
Abstract
Gene duplication followed by nucleotide differentiation is one of the simplest mechanisms to develop new functions for genes. However, the evolutionary processes underlying the divergence of multigene families remain controversial. We used multigene families found within the diversity of toxic proteins in centipede venom to test two hypotheses related to venom evolution: the two-speed mode of venom evolution and the rapid accumulation of variation in exposed residues (RAVER) model. The two-speed mode of venom evolution proposes that different types of selection impact ancient and younger venomous lineages with negative selection being the predominant form in ancient lineages and positive selection being the dominant form in younger lineages. The RAVER hypothesis proposes that, instead of different types of selection acting on different ages of venomous lineages, the different types of selection will selectively contribute to amino acid variation based on whether the residue is exposed to the solvent where it can potentially interact directly with toxin targets. This hypothesis parallels the longstanding understanding of protein evolution that suggests that residues found within the structural or active regions of the protein will be under negative or purifying selection, and residues that do not form part of these areas will be more prone to positive selection. To test these two hypotheses, we compared the venom of 26 centipedes from the order Scolopendromorpha from six currently recognized species from across North America using both transcriptomics and proteomics. We first estimated their phylogenetic relationships and uncovered paraphyly among the genus Scolopendra and evidence for cryptic diversity among currently recognized species. Using our phylogeny, we then characterized the diverse venom components from across the identified clades using a combination of transcriptomics and proteomics. We conducted selection-based analyses in the context of predicted three-dimensional properties of the venom proteins and found support for both hypotheses. Consistent with the two-speed hypothesis, we found a prevalence of negative selection across all proteins. Consistent with the RAVER hypothesis, we found evidence of positive selection on solvent-exposed residues, with structural and less-exposed residues showing stronger signal for negative selection. Through the use of phylogenetics, transcriptomics, proteomics, and selection-based analyses, we were able to describe the evolution of venom from an ancient venomous lineage and support principles of protein evolution that directly relate to multigene family evolution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gene duplication is a well-known mechanism that lays the foundation for genetic novelties contributing to species divergence and species-specific functions (Ohno et al. 1968; Ohta 1991; Zhang 2003; Magadum et al. 2013). However, the type or strength of selection underlying multigene family evolution remains controversial (Eirín-López et al. 2012). Gene duplication is thought to promote diversification as gene copies diverge independently from each other and give rise to novel functions typically under the influence of positive selection (Nei et al. 1997; Nei and Rooney 2005; Tan and Low 2018). However, growing evidence suggests that multigene families not only evolve through strong positive selection, but that negative selection also plays a key role in their evolution (Nei and Rooney 2005; Rooney and Ward 2005; Eirín-López et al. 2012; Sipiczki et al. 2018). This disagreement that has occurred historically in the types of selection and models of multigene evolution highlights the need for further studies describing multigene families and their evolution.
Venom provides an ideal system to study multigene family evolution as this variable cocktail of different venom proteins has evolved numerous times throughout the animal kingdom (Daltry et al. 1996; Casewell et al. 2011; Rokyta et al. 2011; Vonk et al. 2013; Zancolli and Casewell 2020). Venom proteins have been thought to evolve under the influence of strong positive selection (Kordiš and Gubenšek 2000; Gibbs and Rossiter 2008; Juarez et al. 2008; Weinberger et al. 2010; Rokyta et al. 2011; Aird et al. 2017; Casewell et al. 2020). However, a recent review of the evolution of animal venoms summarized two different models that could account for the evolution of paralogous venom genes: the rapid accumulation of variation in exposed residues, or RAVER model, and the two-speed mode of evolution model (Suranse et al. 2018). In addition to positive selection, these models of venom evolution incorporate other factors including purifying selection and physical constraints on protein structure (Sunagar et al. 2013; Sunagar and Moran 2015).
The first model described the evolution of snake three-fingers toxins (a common snake neurotoxin) and proposed that venom genes have a rapid accumulation of variation in exposed residues (RAVER; Sunagar et al. 2013). The RAVER model recognizes and follows the concepts of protein evolution that relate the changes in amino acids to underlying protein structure. Specifically, this model describes the diversification of three-finger toxins by investigating the different types of selection that influence residues based on their three-dimensional location. Sunagar et al. (2013) hypothesized that under the RAVER model, proteins would generally be conserved in the structural and functional areas of the proteins, while those on the exterior of the protein would accumulate more substitutions. The exposed residues that do not serve an essential role in the structure or function of the toxin are expected to be more susceptible to change through positive selection. However, residues found in structural or functional regions of the protein are predicted to be maintained through the influence of negative or purifying selection. Yet, rare mutations can still impact the functional and structural regions of the protein, but only if they substantially improve the potency of the venom and if the strength of positive selection is sufficient to expand these mutations in the population. This structural pattern to changes along a protein supports general principles of protein evolution, which agrees that mutations depend on the physical constraints of the individual residues and their relation to the three-dimensional structure of the protein (Levitt and Chothia 1976; Chothia and Lesk 1986; Thorne et al. 1996; Goldman et al. 1998; Ramsey et al. 2011). Solvent accessibility of residues is known to influence selective pressures across proteins (Conant and Stadler 2009), and the RAVER model claims that changes on the surface of a protein can change the surface chemistry of the proteins and can cause novel, non-specific actions with target proteins through direct interactions. These adaptive advantages can therefore be under the influence of selection without changing the core structure or function of the protein. Thus, the RAVER model predicts that mutations in specific regions of the protein and positive selection play key roles in all predator toxins and not just in snake venom three-finger toxins.
The two-speed mode of venom evolution proposes that different selective pressures act on younger verses ancient venomous lineages (Sunagar and Moran 2015). Venomous lineages are considered younger if they arose around 40 million years ago (advanced snakes and conesnails) and ancient if they arose around 400 million years ago or older (centipedes, cnidarians, coleoids, scorpion, and spiders; Sunagar and Moran 2015). The two-speed mode of evolution proposes that earlier stages of ecological specialization are accompanied by episodic diversifying selection that is then followed by a longer period of purification to retain the potency of the venom repertoire. Therefore, younger lineages, which are thought to be expanding into new ecological niches, maintain venoms under strong diversifying selection, while venom from older lineages are under strong purifying selection to maintain toxic function. Species within the older lineages, however, can re-enter the diversifying stage if they experience pronounced shifts in their environment or their ecology. To test these hypotheses in ancient centipede lineages, Sunagar and Moran (2015) used species across three highly divergent orders of centipedes that have been separated for around 430 million years (Fernández et al. 2014) with only a few individuals from each order. Increasing the representation of individuals within a single lineage would provide a more complete set of toxin paralogs of a specific family which can impact the amount of diversity observed and give more statistical power to subsequently detect incidences of selection acting across the venom repertoire. Species-level evolutionary trends in the venom of giant desert hairy scorpions (Hadrurus) contrast with the broad scale changes hypothesized by the two-speed hypothesis (Nystrom et al. 2023). Scorpions having originated around 430 mya (Waddington et al. 2015) are hypothesized to be predominantly under negative selection. However, in Hadrurus, one of the major toxin families, \(\alpha\) potassium channel toxins, displays strong pervasive and episodic positive selection (Nystrom et al. 2023). Thus, comparing the evolutionary history of animals that are more recently diverged would allow for the detection of a recent shift in ecology or if the venom repertoire of the ancient lineages remains constrained and follows the two-speed hypothesis.
We focus on centipedes, one of the ancient venomous lineages (arose around 430 mya; Anderson and Trewin 2003; Fernández et al. 2014) to test the two evolutionary models proposed. We tested whether centipede venom follows the two-speed hypothesis through incorporating centipede phylogenetics, a field where molecular data are sorely lacking (Edgecombe and Giribet 2019). Recent work integrating both morphology and molecular techniques have uncovered numerous paraphyletic relationships among genera and evidence of cryptic speciation among centipedes (Joshi and Karanth 2012; Vahtera et al. 2013; Edgecombe et al. 2015; Siriwut et al. 2015, 2018). We used transcriptomics to estimate phylogenetic relationships among species and transcriptomics and proteomics to identify the diversity of venom components within each species. We first constructed a phylogeny using transcriptomic data from 26 individual centipedes across six currently recognized species among the order Scolopendromorpha, providing the first multi-locus molecular phylogeny for scolopendromorphs in North America. Using venom-gland transcriptomics and venom proteomics, we characterized the suite of multigene families of venom proteins from each distinct clade in our molecular phylogeny. We tested two distinct hypotheses of venom evolution. We expect to observe more incidences of positive selection on the external structures of different multigene families if centipede venoms evolve through the RAVER hypothesis. Likewise, if centipedes follow the two-speed mode, we would expect that the majority of proteins have a large influence of negative selection unless there has been recent speciation or ecological changes. Our work connects multiple facets of the evolution of venomous organisms using phylogenetics and selection-based analyses across the three-dimensional structure of venom proteins, contributing to our understanding of multigene family evolution.
