
Abstract
In the present work, we carried out a comparative genomic analysis to trace the evolutionary trajectory of the bacterial species that make up the Liquorilactobacillus genus, from the identification of genes and speciation/adaptation mechanisms in their unique characteristics to the identification of the pattern grouping these species. We present phylogenetic relationships between Liquorilactobacillus and related taxa such as Bacillus, basal lactobacilli and Ligilactobacillus, highlighting evolutionary divergences and lifestyle transitions across different taxa. The species of this genus share a core genome of 1023 genes, distributed in all COGs, which made it possible to characterize it as Liquorilactobacillus sensu lato: few amino acid auxotrophy, low genes number for resistance to antibiotics and general and specific cellular reprogramming mechanisms for environmental responses. These species were divided into four clades, with diversity being enhanced mainly by the diversity of genes involved in sugar metabolism. Clade 1 presented lower (< 70%) average amino acid identity with the other clades, with exclusive or absent genes, and greater distance in the genome compared to clades 2, 3 and 4. The data pointed to an ancestor of clades 2, 3 and 4 as being the origin of the genus Ligilactobacillus, while the species of clade 1 being closer to the ancestral Bacillus. All these traits indicated that the species of clade 1 could be soon separated in a distinct genus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The bacterial taxa previously grouped under the Lactobacillus genus was composed of 250 species that were clustered together due their physiological similarities. However, recent genomic analyses led to the separation of this immense taxa to create 23 new genera, with the genus Lactobacillus kept exclusively to accommodate commercial probiotic and dairy-fermenting bacteria (Zheng et al. 2020). In this context, 27 species previously grouped in the cryptic Lactobacillus salivarius clade were further divided in two new genera: Liquorilactobacillus, represented by free-living species, and Ligilactobacillus, composed by host-associated species (Duar et al. 2017; Zheng et al. 2020).
The name Liquorilactobacillus was coined because its members were isolated from liquid sources such as alcoholic beverages and fermented foods. Currently, this genus is composed by Li. mali (type species), Li. aquaticus, Li. sicerae, Li. hordei, Li. cacaonum, Li. vini, Li. capillatus, Li. ghanensis, Li. nagelii, Li. oeni, Li. satsumensis, Li. sucicola and Li. uvarum (Zheng et al. 2020). The first species isolated was Li. mali in 1970 (Carr and Davies 1970) and the last identified species Li. sicerae was proposed by Puertas et al. (Puertas et al. 2014). All these species are rod-shaped Gram-positive, aerotolerant, homofermenters of lactic acid and producers of dextran from sucrose (Zheng et al. 2020). Their cells are typically motile, possessing one flagellum, except Li. cacaonum, Li. hordei and Li. Mali, which possess a flagellum but do not exhibit motility (Zheng et al. 2020). Cells of Li. satsumensis and Li. hordei, isolated from Kefir, were able to produce polysaccharides α-glucans with prebiotic and symbiotic qualities (Fels et al. 2018; Tan et al. 2022) and arabinogalactan, with in vitro antimicrobial properties against pathogenic bacteria such as Klebsiella pneumoniae and Pseudomonas aeruginosa (Zavistanaviciute et al. 2020). Recently, Yetimam and Ortakic (Yetiman and Ortakci 2023) reported for the first time a Liquorilactobacillus strain with probiotic potential namely Liquorilactobacillus nageli AGA58. All these works have revealed the biotechnological potential of this group of bacteria.
Some studies aimed to understand the metabolic traits of the species of the genus Liquorilactobacillus. For instance, the Li. vini is the only known lactic acid bacteria that can homoferment pentoses to lactic acid (Rodas et al. 2006). The species Li. vini was recognized in 2006 from the strain Mont4 isolated in the 1970s from wine in Spain (Rodas et al. 2006). Soon after, this species was identified as the predominant bacterium in the fermentation tanks in ethanol fuel distilleries in Sweden (Passoth et al. 2007), complemented by the study that also identified this bacterium in industrial process for bioethanol production in Brazil (Lucena et al. 2010). Recent studies revealed the role of citrate on the energetic metabolism of this bacteria (da Silva et al. 2019) and the tolerance mechanisms to ethanol production stressing agents, which can be extrapolated to other lactobacilli (Mendonça et al. 2020).
