit time through the nephron is 3–4 min and 30–45 s through the collecting duct. If the higher growth rate is to be believed, crystals nucleating throughout the nephron could have sizes up to 3–8 μm, larger than what we have observed. For the lower growth rate the size would be 0.4–0.6 μm. It is entirely plausible that crystal nucleation only takes place where calcium oxalate reaches its urinary super saturation in the collection duct after water resorption. In this case the higher growth rate would imply crystal sizes approximately 0.5–1.5 μm, in agreement with our observations. The range of sizes from the lower growth rate in the collecting duct would be 0.07–1 μm which is smaller than what we observed. It is therefore plausible, if the higher growth rate is correct, that calcium oxalate kidney stones form by the disordered stacking of plate-like single crystals of COM that nucleate and grow in the collecting duct.
References
Sierakowski R, Finlayson B, Landes RR, Finlayson CD, Sierakowski N (1978) The frequency of urolithiasis in hospital discharge diagnoses in the United States. Invest Urol 15:438–441
Drach GW (1992) Urinary lithiasis etiology, diagnosis, and medical management. In: Campbell MF, Harrison JH (eds) Campbell’s textbook of urology, Sanders: Philadelphia, pp 2085–2156
Iwata H, Iio S, Nishio S, Takeuchi M (1992) Architecture of mixed calcium oxalate dihydrate and monohydrate stones. Scanning Microsc 6:231–238
Tazzoli V, Domeneghetti C (1980) The crystal structures of whewellite and weddellite: re-examination and comparison. Am Mineral 65:327–334
Deganello S (1981) The structure of whewellite, CaC2O4·H2O at 328 K. Acta Crystallogr B37:826–829
Prien EL, Prien EL (1968) Composition and structure of urinary stone. Am J Med 45:654–672
Dorian HH, Rez P, Drach GW (1996) Evidence for aggregation in oxalate stone formation: atomic force and low voltage scanning electron microscopy. J Urol 156:1833–1837
Khan SR, Hackett RL (1987) Crystal-matrix relationships in experimentally induced urinary calcium oxalate monohydrate crystals, an ultrastructural study. Calcif Tissue Int 41:157–163
Khan SR (1997) Calcium phosphate/calcium oxalate crystal association in urinary stones: implications for heterogeneous nucleation of calcium oxalate. J Urol 157:376–383
King JS, Boyce WH (1959) Analysis of renal calculous matrix compared with some other matrix materials with uromucoid. Arch Biochem Biophys 82:455–461
Warpehoski M, Buscemi PJ, Osborn DC, Finlayson B, Goldberg EP (1981) Distribution of organic matrix in calcium oxalate renal calculi. Calcif Tissue Int 33:211–222
Khan SR, Shevock PN, Hackett RL (1988) Presence of lipids in urinary stones: results of preliminary studies. Calcif Tissue Int 42:91–96
Khan SR, Glenton PA, Backov R, Talham DR (2002) Presence of lipids in urine, crystals and stones: implications for the formation of kidney stones. Kidney Int 62:2062–2072
Shaapur F, Rez P, Yaniv DR, Drach GW (1994) Specimen preparation for scanning probe microscopy of materials by ultramicrotomy. Microscopy Society of America, San Francisco Press, New Orleans, pp 878–879
Ryall RL, Chauvet MC, Grover PK (2005) Intracrystalline proteins and urolithiasis: a comparison of the protein content and ultrastructure of urinary calcium oxalate monohydrate and dihydrate crystals. BJU Int 96:654–663
Abramoff MD, Magelhaes PJ, Ram SJ (2004) Image processing with image. J Biophotonics Int 11:36–42
Ryall RL, Fleming DE, Doyle IR, Evans NA, Dean CJ et al (2001) Intracrystalline proteins and the hidden ultrastructure of calcium oxalate urinary crystals: implications for kidney stone formation. J Struct Biol 134:5–14
Holden RE (2005) http://www.herringlab.com/sems/sems2.html
Letellier JR, Lochlear MJ, Campbell AA, Vogel V (1998) Oriented growth of calcium oxalate monohydrate crystals between phospholipid monolayers. Biochim Biophys Acta 1380:31–45
Backov R, Lee CM, Khan SR, Mingotaud C, Fanucci GE et al (2000) Calcium oxalate monohydrate precipitation at phosphatidylglycerol Langmuir monolayers. Langmuir 16:6013–6019
Whipps S, Khan SR, Palko FJ, Backov R, Talham DR (1998) Growth of calcium oxalate monohydrate at phospholipid Langmuir monolayers. J Cryst Growth 192:243–249
Fleming DE, Riessen AV, Chauvet MC, Grover PK, Hunter B et al (2003) Intracrystalline proteins and urolithiais: a synchrotron X-ray diffraction study of calcium oxalate monohydrate. J Bone Miner Res 18:1282–1291
Sheng XX, Jung TS, Wesson JA, Ward MD (2005) Adhesion at calcium oxalate crystal surfaces and the effect of urinary constituents. Proc Natl Acad Sci 102:267–272
Leal JJ, Finlayson B (1977) Adsorption of naturally occurring polymers onto calcium oxalate crystal surfaces. Invest Urol 14:278–283
Vermeulen CW, Lyon ES, Ellis JE, Borden TA (1967) The renal papilla and calculogenesis. J Urol 97:573–582
Kok DJ, Khan SR (1994) Calcium oxalate nephrolithiasis, a free or fixed particle disease. Kidney Int 46:847–854
Finlayson B, Reid F (1978) The expectation of free and fixed particles in stone disease. Invest Urol 15:442–448
Werness PG, Duckworth SC, Smith LH (1979) Calcium oxalate dihydrate crystal growth. Invest Urol 17:230–233
Acknowledgments
We should like to acknowledge funding from the College of Liberal Arts and Sciences, Arizona State University and the use of facilities in the Center for Solid State Science and the Center for Solid State Electronics Research. We would also like to thank Magali Chauvet and Dr. Rosemary Ryall for making available details of their proteolysis protocol. Helpful discussions with Dr. J. Wesson are also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sandersius, S., Rez, P. Morphology of crystals in calcium oxalate monohydrate kidney stones. Urol Res 35, 287–293 (2007). https://doi.org/10.1007/s00240-007-0115-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00240-007-0115-3