Materials and Methods
Sample Collection
Centipedes were collected from across the United States and brought back to Florida State University to be maintained for venom extractions. Venom was collected through electrostimulation of the forcipules as the individuals grasped onto a metal spatula (Ward and Rokyta 2018; Ellsworth et al. 2019). Venom was then transferred from the spatula to a clean vial using a pipette. Venom was lyophilized and stored at \(-80^{\circ }\textrm{C}\) until further use in proteomic analyses. Venom glands were dissected under stereoscopic microscope four days following venom extraction, placed in RNAlater, kept overnight at \(4^{\circ }\textrm{C}\), and then stored at \(-80^{\circ }\textrm{C}\) until RNA extraction. Each centipede specimen following dissection was placed in 95% ethanol and stored at \(-80^{\circ }\textrm{C}\). Species identification was performed using a dichotomous key (Shelley 2002) and the sex of each specimen was determined when possible based on the presence or absence of styles or spinnerets (Bonato et al. 2010).
Venom Proteomics
Individual venom samples were prepared for mass spectrometry (LC-MS/MS), using approximately \(5\mu \hbox {g}\) of whole venom. Whole venom was digested using the Calbiochem ProteoExtract All-in-One Trypsin Digestion Kit (Merch, Darmstadt, Germany) per manufacturer’s instructions. Samples were then dried using a SpeedVac and triplicate LC-MS/MS runs were completed by the College of Medicine Translational Science Laboratory at Florida State University, as previously described (Ward and Rokyta 2018).
Resulting LC-MS/MS data were analyzed using Proteome Discover (version 2.2) and protein and peptide identities were confirmed for each venom sample using Scaffold (version 5.1; Proteome Software Inc., Portland, OR, USA) with a protein false discovery rate set to 1.0% and a minimum number of peptides set to 1. Using the consensus transcriptome for each clade identified in phylogenetic analysis, peptide abundances for each individual were calculated using the normalized number of total spectra counts across the three LC-MS/MS replicates, as described by Ward and Rokyta (2018).
Transcriptome Sequencing
Total RNA from the venom glands was prepared by removing glands from RNAlater and performing a TRIZol-chloroform (Invitrogen) extraction, as previously described (Ward and Rokyta 2018). Total RNA content of the samples was quantified using the Qubit RNA Broad-range kit (Thermo Fisher Scientific) and an RNA 6000 Pico Bioanalyzer chip (Agilent Technologies), per the manufacturer’s instructions. To isolate only the mRNA and to prepare the sequencing libraries, a NEBNext Poly(A) mRNA Magnetic Isolation Module, a NEBNext Ultra RNA Library Prep Kit, High Fidelity 2\(\times\) Hot Start PCR Mix, AMPure XP beads (Agencourt) for purification the PCR reaction, and Illumina Multiplex Oligos were used (New England Biolabs). Samples were fragmented for 15.5 min to obtain an average fragment size of approximately 370 base pairs. Samples were then quantified and quality checked using KAPA PCR (performed by the Molecular Cloning Facility at Florida State University Department of Biological Science) and a High Sensitivity DNA Bioanalyzer chip (Aglient Technologies). RNA-seq libraries were pooled with other sequencing libraries and sequenced with 150PE on an Illumina HiSeq 2500 at the Florida State University College of Medicine Translational Science Laboratory.
Transcriptome Assembly and Analysis
Transcriptomes were analyzed and assembled using the raw 150 paired-end (PE) sequencing reads as previously described (Holding et al. 2018; Ward and Rokyta 2018). Raw reads from three previously published centipede venom glands from North America were downloaded from the National Center for Biotechnology Information (NCBI) and processed alongside the samples we produced. We downloaded and analyzed raw reads for Hemiscolopendra marginata (SRR8188011, SRR8188012, SRR8188013, SRR8188014; Nystrom et al. 2019), Scolopocryptops sexspinosus (SRR8188015, SRR8188016, SRR8188017, SRR8188018; Ellsworth et al. 2019), and Scolopendra viridis (SRR7102113, SRR7102114; Ward and Rokyta 2018). Using a custom python script and FASTQC (version 0.11.5; Andrews et al. 2010), raw reads were filtered and quality controlled to find and remove cross contamination from other samples. Trim Galore! (version 0.4.4; Krueger 2015) and PEAR (version 0.9.6; Zhang et al. 2014) were then used to quality trim and merge the filtered reads, respectively. De novo transcriptome assemblies were performed using multiple assemblers to help identify the greatest number of unique toxin transcripts (Holding et al. 2018). The following three assemblers were used: DNAStar NGen (version 12.3.1), Extender (version 1.04; Rokyta et al. 2012), and Trinity (version 2.4.0; Grabherr et al. 2011). DNAStar NGen and Trinity (k-mer size of 31) utilized both the merged and unmerged reads but considered all of the reads as unpaired. Extender ran in replicates of 20 using only the merged reads, a minimum phred of 30, and an overlap of 20 nucleotides.
Using custom python scripts, contigs from the assembled transcriptomes were filtered and annotated based on homologous toxins downloaded from the Uniprot (UPT) toxin database. Toxins were filtered out if they had at least a 90% match to the total length of the curated toxin match or if they did not have a signal peptide. The open reading frame (ORF) was determined based on the primary BLAST hit and signal peptides were checked using SignalP (version 4.1; Petersen et al. 2011) under the sensitive settings. Sequences that had a signal peptide, a stop codon, and a 90% match to a known curated toxin were added to the putative toxins list for each species. However, because the current curated toxin database for centipedes is still growing, we chose to also do a proteomic-based annotation of the toxins to fully capture the diversity of toxins in each of these species.
Proteomic annotations started with getting the ORF from each of the sequences in the assembled transcriptomes. The getorf function in Emboss (version 6.6.0.0; Rice et al. 2000) was used to identify all available ORFs from each assembly. The proteomic data for each individual used the identified ORFs as a database to search against using Proteome Discoverer and Scaffold. Custom python scripts then filtered the contigs identified in Proteome Discoverer and Scaffold based on the presence of a signal peptide and a valid stop codon. These sequences that have proteomic evidence were then named according to a BLAST search of the Chilopoda transcriptome shotgun assemble database. If a sequence did not have a hit to this database, it was named as a venom protein (VP) in the final transcriptome set.
Putative toxins from both methods of annotations were combined within an individual and were clustered at 100% sequence identity. Sequences were then aligned to merged and unmerged reads using bwa (version 0.7.12; Li 2013) to check for potential chimeric sequences. A sliding window of 151 was used to determine if any place along the sequence did not have any coverage and those sequences were removed from the data set. The remaining sequences were marked to be hand checked if they had \(> 20\)-fold difference based on read coverage between any two of the sliding windows. All marked sequences were checked for chimeric properties using the distribution of reads mapped across the sequence. The remaining toxins were then clustered across the clades identified in phylogenetic analysis below using cd-hit at 98% sequence identity.
Due to the large data set of 26 individuals, a final search for homologous toxins of each of the assembled transcriptomes was completed using a combined database of all of the toxins identified above. The database was then used to search the three assemblies described above and three additional assemblies from BinPacker version 1.0 (Liu et al. 2016), SOAPdenovo-trans version 1.03 (Xie et al. 2014), and rnaSPAdes version 3.10.1 (Bankevich et al. 2012). Matches against this database for each individual were then annotated as described above and appended onto the clean toxin set for each individual. These toxins were then clustered at 98% using cd-hit and checked again for chimeric sequences using the same process as before. To generate the final consensus toxin transcriptomes, final toxins identified from each clade were then clustered at 98%. Because the current curated toxin database for centipedes is still growing, we only considered putative toxins as toxins if they were identified in both the transcriptome and the proteome.
To identify the non-toxin transcripts from each individual contigs generated from Trinity assembly were processed using BUSCO (version 3.1.0; Waterhouse et al. 2018) under the genomics settings and using the arthropoda_odb9 database. Custom scripts were then used to process the single-copy BUSCO matches for each individual and annotate them using the same criteria as the homologous annotations above. All sequences both toxins and non-toxins were then clustered at 98% using cd-hit to generate the final consensus transcriptome. Individual transcriptomic abundances were then calculated using RSEM with bowtie2 (version 2.3.0; Langmead and Salzberg 2012) with the consensus transcriptomes for each clade.