The genetic divergences within this group, as well as its evolutionary process, were proposed being induced by mobile genetic elements, such as bacteriophages, responsible for genomic plasticity based on gene gains and losses in the contribution to niches specific adaptation (Liu et al. 2023). However, the evolutionary forces that drove the evolution of Liquorilactobacillus to the liquid environment are still unknown. In this context, the present work aimed to deepen the analyses of the phylogenetic and evolutionary relationships between Liquorilactobacillus species, focusing on aspects that received limited or no exploration in the recent work by Liu et al. (Liu et al. 2023). For references, we employed the closest phylogenetic groups: Ligilactobacillus, Lacticaseibacillus and Latilactobacillus. These last two genera would represent species constituting the basal lactobacilli group, forming the base of the phylogenetic tree of the genus previously recognized as Lactobacillus (Duar et al. 2017; Zheng et al. 2020). In addition, the genomes of Bacillus species were added to the analysis as representative of the ancestral species of this evolutionary branch of bacteria. We also aggregated the newly sequenced genome of Li. sicerae described as the new component of the Liquorilactobacillus group. The results both supported recent information about the evolutionary history of this genus and uncovered contradictory findings, highlighting new aspects concerning the origin of its species, genomic evolution and the coherence of the genus itself. Finally, the results pointed for the possibility of separation of some species for the establishment of a new genus formed by a group of bacteria that kept similarities with the ancient Bacillus.
Material and Methods
Genomic Dataset and Gene Annotation
The genome sequences of nomenclatural type of species from the thirteen Liquorilactobacillus species available in the ENA (European Nucleotide Archive) and NCBI (National Center for Biotechnology Information) public databases, only complete assembled genomes and high coverage were selected. As related groups, fourteen genomes of Ligilactobacillus genus, three genomes of Latilactobacillus and 3 genomes of Lacticaseibacillus were included together with six genomes of Bacillus genus species as representative of the most reliable common ancestor (Makarova et al. 2006; Makarova and Koonin 2007). All these genomes were selected using the same criteria as above (supplementary material Table S1) and the gene annotation was performed by Bakta Web 1.9.1 (Schwengers et al. 2021) at http://bakta.computational using default settings.
Phylogeny
Phylogeny relationship among the 44 species (Baciilus, Latilactobacillus, Lacticaseibacillus, Liquorilactobacillus, Ligilactobacillus) was inferred with 16 s rDNA gene by Phylogeny Server Gene phylogenies and similarities into GGDC web server available at http://ggdc.dsmz.de/ using the DSMZ phylogenomics pipeline adapted to single genes (Meier-Kolthoff et al. 2022). A multiple sequence alignment was created with MUSCLE. Maximum likelihood (ML) and maximum parsimony (MP) trees were constructed from the sequence alignment with RAxML and TNT, respectively. For ML tree, it was employed the rapid bootstrapping in conjunction with the autoMRE bootstopping for topology-branch test. Regarding to MP, it was employed a topology-branch test bootstrapping with 1000 replicates. The sequences were checked for a compositional bias using the Χ2 test as implemented in PAUP. For ANI analyses by Edgar 3.0, a BLASTN comparison of the genome sequences was performed as described by Goris et al. (2007). For phylogenetic inference by reconciliation, 28 sequences of complete genomes of Liquorilactobacillus and Ligilactobacillus species and Lacticaseibacillus casei as outgroup were uploaded to the Type (Strain) Genome Server (TYGS), available under https://tygs.dsmz.de, for a whole-genome-based taxonomic analysis (Meier-Kolthoff and Göker 2019). For the phylogenomic inference, all pairwise comparisons among the set of genomes were conducted using GBDP (Genome BLAST Distance Phylogeny) under the algorithm 'coverage' and distance formula d5 (Meier-Kolthoff et al. 2013) and accurate intergenomic distances inferred under the algorithm ‘trimming’ and distance formula d5 (Camacho et al. 2009). Hundred distance replicates were calculated. Digital DDH values and confidence intervals were calculated using the recommended settings of the GGDC 4.0 (Camacho et al. 2009; Meier-Kolthoff et al. 2022). The resulting intergenomic distances were used to infer a balanced minimum evolution tree with branch support via FASTME 2.1.6.1 including SPR postprocessing (Lefort et al. 2015). Branch support was inferred from 100 pseudo-bootstrap replicates. The trees were rooted at the midpoint (Farris 1972). Two phylogenetic trees were generated, based on the complete genome and the 16S rDNA gene. Newick format trees were added in Notung 2.9 (Chen et al. 2000), a gene tree-species tree reconciliation software package that supports duplication-loss (DL) and duplication-transfer-loss (DTL) event models with a parsimony-based optimization criterion. Program default parameters were used. The final tree model was obtained from interactive tree of life (iTOL) v6 (Letunic and Bork 2024). The Timetree tool (http://timetree.org/) was used to indicate the divergency time of the Liquorilactobacillus among the species (Kumar et al. 2022).
Genome Cluster, Synteny and Gene Sharing
The decay function was predicted to basal lactobacilli, Ligilactobacillus and Liquorilactobacilus in the development of the core genome with increasing genome number (using a nonlinear least squares model fit). Calculation of pan genome, core genome identification, singletons genes, COG calculation and the analysis of average amino acid identity (AAI) were performed using the online Edgar 3.0 platform (Dieckmann et al. 2021) using the respective tools (https://edgar3.computational.bio.uni-giessen.de/cgi-bin/edgar.cgi). The pan, core genome and singletons genes were calculated using Li. mali as standard, the type strain of the species according to Zheng et al. (2020) using default settings. The functional category annotation (COG) for the complete genome was calculated using datasets from KEGG, COG and GO. Amino acid identity was calculated by blast hits from the orthologous genes present in the complete core of the selected genome according to their mean/median percent identity values. The Upsetplot was generated to show the genetic sharing of Liquorilacbacillus species. Statistical analysis was performed using the R software. Whole-genome multiple alignments were performed in Progressive Mauve software (Darling et al. 2004).
Phages and Resistance Antibiotic Genes
Phage presence was detected and annotated by PHIGARO 2.3.0 (Starikova et al. 2020) into Prokasee (https://proksee.ca/) using the complete genome as input. Open-reading frames are predicted from the input FASTA file using Prodigal. Phage genes are annotated with prokaryotic viral orthologous groups (pVOGs) profile, hidden Markov models (HMMs). Each contig is represented as a sequence of phage and non-phage genes. A smoothing window algorithm (a triangular window function) determines regions with a high density of phage genes and therefore the prophage regions and boundaries, considering the pVOG annotations and the GC content.
The FASTA file generated by the PHIGARO 2.3.0 program served as input for the synteny was drawn by Viptree 4.0: the viral proteomic tree server (https://www.genome.jp/viptree/). Viral genomic similarity was calculated from all-versus-all (SG) genomic similarity scores from tBLASTx results (Bhunchoth et al. 2016). The antibiotic resistance genes were identified from the Comprehensive Antibiotic Resistance Database (https://card.mcmaster.ca/) using the resistance gene identifier (RGI) in default parameters (McArthur et al. 2013). The presence of pathogenic islands was tested with the aid of the IslandViewer 4 software (Bertelli et al. 2017) in default parameters. IslandViewer integrates two sequence composition GI prediction methods, SIGI-HMM and IslandPath-DIMOB, and a single comparative GI prediction method, IslandPick (https://www.pathogenomics.sfu.ca/islandviewer/).
Carbohydrates, Glycogen and Amino Acids Metabolism and Genes with Biotechnological Potential
The assimilation of sugars by the 13 Liquorilactobacillus species was grouped based on the physiological data for API® test deposited at the BacDive-The Bacterial Diversity Metadatabase (https://bacdive.dsmz.de/) and by genes/pathways located in current database in the BioCyc Pathway/Genome Database Collection (https://biocyc.org/). The ByoCyc database was also used to analyse the amino acid biosynthetic pathways and glycogen metabolism, as well as to verify the genes with biotechnological potential. Metabolic pathways were confirmed in KEGG PATHWAY Database (Kanehisa 2000).