Phylogenetic Analysis
To infer a phylogeny for our taxa, we first retrieved additional venom-gland transcriptomes from NCBI to function as outgroups. Specifically, we retrieved data for Theatops posticus (SRR24351575, SRR24351576), Scutigera coleoptrata (SRR8998264), Lithobius forficatus (SRR8998265), and Strigamia maritima (SRR8998266; Jenner et al. 2019; Lane et al. 2023). All outgroups were processed as above and assembled using Trinity. Next, we used BUSCO (v4.0.6; –mode genome) to extract single-copy orthologs from each Trinity assembly (Manni et al. 2021).
We aligned each BUSCO locus using MAFFT (–auto –adjustdirectionaccurately; Katoh and Standley 2013) and used CIAlign (Tumescheit et al. 2022) to clean the alignment removing divergent sequences, short sequences, insertions, and cropping ends with default settings. We then used trimal (-gappyout; Capella-Gutiérrez et al. 2009) to remove gappy sites from the alignment and removed sequences where > 30% of the total alignment length consisted of gaps with a custom script. We used these preliminary alignments to infer gene trees using IQTree2 with default settings and 1000 UltraFast bootstraps (Minh et al. 2013; Kalyaanamoorthy et al. 2017; Minh et al. 2020). We used the gene trees as input to TreeShrink (Mai and Mirarab 2018), which searches for abnormally long branches across trees and removes these taxa from the tree/alignment.
We re-cleaned the alignments with trimal (-gappyout) and filtered the alignments for missing data. Specifically, we removed (1) sequences with \(< 50\%\) of the samples represented and (2) samples that had \(< 5\%\) of the loci represented. The final alignments were used to infer gene trees using IQTree2 as above. We collapsed low support branches (i.e., bootstrap support \(< 10\)%) from each gene tree and then used Astral-III (Zhang et al. 2018; Yin et al. 2019; Rabiee et al. 2019) to infer the final species tree.
Selection Analyses
Sequences from all of the major families of toxins were combined together without their signal peptides and translationally aligned in Geneious Prime (version 2022.1.1) using Clustal Omega (version 1.2.3) under default parameters. Each toxin family was then further separated into groups based on their percent sequence similarity. Groups were created for sequences that showed at least 40% similarity with at least one other sequence in the same toxin family. The new groups that contained at least four sequences were then translationally aligned as described above and the sequence alignments for each group were then used to generate trees with IQTree (version 2.0.3) using a codon model. HyPhy (version 2.5) was then used to run selection analyses using the maximum likelihood tree from IQTree under the following different models; aBSREL, BUSTED, FEL, and MEME (Kosakovsky Pond et al. 2020). Using all of the these models, we were able to test for evidence of episodic positive selection (MEME), pervasive positive and negative selection (FEL), positive selection across the whole toxin (BUSTED), and positive selection across the gene tree (aBSREL).
Protein Structure and Solvent Accessibility
Three-dimensional structures were obtained from predicted AlphaFold structures deposited in the Uniprot database through finding the closest BLAST match of the six most diverse groups of venom proteins (Jumper et al. 2021; Varadi et al. 2022; The UniProt Consortium 2023).We searched the consensus sequence for \(\beta\)-Pore forming toxins (\(\beta\)PFTX) group 5, \(\beta\)PFTX group 6, cysteine-rich, allergen, and pathogenesis-related proteins (CAP) in the CAP2 family (CAP2), \(\gamma\)-Glutamyl transferases (GGT), Scoloptoxins (SLPTX) in SLPTX10 group 3, and SLPTX15 group 4 for the best BLAST match and downloaded the following structures, respectively, T1IT04, A0A4D5R9P5, A0A646QDI4, P0DPU3, A0A4D5R9T6, A0A4D5R9Z4. To determine the solvent accessible surface area of each protein residue, we used the EDTSurf algorithm (Xu and Zhang 2009) implemented in iCn3D (Wang et al. 2022, 2020). Sites of positive and negative selection identified above using MEME and FEL were plotted onto the three-dimensional structures in Geneious Prime. Sites that were identified as positive in either test were mapped as positive onto the model regardless of whether they were identified as either neutral or negative in the other test. Because we noted a difference in the number of sites that were accessible versus inaccessible as well as potential differences in the numbers of sites under positive and negative selection, we used a \(\chi ^2\) test to determine whether the proportion of sites of selection was correlated with solvent accessibility.
Data Availability
Raw sequencing reads were submitted to the National Center for Biotechnology Information’s (NCBI) Sequence Read Archive (SRA) under BioProject PRJNA340270 and BioSample accessions SAMN38699543–SAMN38699562 and SRA accessions SRR27151397–SRR27151416. Raw reads for Hemiscolopendra marginata, Scolopocryptops sexspinosus, and Scolopendra viridis were submitted previously under the BioSample accessions SAMN10423645,SAMN10423646, SAMN10423647,SAMN10423648, SAMN09042581, and SAMN09042582. We submitted the assembled consensus transcriptomes for each clade to the NCBI Transcriptome Shotgun Assembly (TSA) database at DDBJ/EMBL/GenBank under the accessions GKRH00000000 (H. marginata), GKRI00000000 (S. sexspinosus), GKRJ00000000 (S. viridis), GKRB00000000 (S. alternans), GKRC00000000 (S. heros Clade 1), GKRD00000000, (S. heros Clade 2), GKRE00000000 (S. polymorpha Clade 1), GKRF00000000 with GKMY01000000 (S. polymorpha Clade 2), and GKRG00000000 (S. polymorpha Clade 3). The versions described in this paper are the first versions, GKRH01000000 (H. marginata), GKRI01000000 (S. sexspinosus), GKRJ01000000 (S. viridis), GKRB01000000 (S. alternans), GKRC01000000 (S. heros Clade 1), GKRD01000000, (S. heros Clade 2), GKRE01000000 (S. polymorpha Clade 1), GKRF01000000 with GKMY01000000 (S. polymorpha Clade 2), and GKRG01000000 (S. polymorpha Clade 3). Raw mass spectrometry reads and consensus proteome files were deposited in the ProteomeXchange Consortium with the PRIDE partner repository (Vizcaíno et al. 2016) under the PXD048308 dataset identifier.
Results and Discussion
Centipede Phylogenetics
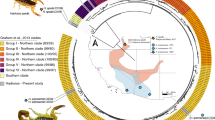
Our phylogeny of centipedes from North America shows paraphyly among the genus Scolopendra and provides evidence for cryptic diversity for widely ranging species (Fig. 1). We found Plutoniumidae and Scolopocryptopidae form a sister clade with respect to Scolopendridae (Fig. 1). Within Scolopendridae, Scolopendra alternans forms the most basal taxa. Scolopendra heros from Arizona and Texas form a moderately divergent, but monophyletic group sister to the remaining Scolopendridae. Interestingly, Hemiscolopendra marginata causes the genus Scolopendra to be paraphyletic as this genus/species forms the sister group to S. viridis and S. polymorpha. Additionally, we found that S. polymorpha is highly paraphyletic, forming three deeply divergent and independent clades. The first clade occurs in Arizona and, surprisingly, is sister to S. viridis from Florida. The remaining two clades of S. polymorpha are sister to one another with one clade occurring in northern California and the other occurring in southeastern Arizona. Cryptic diversity exists within this group, given that we described two paraphyletic southeastern Arizona lineages. However, we are unable to address this cryptic diversity taxonimically as we had limited sampling across each species’ range.

Phylogenic analyses revealed paraphyletic relationships within Scolopendra and provided evidence for cryptic species. The phylogeny was generated using Astral-III and single-copy orthologs identified using BUSCO. Support for individual nodes across the tree are from UltraFast bootstrap values. Hemiscolopendra marginata causes the genus of Scolopendra to be paraphyletic because this clade forms a sister group to S. viridis and S. polymorpha. We also observed evidence for cryptic species within S. heros and S. polymorpha. Scolopendra heros has a moderately divergent, yet monophyletic group, and S. polymorpha shows a divergent relationship that is paraphyletic, where clade one is sister to S. viridis. The map shows the distribution of centipedes sampled across the southern portion of the United States. Colored dots associated with each clade of the phylogeny correspond to the points on the distribution map. Branch lengths are measured in coalescent units (CU)
Centipede systematics is in need of more advanced molecular analyses to describe the diversity that is present within this group (Edgecombe and Giribet 2019). Previous work on the classification of centipede orders generally agree on their phylogenetic placement whether looking at molecular or morphological evidence (Fernández et al. 2014). However, examination of generic relationships have revealed rampant paraphyly among genera (Vahtera et al. 2013; Edgecombe et al. 2015; Joshi and Karanth 2012; Siriwut et al. 2015, 2018). Our results support these studies by showing monophyletic relationships of centipede orders and families, but paraphyly in geographically proximal members of the Scolopendra genus in North America. Even with limited sampling, our phylogeny provides evidence of potential cryptic species occurring in two species complexes (Fig. 1). This work further supports the need for a more rigorous sampling of centipedes across North America using molecular techniques to accurately describe and classify the diversity of centipedes. The relationship between S. viridis and S. polymorpha especially needs denser sampling across their range to resolve any cryptic species and to resolve species and population distributions. The large distribution of both S. polymorpha and S. viridis and their considerable range overlap (Shelley 2002) supports the relationship that we observed between these species and the need for further investigation.