Alarmone Biosynthesis Genes and Proteins
The presence of alarmone genes/pathways were located in BioCyc Pathway/Genome Database Collection (https://biocyc.org/) and were confirmed in KEGG PATHWAY Database (Kanehisa 2000). The Hmmer (biosequence analysis using profile hidden Markov models) (https://www.ebi.ac.uk/Tools/hmmer/) was used to annotate protein motifs of RelA, SAS and SpoT proteins using query sequences (Potter et al. 2018).
Results
The Evolutionary Lineage of the Liquorilactobacillus: From Bacillus to Ligilactobacillus
The first analysis in this work was to establish the phylogenetic tree topology of the clade composed by species in the genera Liquorilactobacillus, Latilactobacillus, Laticaseibacillus and Ligilactobacillus, using Bacillus species for rooting (Fig. 1). Bacillus is widely accepted as the ancestral group of Lactobacillus sensu lato (Makarova et al. 2006; Makarova and Koonin 2007; Duar et al. 2017). The isolation source of each species of the test groups and the Bacillus was included in the tree, which points out the lifestyle adaptation and the transition from the ancestral free-living ecological niche to new environments along the speciation process, such as plant surfaces or vertebrate-host association (Fig. 1). The phylogenetic analysis showed that the group of basal lactobacilli sensu lato formed by the genera Latilactobacillus and Laticaseibacillus diverged from the ancestral group 1.8 billion years ago, while the genus Liquorilactobacillus diverged from the basal lactobacilli 398 million years ago (Fig. 1). Henceforth, the present analysis will use the term basal lactobacilli to define the set of bacterial species belonging to the Latilactobacillus and Laticaseibacillus that were in the basis of the phylogenetic tree (Fig. 1).

Maximum likelihood phylogenetic tree of Liquorilactobacillus, Ligilactobacillus, the basal lactobacilli (Lacticaseibacillus and Latilactobacillus) and their ancestral group Bacillus. Bootstrap percentages (1000 replicates) are represented by circles on the branches with the corresponding legend. Bar 0.1 represents nucleotide position substitutions. The evolutionary divergence time is shown below the phylogenetic tree and the dark dots represent the divergence time between two genera in millions of years. The isolation source materials of each species are represented as fields of green grass and grass, meat, milk carton, female genital tract, gastrointestinal tract, wine, cocoa, beer, tofu, tree, spring water, grapes, animals, fungi and oral cavity (Color figure online)
The segment of the tree that leads to Liquorilactobacillus was composed of two main branches, one that leads to the clade 1 (Li. vini, Li. sicerae, Li. nagelii and Li. ghanensis) and the second composed by clade 2 (Li. satsumensis and Li. oeni), clade 3 (Li. mali, Li. hordei and Li. cacaonum) and clade 4 (Li. aquaticus, Li. capillatus, Li. sucicola and Li. uvarum). The clade 1 diverged 117 million years ago from another three clades, while clade 3 and 4 seemed sister groups derived from clade 2. The position of each of the four Liquorilactobacillus clades in the phylogenetic tree (Fig. 1) was confirmed with average nucleotide identity (ANI) analysis (Supplementary Figure S1). Furthermore, the phylogenetic tree with reconciliation for Liquorilactobacillus, Ligilactobacillus and as an outgroup Lacticaseibacillus casei (Fig. 2) showed that the evolutionary events of speciation within these two genera were mainly due to the presence of mobile genetic elements and gene loss, mainly in Ligilactobacillus. No gene duplication events were observed (Fig. 2).