Before the separation of H. marginata from the other members of Scolopendra, Scolopendra marginata was described as the most problematic name in North American Scolopendromorph centipedes (Hoffman and Shelley 1996). The designation to move H. marginata from Scolopendra was based on a single key morphological feature (lack of the distoventral spur on the proximotarsi; Hoffman and Shelley 1996). Hemiscolopendra marginata is currently described as a monotypic genus based on 21 anatomical features and is found across the southern United States and extends southward into Mexico (Shelley 2008). More recently, Vahtera et al. (2013) included H. marginata, S. alternans, S. polymorpha, and S. viridis in an analysis that used morphological traits as well as four molecular markers (two nuclear and two mitochondrial). Based solely on morphological data, H. marginata clustered within a clade containing species from Scolopendra sampled within this study and other more distantly related Scolopendra species. However, when morphological traits are combined with the molecular markers, the phylogeny placed H. marginata in the same clade as the Scolopendra species sampled in the current study and also showed rampant paraphyly among the genus Scolopendra. Based on the singular key morphological feature to distinguish the two genera (Hoffman and Shelley 1996), the additional support showing their clustering within Scolopendra (Vahtera et al. 2013), and our results, we advise the taxonomic change to reinstate Hemiscolopendra marginata to Scolopendra marginata.
General Venom Composition
We sequenced 26 venom-gland transcriptomes from six currently recognized species of scolopendromorph centipedes and obtained 6–44 million reads pairs for each individual transcriptome with at least two representatives for each species (Table 1). From the assembled transcriptomes, we were able to identify 824–1142 total proteins in each clade with the lowest number of proteomically confirmed toxins being 38 and the highest being 114. Toxins contributed between 22.0% and 93.4% of the total transcript abundance, with an average of 75.3% (Table 1).
Through comparing the venom composition of multiple lineages of scolopendromorph centipedes, we found that the overall abundance of the different toxin families is very similar across all species with the exception of S. sexspinosus and H. marginata (Fig. 2). Scolopocryptops sexspinosus has an abundant and distinct toxin class separate from all of the other venoms, adamalysin-like metalloproteases. The novel recruitment of adamalysin-like metalloproteases in centipede venoms was described previously from these two individuals highlighting the similarity between this family of toxins and the well-characterized snake venom metalloproteases (Ellsworth et al. 2019). This family of toxins is only known to occur within the family Scolopocryptopidae and is not present in any of the other species within Scolopendridae or Plutoniumidae. However, because S. sexspinosus is the only representative from Scolopocryptopidae, we cannot determine if this toxin family is found within other members of Scolopocryptopidae or if this recruitment was unique to this species.

The relative transcriptomic and proteomic abundance of each major toxin family. Each bar represents an individual transcriptome or proteome and groups of bars are separated into clades based on the phylogenetics described in this paper. The most expressed toxin families in the transcriptome across all taxa were the \(\beta\)-pore forming toxins (BPFTXs), low-density lipoprotein receptor Class A repeat domain (LDLAs), and the Scoloptoxins (SLPTXs). The most highly expressed toxin families in the proteome were \(\gamma\)-Glutamyl transferases (GGTs), cysteine-rich, allergen, and pathogenesis-related proteins (CAPs), and Scoloptoxins (SLPTXs)
Hemiscolopendra marginata displays a distinct venom between the two individuals within the same species (Fig. 2). The variation seen in the venom for H. marginata was previously identified as a sex-biased venom expression and is so far the only centipede venom to show sex-biased venom variation (Nystrom et al. 2019). Nystrom et al. (2019) previously used the same transcriptomic and proteomic data that we used; however, they increased proteomic sampling between sexes of H. marginata and found a statistical sex-based difference in venom composition. While this apparent difference was described in this species through increased samples of the sexes, we could not account for sex-based difference in venom composition for the other eight clades due to our limited sampling. However, such a dramatic shift in venom composition was not identified among the sexes of the other clades where the sex of the individuals could be identified. This trend, however, should be investigated further with denser sampling of male and female centipedes to fully categorize other potential instances of sex-based venom variation in centipedes.
Even with the two distinct venoms discussed above, the overall venom composition within Scolopendromorpha appears to be conserved with major toxin families showing a similar abundance across all species. Looking at the average percent abundance across all individuals showed that three families had above average expression in the transcriptome and three families had above average expression in the proteome. The highest expressed families in the transcriptome were the \(\beta\)-Pore forming toxins (\(\beta\)PFTXs), proteins containing a low-density lipoprotein receptor Class A repeat domain (LDLAs), and the Scoloptoxins (SLPTXs). The proteome, however, revealed that cysteine-rich, allergen, and pathogenesis-related proteins (CAPs), \(\gamma\)-Glutamyl transferases (GGTs), and Scoloptoxins (SLPTXs) were the most abundant families.
Discrepancies between the venom proteome and the venom-gland transcriptome have been reported in a variety of other taxa (Rokyta et al. 2015; Biass et al. 2015; Gonçalves-Machado et al. 2016; Xu et al. 2017; Ward and Rokyta 2018; Nystrom et al. 2019). Correlative differences between transcriptomic and proteomic expression are thought to be more common than not due to a variety of different biological and technical processes (Maier et al. 2009; Diz et al. 2012). Venom and venom glands have been found to have high correlations between proteomic and transcriptomic abundances (Rokyta et al. 2015), in contrast to weaker correlations found in other systems (Schrimpf et al. 2009; Ghazalpour et al. 2011; Bantscheff et al. 2012). Differences in expression are often attributed to post-translational modifications that can influence detection and expression levels (Fox and Serrano 2008; Rokyta et al. 2015) and to mapping biases that exist within the bioinformatic pipeline (Wang et al. 2009; Fang and Cui 2011; Rokyta et al. 2012). The differences we found could also be caused by the timing between venom extraction and venom-gland dissections that we performed on the individual centipedes. Four days between extracting the venom and dissecting, the venom glands were shown to be the ideal timing to receive maximal transcriptomic output in snakes (Rotenberg et al. 1971); however, this timing has not been documented in centipedes. Asynchronous regeneration is known for some centipedes (Cooper et al. 2014; Nystrom et al. 2022) and could explain the proteomic and transcriptomic differences we found since venom family replacement happening near the beginning or end of venom regeneration would lead to a mismatch of expression patterns.
Even though expression patterns between the transcriptome and the proteome are not entirely consistent, we have identified the same principal venom components found in other centipede venoms (Undheim et al. 2014, 2015; Smith and Undheim 2018; Jenner et al. 2019). The vast majority of centipede venom components have not been functionally characterized. However, broad categories of functions can be assigned through homology. \(\beta\)PFTXs and some of the SLPTX superfamily of toxins (SLPTX01) are likely involved with cytotoxic functions in the venoms since they form transmembrane pores lysing cells and act as chitinases destroying the exoskeleton of their prey, respectively. The other members of the SLPTX superfamily that were found to have the highest expression (SLPTX10, SLPTX15) are mainly considered neurotoxins because of their effect on calcium, potassium, and sodium channels (Undheim et al. 2015; Smith and Undheim 2018). CAP2 proteins are also shown to be neurotoxic effecting calcium channels but also can play a role trypsin inhibition (Rates et al. 2007; Fry et al. 2009; Undheim et al. 2015). The GGTs and LDLAs, however, currently do not have a described function in centipede venoms but their high expression in the venom likely means that they have important functional or maintenance roles in the venom and venom gland.