The ancestral states of COG genes were inferred on the parsimony tree using the parsimony ancestral state reconstruction method and the MK1 model (Markov 1 parameter) for species of Bacillus, Lacticaseibacillus, Latilactobacillus, Liquorilactobacillus, and Ligilactobacillus. The colors represent the number of genes in each COG, where red represents the largest number of genes and dark blue represents the smallest. Each COG was represented by a letter from A to Q, and the circles on the branches represent the variation in gene numbers (Color figure online)
The Carbohydrate-Driven Evolution
The genome evolution of these five bacterial genera (Liquorilactobacillus, Ligilactobacillus, Lacticaseibacillus Latilactobacillus and Bacillus) was investigated regarding the changes in the COG patterns (Fig. 3). The results indicated that genes of maintenance and related to the biological basic processes, such as cell cycle control, cell wall organization, DNA replication and gene expression, achieved lower level of variation in all five groups (Fig. 3). On the other hand, genes involved in the metabolism of carbohydrates and amino acids showed the highest level of variation in lactobacilli, but not in the Bacillus genomes (Fig. 3).

Clusters Orthologs Group (COG) showing the distribution of genes in different metabolic processes and mechanisms identified in the complete genome of Bacillus (a), basal lactobacilli (b), Liquorilactobacillus (c), and Ligillactobacillus (d)
The species of Bacillus used in this work showed the lowest level of COG variation regarding the metabolism of carbohydrates, which denoted its free-living style that requires a genetic diversity and functionality to cope the environment conditions fluctuations (Fig. 1). The divergency of the lactobacilli ancestor group from the Bacillus seemed to also relay on genomic reduction events, possibly driving the speciation process to a nutrient-rich environment.
The reconciliation tree indicated the loss of genes as an important evolutionary force acting in this group of bacteria (Fig. 2). The genomic reduction event in the divergency of lactobacilli ancestor from the bacilli can be observed when applying the method of linear genomic regression model (Fig. 4). This approach defines the number of gene shared by all genomes studied by excluding the unique gene in each genome. Therefore, the process of loss and gain of genes in the bacterial groups acted as a core genome retraction force and increased the steeping degree of the curve by the angular coefficient. This approach showed that basal lactobacilli shared 620 genes in the core genome with bacilli (angular coefficient of −x/1.115) (Fig. 4a), while the Liquorilactobacillus group shared 655 genes (angular coefficient of −x/2.421) (Fig. 4b) and Ligilactobacillus shared 494 genes (angular coefficient of −x/2.107) (Fig. 4c). Liquorilactobacillus shared more genes in the core genome with basal lactobacilli (715 genes angular and coefficient of −x/2.853) (Fig. 4d) than with its sibling genus Ligilactobacillus (627 genes and angular coefficient of −x/3.696) (Fig. 4f). Finally, Ligilactobacillus shared only 593 genes with basal lactobacilli (angular coefficient of −x/2.741) (Fig. 4e). These results suggest that Liquorilactobacillus retained more genes from the Bacillus ancestor in addition to the gene gain/loss related above during the Bacillus-to-Lactobacillus transition (Fig. 4). Noteworthy, the COG of transposase in the basal lactobacilli showed a variation expansion (Fig. 3b), indicating that the Bacillus-to-Lactobacillus passage might be related to the genomic plasticity conferred by these mobile elements. Interestingly, these elements were reduced in the genome of Liquorilactobacillus more than in the genome of Ligilactobacillus (Fig. 3c, d). Therefore, the transposons may have had a key role in shaping the genomic evolution of basal lactobacilli and Ligilactobacillus. Furthermore, there were gene losses in Ligilactobacillus, as shown in Fig. 2.

Linear regression model of genomic retraction analysis based on the core genome comparing the ancestral group of Bacillus with the basal lactobacilli (a), with the Liquorilactobacillus (b) and with the Ligilactobacillus (c), comparing the basal lactobacilli with Liquorilactobacillus (d) and Ligilactobacillus (e) and comparing Liquorilactobacillus with Ligilactobacillus (f)
A genomic shortening of the median number of genes was detected during the passage from Bacillus to Lactobacillus (Fig. 4). Meanwhile, there was an increasing in gene number variation within the clusters for carbohydrates assimilation and general metabolism in the lactobacilli sensu lato (Fig. 3), in agreement with the detected whole-genome reduction (Fig. 4). However, this reduction was not uniform among the species, which explain the high variation in genes as well as in the carbohydrate assimilation phenotype (Fig. 3). Therefore, this evolutionary phenomenon might be responsible for the trait of environmental specialization.