Modes of Centipede Venom Evolution
Grouping toxins families based on their percent identity resulted in 33 toxin groups that contained at least four different sequences from across all consensus transcriptomes. Toxin families were grouped based on percent similarity due to the extreme divergence that we see in the different toxin families based on current naming conventions. We grouped the toxins using a cut off of 40% similarity with at least one other sequence in the same group because some families had sequences that were less than 10% similar with other similarly named sequences. The diversity of toxins and number of similar types of toxins in venomous systems is thought to have arisen from gene duplication events (Conant and Wolfe 2008; Wong and Belov 2012). These duplication events have been thought to give an immediate benefit to venom components through increased expression due to venom dosage effects and could later lead to gene subfunctionalization or neofunctionalization (Conant and Wolfe 2008; Fry et al. 2009; Casewell et al. 2013; Margres et al. 2017). Duplicated genes, however, require the protection of natural selection to support neofunctionalization and subfunctionalization and to prevent the removal of duplicated genes from the accumulation of degenerative mutations (Conant and Wolfe 2008). Most venom genes are thought to have arisen under the influence of positive selection (Kordiš and Gubenšek 2000; Gibbs and Rossiter 2008; Aird et al. 2017; Casewell et al. 2020). To test for the evidence of selection acting on these toxins genes, we examined the genes first for evidence of positive selection across the entirety of the gene for at least one site or one branch. Sixteen of the 33 groups showed evidence of positive selection across the gene (Table 2) which supports the evolution of venom genes through positive selection. To look closer at the patterns of selection at the site and branch level, we focused on six groups of toxins that had the most paralogous sequences (18 or more sequences); however, all of the results are included in Tables 2 and 3.
The toxin groups that included at least 18 sequences were two groups of \(\beta\)PFTXs, all of the CAP2 proteins, all of the GGTs, one group from the SLPTX10, and one group of the SLPTX15 (Figs. 3, 4, 5, 6, 7, and 8). All of these groups, except for the SLPTX15, showed at least four sites that are under episodic positive selection, while only three of the six groups had evidence for pervasive positive selection. The vast majority of sites among these six toxin groups showed evidence for negative selection (13–274 sites; Table 2). Five of the six groups all had one to three branches that displayed evidence for positive selection, while the majority of branches did not show evidence for positive selection. Between 91.3% and 97.0% of the total sites under selection were classified as negative. We found 17 total sites among all of the different toxin families that were under pervasive positive selection (Table 2), which is similar to what was previously reported in centipedes (Sunagar and Moran 2015). Sunagar and Moran (2015) also found a high percentage of negative selection among centipede toxins. Between 92.3% and 100% of the total sites under pervasive selection were classified as negative. The lack of sites under positive selection is further contrasted with analyses performed on evolutionary younger lineages like cone snails and snakes, where 166 and 531 sites were reported, respectively, compared to only eight sites in centipedes (Sunagar and Moran 2015). Thus, the high prevalence of negative selection supports the two-speed mode of venom evolution hypothesis proposed by Sunagar and Moran (2015), where venom evolution is first dominated by positive selection and rapid diversification followed by strong negative selection preserving the toxic arsenal. Sunagar and Moran (2015) proposed that older venomous lineages like centipedes, scorpions, spiders, coleoids, and cnidarians would be dominated by negative selection due to their evolutionary history. A relatively stable ecological state could offer the opportunity for negative selection to prevail, preserving toxicity found to be beneficial in that state. However, if a shift in the trophic ecology or environment of an ancient lineage disrupts that stable ecological state, they could re-enter the diversifying phase of selection. Additionally, diversifying selection can occur if the venomous species is involved in an evolutionary arms race facilitating the rapid reciprocal evolution between two coevolving traits (Van Valen 1977). Venom, however, does not necessarily always evolve under an arms-race scenario, but can have selective pressures from a vast community of predators or prey that can lead to diffuse selection or non-directional selection of venom (Strauss et al. 2005; Hall et al. 2020). The high prevalence of negative selection that we observed in some of the toxin families could also be explained by the high specificity of which some venom components interact with their targets. For example, the \(\beta\)PFTXs are hypothesized to undergo oligomerization which would constrain the amount of change that can occur on the protein and still result in a functional protein (Sunagar and Moran 2015; Jouiaei et al. 2015). Furthermore, this protein family is thought to bind to cellular membranes which may increase purifying selection to maintain interactions with these highly conserved targets.

Selection analyses performed on \(\beta\)-pore forming toxin group 5. We found evidence for positive selection across the entire protein according to HYPHY BUSTED (p value \(\ll 0.001\)). Selection was then tested across the branches of the gene tree showing evidence for one branch under positive selection. The abundances for the transcriptome and the proteome are shown to the side of the gene tree and represent the average expression of all individuals in a single clade. Transcriptomic and proteomic abundances have been standardized to each clade to display the abundance of paralogs. Evidence for episodic positive selection is shown on the top part of the graph with red points showing signs for positive selection using MEME. The lower part of the graph displays the FEL results that provide evidence for pervasive negative and positive selection with blue points indicating negative selection and red points indicating positive selection. The positive and negative selection identified were plotted onto the predicted three-dimensional structures in blue and red. The solvent accessible regions are plotted onto the same structure with solvent inaccessible sites being shown in green and solvent accessible sites being shown in orange. A \(\chi ^2\) test for independence showed no significant difference in the proportion of sites under selection and the accessibility of the sites (\(\chi ^2 = 0.11\), \(p = 0.74\))

Selection analyses performed on \(\beta\)-pore forming toxin group 6. We found evidence for positive selection across the entire protein according to HYPHY BUSTED (p value \(\ll 0.001\)). Selection was then tested across the branches of the gene tree showing evidence for three branches under positive selection. The abundance for the transcriptome and the proteome is shown to the side of the gene tree, and represents the average expression of all individuals in a single clade. Transcriptomic and proteomic abundances have been standardized to each clade to display the abundance of paralogs. Evidence for episodic positive selection is shown on the top part of the graph with red points showing signs for positive selection using MEME. The lower part of the graph displays the FEL results that provide evidence for pervasive negative and positive selection with blue points indicating negative selection and red points indicating positive selection. The positive and negative selection identified were plotted onto the predicted three-dimensional structures in blue and red. The solvent accessible regions are plotted onto the same structure with solvent inaccessible sites being shown in green and solvent accessible sites being shown in orange. A \(\chi ^2\) test for independence showed no significant difference in the proportion of sites under selection and the accessibility of the sites (\(\chi ^2 = 0.18\), \(p = 0.67\))

Selection analyses performed on cysteine-rich, allergen, and pathogenesis-related proteins (CAP) in the CAP2 family. We found evidence for positive selection across the entire protein according to HYPHY BUSTED (p value \(\ll 0.001\)). Selection was then tested across the branches of the gene tree showing evidence for three branches under positive selection. The abundance for the transcriptome and the proteome is shown to the side of the gene tree, and represents the average expression of all individuals in a single clade. Transcriptomic and proteomic abundances have been standardized to each clade to display the abundance of paralogs. Evidence for episodic positive selection is shown on the top part of the graph with red points showing signs for positive selection using MEME. The lower part of the graph displays the FEL results that provide evidence for pervasive negative and positive selection with blue points indicating negative selection and red points indicating positive selection. The positive and negative selection identified were plotted onto the predicted three-dimensional structures in blue and red. The solvent accessible regions are plotted onto the same structure with solvent inaccessible sites being shown in green and solvent accessible sites being shown in orange. A \(\chi ^2\) test for independence showed no significant difference in the proportion of sites under selection and the accessibility of the sites (\(\chi ^2 = 0.42\), \(p = 0.52\))

Selection analyses performed on \(\gamma\)-Glutamyl transferases. We found evidence for positive selection across the entire protein according to HYPHY BUSTED (p value = 0.04). Selection was then tested across the branches of the gene tree showing evidence for three branches under positive selection. The abundance for the transcriptome and the proteome is shown to the side of the gene tree, and represents the average expression of all individuals in a single clade. Transcriptomic and proteomic abundances have been standardized to each clade to display the abundance of paralogs. Evidence for episodic positive selection is shown on the top part of the graph with red points showing signs for positive selection using MEME. The lower part of the graph displays the FEL results that provide evidence for pervasive negative and positive selection with blue points indicating negative selection and red points indicating positive selection. The positive and negative selection identified were plotted onto the predicted three-dimensional structures in blue and red. The solvent accessible regions are plotted onto the same structure with solvent inaccessible sites being shown in green and solvent accessible sites being shown in orange. A \(\chi ^2\) for independence showed significant differences in the proportion of sites under selection and the accessibility of the sites (\(\chi ^2 = 17.14\), p value \(\ll 0.001\))

Selection analyses performed on family 10 of Scoloptoxins group 3. We found evidence for positive selection across the entire protein according to HYPHY BUSTED (p value \(\ll 0.001\)). Selection was then tested across the branches of the gene tree showing evidence for one branch under positive selection. The abundance for the transcriptome and the proteome is shown to the side of the gene tree, and represents the average expression of all individuals in a single clade. Transcriptomic and proteomic abundances have been standardized to each clade to display the abundance of paralogs. Evidence for episodic positive selection is shown on the top part of the graph with red points showing signs for positive selection using MEME. The lower part of the graph displays the FEL results that provide evidence for pervasive negative and positive selection with blue points indicating negative selection and red points indicating positive selection. The positive and negative selection identified were plotted onto the predicted three-dimensional structures in blue and red. The solvent accessible regions are plotted onto the same structure with solvent inaccessible sites being shown in green and solvent accessible sites being shown in orange. We were unable to perform a \(\chi ^2\) test for independence because we did not detect any sites of positive selection in either solvent accessible or solvent inaccessible regions

Selection analyses performed on family 15 of Scoloptoxins group 4. We found evidence for positive selection across the entire protein according to HYPHY BUSTED (p value = 0.01). Selection was then tested across the branches of the gene tree and showed no evidence for specific branches being under positive selection. The abundance for the transcriptome and the proteome is shown to the side of the gene tree, and represents the average expression of all individuals in a single clade. Transcriptomic and proteomic abundances have been standardized to each clade to display the abundance of paralogs. Evidence for episodic positive selection is shown on the top part of the graph with red points showing signs for positive selection using MEME. The lower part of the graph displays the FEL results that provide evidence for pervasive negative and positive selection with blue points indicating negative selection and red points indicating positive selection. The positive and negative selection identified were plotted onto the predicted three-dimensional structures in blue and red. The solvent accessible regions are plotted onto the same structure with solvent inaccessible sites being shown in green and solvent accessible sites being shown in orange. A \(\chi ^2\) for independence showed significant differences in the proportion of sites under selection and the accessibility of the sites (\(\chi ^2 = 5.00\), \(p = 0.03\))
Solvent accessibility provides important information on the structure and function of proteins and how these molecules can interact with their environment (Ahmad 2005). We mapped the solvent accessible regions for the six toxins that we used for our analyses in selection and the sites under positive and negative selection onto the three-dimensional structure of the protein (Figs. 3, 4, 5, 6, 7, and 8). Consistent with the RAVER model of venom protein evolution, we observed that solvent inaccessible sites corresponded with sites under negative selection. However, some of the proteins did not show as strong of a relationship with the solvent accessible sites and positive selection. Because of differences in numbers of accessible and inaccessible sites as well as differences in those sites under positive and negative selection, we used a \(\chi ^2\) test to determine whether sites of selection were correlated with solvent accessibility. Three of the six groups (\(\beta\)PFTX-Group5, \(\beta\)PFTX-Group6, CAP2) had no significant association between the proportion of sites under selection and the accessibility of the sites. Out of the 20 sites under negative selection for SLPTX-Group 3, 19 were found in solvent inaccessible regions and only one was found in a solvent accessible region. No sites of positive selection corresponded with sites that were determined to be either solvent accessible or inaccessible. Two of the six groups (GGT and SLPTX15-Group4) had a significant association between the proportion of sites under selection and the accessibility of those sites, where most of the sites under negative selection were found in the inaccessible regions of the proteins (GGT: \(\chi ^2 = 17.14\), \(p \ll 0.01\) and SLPTX15-Group 4: \(\chi ^2 = 5.00\), \(p = 0.02\)).
This set of analyses allow us to directly compare two models for the evolution of venom using a closely related clade within an ancient venomous lineage. We found support for both the two-speed mode of venom evolution and the RAVER model. Negative selection was the dominant force of selection across the six toxin groups that we analyzed with only a few residues being detected under either pervasive or episodic positive selection. These results directly align with the two-speed model of evolution. Under this model, the venom proteins are thought to be under the conservation of negative selection to retain their potency and overarching positive selection will only become dominant after ecological shifts in the venomous lineage. Most of the larger centipedes, including Scolopendromorpha, have a very generalist diet and corresponding ecology (Alessandro 2011). Although a broad range of habitat types exist for scolopendromorphs, most centipedes are soil inhabitants that rely on moist microenvironments and forage during the night (Alessandro 2011). However, little is still known about the individual ecology of each of these species and more work is needed to be undertaken to fully understand the diversity of behaviors and potential differences in ecology. Similarities in ecology and preference for moist microhabitats could contribute to venom being conserved in centipedes under negative selection.
The location of negative selection along the three-dimensional structure of the protein coincided with solvent inaccessible regions in two of the toxin groups. Three of the other four toxin groups did not show an association between solvent accessibility and selection and one group only had negative selection detected across the protein. Therefore, we could not test to see if there was an association between the type of selection and the solvent accessibility. These results directly support that some venom proteins might be experiencing selection similar to what was predicted in the RAVER model of evolution and general protein evolution (Goldman et al. 1998; Conant and Stadler 2009; Sunagar et al. 2013). However, sites identified as being under positive selection were still detected on surface inaccessible residues. This does not necessarily disagree with the RAVER model since that model proposed that areas under positive selection could still be in inaccessible regions of the proteins and that those mutations could give rise to new structure and potentially new functions or specificity. Substitutions in these inaccessible and functionally or structurally important regions would typically occur less often.
Conclusions
Using transcriptomics and proteomics, we characterized venom protein diversity across multiple species of Scolopendromorpha and estimated their phylogenetic relationships using transcriptomics. From our phylogeny, we found Scolopendra to be paraphyletic since it included the genus Hemiscolopendra, and we found evidence of cryptic species occurring in two widely distributed species (Fig. 1). These results demonstrate the need for future phylogenetic studies within centipedes especially since our phylogeny is the first molecular phylogeny for centipedes found in North America. From the clades that we described, we were able to describe a similar venom composition across all of the centipedes which is mainly composed of \(\beta\)PFTXs, CAPs, GGTs, LDLAs, and SLPTXs (Fig. 2). Identifying the type of selection and the location of selection along the protein allowed us to test two different hypotheses for venom evolution: the two-speed model of venom evolution and the RAVER model. We found more sites of negative selection compared to positive selection across all toxin families (Figs. 3, 4, 5, 6, 7, and 8); however, when we did find evidence for positive selection, it often corresponded with the exposed residues of the protein, supporting both models of venom evolution. Through the use of phylogenetics, transcriptomics, proteomics, and selection-based analyses, we were able to describe the evolution of venom from an ancient venomous lineage and support longstanding principles of protein evolution that directly relate to multigene family evolution.
References
Ahmad S (2005) Role of solvent accessibility in structure based drug design. Curr Comput Aided Drug Des 1:223–235
Aird SD, Arora J, Barua A, Qiu L, Terada K, Mikheyev AS (2017) Population genomic analysis of a Pit viper reveals microevolutionary forces underlying venom chemistry. Genome Biol Evol 9:2640–2649
Alessandro M (2011) Treatise on zoology—anatomy, taxonomy, biology. The Myriapoda, Volume 1. Treatise on Zoology: Anatomy, Taxonomy, Biology. Brill
Anderson LI, Trewin NH (2003) An early Devonian arthropod fauna from the Windyfield cherts, Aberdeenshire, Scotland. Palaeontology 46:467–509
Andrews S et al (2010) FastQC: a quality control tool for high throughput sequence data. Cambridge
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD et al (2012) Spades: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477
Bantscheff M, Lemeer S, Savitski MM, Kuster B (2012) Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem 404:939–965
Biass D, Violette A, Hulo N, Lisacek F, Favreau P, Stöcklin R (2015) Uncovering intense protein diversification in a cone snail venom gland using an integrative venomics approach. J Proteome Res 14:628–638
Bonato L, Edgecombe GD, Lewis JG, Minelli A, Pereira LA, Shelley RM, Zapparoli M (2010) A common terminology for the external anatomy of centipedes (Chilopoda). ZooKeys 69:17
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973
Casewell NR, Wagstaff SC, Harrison RA, Renjifo C, Wüster W (2011) Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol Biol Evol 28:2637–2649
Casewell NR, Wüster W, Vonk FJ, Harrison RA, Fry BG (2013) Complex cocktails: the evolutionary novelty of venoms. Trends Ecol Evol 28:219–229
Casewell NR, Jackson TN, Laustsen AH, Sunagar K (2020) Causes and consequences of snake venom variation. Trends Pharmacol Sci 41:570–581
Chothia C, Lesk AM (1986) The relation between the divergence of sequence and structure in proteins. EMBO J 5:823–826
Conant GC, Stadler PF (2009) Solvent exposure imparts similar selective pressures across a range of yeast proteins. Mol Biol Evol 26:1155–1161
Conant GC, Wolfe KH (2008) Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet 9:938–950
Cooper AM, Kelln WJ, Hayes WK (2014) Venom regeneration in the centipede Scolopendra polymorpha: evidence for asynchronous venom component synthesis. Zoology 117:398–414
Daltry JC, Wüster W, Thorpe RS (1996) Diet and snake venom evolution. Nature 379:537–540
Diz AP, Martínez-Fernández M, Rolán-Alvarez E (2012) Proteomics in evolutionary ecology: linking the genotype with the phenotype. Mol Ecol 21:1060–1080
Edgecombe GD, Giribet G (2019) The molecularization of centipede systematics. In: Fusco G (ed) Perspectives on evolutionary and developmental biology: essays for Alessandro Minelli. Padova University Press
Edgecombe GD, Vahtera V, Giribet G, Kaunisto P (2015) Species limits and phylogeography of Newportia (Scolopendromorpha) and implications for widespread morphospecies. ZooKeys 510:65
Eirín-López J, Rebordinos L, Rooney A, Rozas J (2012) The birth-and-death evolution of multigene families revisited. Repetitive DNA 7:170–196
Ellsworth SA, Nystrom GS, Ward MJ, de Sousa LAF, Hogan MP, Rokyta DR (2019) Convergent recruitment of adamalysin-like metalloproteases in the venom of the red bark centipede (Scolopocryptops sexspinosus). Toxicon 168:1–15
Fang Z, Cui X (2011) Design and validation issues in RNA-Seq experiments. Brief Bioinform 12:280–287
Fernández R, Laumer CE, Vahtera V, Libro S, Kaluziak S, Sharma PP, Pérez-Porro AR, Edgecombe GD, Giribet G (2014) Evaluating topological conflict in centipede phylogeny using transcriptomic data sets. Mol Biol Evol 31:1500–1513
Fox JW, Serrano SM (2008) Exploring snake venom proteomes: multifaceted analyses for complex toxin mixtures. Proteomics 8:909–920
Fry BG, Roelants K, Champagne DE, Scheib H, Tyndall JD, King GF, Nevalainen TJ, Norman JA, Lewis RJ, Norton RS et al (2009) The toxicogenomic multiverse: convergent recruitment of proteins into animal venoms. Annu Rev Genomics Hum Genet 10:483–511
Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, Farber CR, Sinsheimer J, Kang HM, Furlotte N et al (2011) Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet 7:e1001393
Gibbs HL, Rossiter W (2008) Rapid evolution by positive selection and gene gain and loss: PLA2 venom genes in closely related Sistrurus rattlesnakes with divergent diets. J Mol Evol 66:151–166
Goldman N, Thorne JL, Jones DT (1998) Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 149:445–458
Gonçalves-Machado L, Pla D, Sanz L, Jorge RJB, Leitão-De-Araújo M, Alves MLM, Alvares DJ, De Miranda J, Nowatzki J, de Morais-Zani K et al (2016) Combined venomics, venom gland transcriptomics, bioactivities, and antivenomics of two Bothrops jararaca populations from geographic isolated regions within the Brazilian Atlantic rainforest. J Proteomics 135:73–89
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Hall AR, Ashby B, Bascompte J, King KC (2020) Measuring coevolutionary dynamics in species-rich communities. Trends Ecol Evol 35:539–550
Hoffman RL, Shelley RM (1996) The identity of Scolopendra marginata Say (Chilopoda: Scolopendromorpha: Scolopendridae). Virginia Museum of Natural History
Holding ML, Margres MJ, Mason AJ, Parkinson CL, Rokyta DR (2018) Evaluating the performance of de novo assembly methods for venom-gland transcriptomics. Toxins 10:249
Jenner RA, von Reumont BM, Campbell LI, Undheim EA (2019) Parallel evolution of complex centipede venoms revealed by comparative proteotranscriptomic analyses. Mol Biol Evol 36:2748–2763
Joshi J, Karanth KP (2012) Coalescent method in conjunction with niche modeling reveals cryptic diversity among centipedes in the Western Ghats of South India. PLoS ONE 7:e42225
Jouiaei M, Sunagar K, Federman Gross A, Scheib H, Alewood PF, Moran Y, Fry BG (2015) Evolution of an ancient venom: recognition of a novel family of cnidarian toxins and the common evolutionary origin of sodium and potassium neurotoxins in sea anemone. Mol Biol Evol 32:1598–1610
Juarez P, Comas I, Gonzalez-Candelas F, Calvete JJ (2008) Evolution of snake venom disintegrins by positive Darwinian selection. Mol Biol Evol 25:2391–2407
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589
Kalyaanamoorthy S, Minh BQ, Wong TK, Von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Kordiš D, Gubenšek F (2000) Adaptive evolution of animal toxin multigene families. Gene 261:43–52
Kosakovsky Pond SL, Poon AF, Velazquez R, Weaver S, Hepler NL, Murrell B, Shank SD, Magalis BR, Bouvier D, Nekrutenko A et al (2020) HyPhy 2.5-a customizable platform for evolutionary hypothesis testing using phylogenies. Mol Biol Evol 37:295–299
Krueger F (2015) Trim galore. A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files 516
Lane AN, Nash PD, Ellsworth SA, Nystrom GS, Rokyta DR (2023) The arylsulfatase-and phospholipase-rich venom of the plutoniumid centipede Theatops posticus. Toxicon 233:107231
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Levitt M, Chothia C (1976) Structural patterns in globular proteins. Nature 261:552–558
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997
Liu J, Li G, Chang Z, Yu T, Liu B, McMullen R, Chen P, Huang X (2016) BinPacker: packing based de novo transcriptome assembly from RNA-Seq data. PLoS Comput Biol 12:e1004772
Magadum S, Banerjee U, Murugan P, Gangapur D, Ravikesavan R (2013) Gene duplication as a major force in evolution. J Genet 92:155–161
Mai U, Mirarab S (2018) TreeShrink: fast and accurate detection of outlier long branches in collections of phylogenetic trees. BMC Genomics 19:23–40
Maier T, Güell M, Serrano L (2009) Correlation of mRNA and protein in complex biological samples. FEBS Lett 583:3966–3973
Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM (2021) BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol 38:4647–4654
Margres MJ, Wray KP, Hassinger AT, Ward MJ, McGivern JJ, Moriarty Lemmon E, Lemmon AR, Rokyta DR (2017) Quantity, not quality: rapid adaptation in a polygenic trait proceeded exclusively through expression differentiation. Mol Biol Evol 34:3099–3110
Minh BQ, Nguyen MAT, Von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30:1188–1195
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, Lanfear R (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152
Nei M, Gu X, Sitnikova T (1997) Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci 94:7799–7806
Nystrom GS, Ward MJ, Ellsworth SA, Rokyta DR (2019) Sex-based venom variation in the eastern bark centipede (Hemiscolopendra marginata). Toxicon 169:45–58
Nystrom GS, Fry LG, Ellsworth SA, Rokyta DR (2022) Contrasting patterns of venom regeneration in a centipede (Scolopendra viridis) and a scorpion (Centruroides hentzi). Toxicon 210:132–140
Nystrom GS, Ellsworth SA, Ward MJ, Rokyta DR (2023) Varying modes of selection among toxin families in the venoms of the giant desert hairy scorpions (Hadrurus). J Mol Evol 91:935–962
Ohno S, Wolf U, Atkin NB (1968) Evolution from fish to mammals by gene duplication. Hereditas 59:169–187
Ohta T (1991) Multigene families and the evolution of complexity. J Mol Evol 33:34–41
Petersen TN, Brunak S, Von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Rabiee M, Sayyari E, Mirarab S (2019) Multi-allele species reconstruction using ASTRAL. Mol Phylogenet Evol 130:286–296
Ramsey DC, Scherrer MP, Zhou T, Wilke CO (2011) The relationship between relative solvent accessibility and evolutionary rate in protein evolution. Genetics 188:479–488
Rates B, Bemquerer MP, Richardson M, Borges MH, Morales RA, De Lima ME, Pimenta AM (2007) Venomic analyses of Scolopendra viridicornis nigra and Scolopendra angulata (Centipede, Scolopendromorpha): shedding light on venoms from a neglected group. Toxicon 49:810–826
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277
Rokyta DR, Wray KP, Lemmon AR, Lemmon EM, Caudle SB (2011) A high-throughput venom-gland transcriptome for the eastern diamondback rattlesnake (Crotalus adamanteus) and evidence for pervasive positive selection across toxin classes. Toxicon 57:657–671
Rokyta DR, Lemmon AR, Margres MJ, Aronow K (2012) The venom-gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genomics 13:312
Rokyta DR, Margres MJ, Calvin K (2015a) Post-transcriptional mechanisms contribute little to phenotypic variation in snake venoms. G3: Genes Genomes Genetics 5:2375–2382
Rokyta DR, Wray KP, McGivern JJ, Margres MJ (2015b) The transcriptomic and proteomic basis for the evolution of a novel venom phenotype within the timber rattlesnake (Crotalus horridus). Toxicon 98:34–48
Rooney AP, Ward TJ (2005) Evolution of a large ribosomal RNA multigene family in filamentous fungi: birth and death of a concerted evolution paradigm. Proc Natl Acad Sci 102:5084–5089
Rotenberg D, Bamberger E, Kochva E (1971) Studies on ribonucleic acid synthesis in the venom glands of Vipera palaestinae (Ophidia, Reptilia). Biochem J 121:609–612
Schrimpf SP, Weiss M, Reiter L, Ahrens CH, Jovanovic M, Malmström J, Brunner E, Mohanty S, Lercher MJ, Hunziker PE et al (2009) Comparative functional analysis of the Caenorhabditis elegans and Drosophila melanogaster proteomes. PLoS Biol 7:e1000048
Shelley RM (2002) A synopsis of the North American centipedes of the order Scolopendromorpha (Chilopoda). Virginia Museum of Natural History
Shelley R (2008) Revision of the centipede genus Hemiscolopendra Kraepelin, 1903: Description of H. marginata (Say, 1821) and possible misidentifications as Scolopendra spp.; proposal of Akymnopellis, n. gen., and redescriptions of its South American components (Scolopendromorpha: Scolopendridae: Scolopendrinae). Int J Myriapodol 1:171–204
Sipiczki M, Horvath E, Pfliegler WP (2018) Birth-and-death evolution and reticulation of ITS segments of Metschnikowia andauensis and Metschnikowia fructicola rDNA repeats. Front Microbiol 9:1193
Siriwut W, Edgecombe GD, Sutcharit C, Panha S (2015) The centipede genus Scolopendra in mainland Southeast Asia: molecular phylogenetics, geometric morphometrics and external morphology as tools for species delimitation. PLoS ONE 10:e0135355
Siriwut W, Edgecombe GD, Sutcharit C, Tongkerd P, Panha S (2018) Systematic revision and phylogenetic reassessment of the centipede genera Rhysida Wood, 1862 and Alluropus Silvestri, 1912 (Chilopoda: Scolopendromorpha) in Southeast Asia, with further discussion of the subfamily Otostigminae. Invertebr Syst 32:1005–1049
Smith JJ, Undheim EA (2018) True lies: using proteomics to assess the accuracy of transcriptome-based venomics in centipedes uncovers false positives and reveals startling intraspecific variation in Scolopendra subspinipes. Toxins 10:96
Strauss SY, Sahli H, Conner JK (2005) Toward a more trait-centered approach to diffuse (co) evolution. New Phytol 81–89
Sunagar K, Moran Y (2015) The rise and fall of an evolutionary innovation: contrasting strategies of venom evolution in ancient and young animals. PLoS Genet 11:e1005596
Sunagar K, Jackson TN, Undheim EA, Ali SA, Antunes A, Fry BG (2013) Three-fingered RAVERs: rapid accumulation of variations in exposed residues of snake venom toxins. Toxins 5:2172–2208
Suranse V, Srikanthan A, Sunagar K (2018) Animal venoms: origin, diversity and evolution. eLS 1–20
Tan HM, Low WY (2018) Rapid birth-death evolution and positive selection in detoxification-type glutathione s-transferases in mammals. PLoS ONE 13:e0209336
The UniProt Consortium (2023) Uniprot: the universal protein knowledgebase in 2023. Nucleic Acids Res 51:D523–D531
Thorne JL, Goldman N, Jones DT (1996) Combining protein evolution and secondary structure. Mol Biol Evol 13:666–673
Tumescheit C, Firth AE, Brown K (2022) CIAlign: a highly customisable command line tool to clean, interpret and visualise multiple sequence alignments. PeerJ 10:e12983
Undheim EA, Jones A, Clauser KR, Holland JW, Pineda SS, King GF, Fry BG (2014) Clawing through evolution: toxin diversification and convergence in the ancient lineage Chilopoda (Centipedes). Mol Biol Evol 31:2124–2148
Undheim EA, Fry BG, King GF (2015) Centipede venom: recent discoveries and current state of knowledge. Toxins 7:679–704
Vahtera V, Edgecombe GD, Giribet G (2013) Phylogenetics of scolopendromorph centipedes: can denser taxon sampling improve an artificial classification? Invertebr Syst 27:578–602
Van Valen L (1977) The red queen. Am Nat 111:809–810
Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A et al (2022) AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50:D439–D444
Vizcaíno JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T et al (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44:D447–D456
Vonk FJ, Casewell NR, Henkel CV, Heimberg AM, Jansen HJ, McCleary RJ, Kerkkamp HM, Vos RA, Guerreiro I, Calvete JJ et al (2013) The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc Natl Acad Sci 110:20651–20656
Waddington J, Rudkin DM, Dunlop JA (2015) A new mid-Silurian aquatic scorpion-one step closer to land? Biol Let 11:20140815
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wang J, Youkharibache P, Zhang D, Lanczycki CJ, Geer RC, Madej T, Phan L, Ward M, Lu S, Marchler GH et al (2020) iCn3D, a web-based 3D viewer for sharing 1D/2D/3D representations of biomolecular structures. Bioinformatics 36:131–135
Wang J, Youkharibache P, Marchler-Bauer A, Lanczycki C, Zhang D, Lu S, Madej T, Marchler GH, Cheng T, Chong LC et al (2022) iCn3D: from web-based 3D viewer to structural analysis tool in batch mode. Front Mol Biosci 9:831740
Ward MJ, Rokyta DR (2018) Venom-gland transcriptomics and venom proteomics of the giant Florida blue centipede, Scolopendra viridis. Toxicon 152:121–136
Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, Kriventseva EV, Zdobnov EM (2018) BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 35:543–548
Weinberger H, Moran Y, Gordon D, Turkov M, Kahn R, Gurevitz M (2010) Positions under positive selection-Key for selectivity and potency of scorpion \(\alpha\)-toxins. Mol Biol Evol 27:1025–1034
Wong ES, Belov K (2012) Venom evolution through gene duplications. Gene 496:1–7
Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S, Zhou X, Lam T-W, Li Y, Xu X, Wong GK-S, Wang J (2014) SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30:1660–1666
Xu D, Zhang Y (2009) Generating triangulated macromolecular surfaces by Euclidean distance transform. PLoS ONE 4:e8140
Xu N, Zhao H-Y, Yin Y, Shen S-S, Shan L-L, Chen C-X, Zhang Y-X, Gao J-F, Ji X (2017) Combined venomics, antivenomics and venom gland transcriptome analysis of the monocoled cobra (Naja kaouthia) from China. J Proteomics 159:19–31
Yin J, Zhang C, Mirarab S (2019) ASTRAL-MP: scaling ASTRAL to very large datasets using randomization and parallelization. Bioinformatics 35:3961–3969
Zancolli G, Casewell NR (2020) Venom systems as models for studying the origin and regulation of evolutionary novelties. Mol Biol Evol 37:2777–2790
Zhang J (2003) Evolution by gene duplication: an update. Trends Ecol Evol 18:292–298
Zhang J, Kobert K, Flouri T, Stamatakis A (2014) PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30:614–620
Zhang C, Rabiee M, Sayyari E, Mirarab S (2018) ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform 19:15–30
Acknowledgements
This work was supported by the National Science Foundation (NSF DEB 1638902) and the Florida State University Council on Research and Creativity. We would also thank the FSU Translation Sciences Core Facility, especially Cynthia Vied for help with transcriptome sequencing and Rakesh Singh for processing the mass spectrometry samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no Conflict of interest.
Additional information
Handling editor: Willie Swanson.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ellsworth, S.A., Rautsaw, R.M., Ward, M.J. et al. Selection Across the Three-Dimensional Structure of Venom Proteins from North American Scolopendromorph Centipedes. J Mol Evol 92, 505–524 (2024). https://doi.org/10.1007/s00239-024-10191-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-024-10191-y