The distribution of minimal genome within the ontological categories was very similar between the different groups of bacteria when compared with the ancestral group of Bacillus. The genes involved in the processes like cell cycle control, transcription and translation were highly conserved and shared among the species of the four genera (Fig. 5). On the other hand, genes involved in the biosynthesis of secondary compounds, extracellular structures and encoding transposases were the least conserved, therefore the most diverse among species (Fig. 5). The first group of conserved genes must reflect the common evolutionary history of this entire set of species of Liquorilactobacillus, Latilactobacillus, Laticaseibacillus and Ligilactobacillus. Meanwhile, the transposase genes represented markers of divergence occurred during speciation events (Figs. 2 and 3).

Distribution of the genes of the minimum genome into different COG categories for basal lactobacilli (blue), Liquorilactobacillus (orange) and Ligilactobacillus (red) (Color figure online)
The Diversity Among the Liquorilactobacillus Species
The 13 species that compose the genus Liquorilactobacillus showed a core genome composed by 1023 genes (Fig. 6a). Moreover, the analyses revealed a set of unique genes in each species with the median of 177 unique genes ranging from 89 in Li. cacaonum to 328 unique genes in Li. sucicola (Fig. 6a). The most exclusive genes were classified for carbohydrate metabolism, transposases and transcription factors. This indicated that the variation in carbohydrate metabolism was an important mechanism in the evolution of these bacterial species (Fig. 6b). These core genome genes were further separated into ontology groups related to the metabolism of amino acids, vitamins, carbohydrates and lipids, to the processes of transcription and translation, to the mechanisms of gene regulation and to the stress response (Fig. 5b). The genes involved in the processes of translation, cell cycle control and cell motility were conserved in the core genome (Fig. 6b).

Gene diversity and core genome within the Liquorilactobacillus genus. a Number of genes shared and unique in each of the 13 species of the genus. b Number of genes that are part of the core genome, unique or accessory in different COG categories of the 13 species of the genus
Moreover, genes related to the stress response, like the usp genes (universal stress protein), and to oxidative (peroxidases) and alkaline stress responses (alkaline shock proteins), as well as those encoding the transcriptional regulators CtsR and TetR were present in all strains scrutinized (data not shown). This might indicate that stress response mechanism may function similar among the species of Liquorilactobacillus. These species also showed differences in genes that encode transcription sigma factors. All species presented the genes encoding the sigma factor 70 (rpoD) and the alternative sigma factors 54 (rpoN), 24 (rpoE) and 28 (fliA). Only Li. vini (clade 1) and Li. hordei (clade 3) harboured the gene encoding the sigma factor V (sigV) in their genomes, which are associated with lysozyme resistance in Bacillus (data not shown).
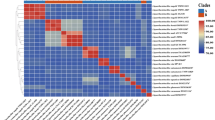
Further analysis involved the quantification of amino acid identity of the translated genomes. This approach retrieved the same tree topology of the phylogenetic clades described above (Fig. 1), with the Liquorilactobacillus being divided into four distinct groups (Fig. 7). However, the species of Liquorilactobacillus were separated into only two big clades when considering the average identity of 70% in the amino acids of the predicted proteome. Thus, the clade 1 remained separated as predicted by phylogenetic analysis (Fig. 1), while the species of other three clades coalesced into a large group, although remained independent (Supplementary Figure S2).

Average of amino acid identity (AAI) among the species of Liquorilactobacillus genus
Mobile Elements and Antibiotic Resistance Genes: Agents that Might Shape the Evolution of Liquorilactobacillus
The genomes of Liquorilactobacillus species were searched for the presence of intact phages (Fig. 8). The only bacteria that not have phage incorporated into their genome were the type strains Li. sicerae CECT 8227, Li. vini DSM 20605 and Li. nagelii DSM 13675. Therefore, these strains were replaced by their counterparts JP7.8.9 of Li. vini and ARG58 of Li. nagelii, to address the type of phages that are prone to infect these species.

Diversity of phages found in the Liquorilactobacillus genomes. The phages were aligned by synteny according to the clade separation indicated by the phylogenetic tree (Fig. 1)
Unfortunately, it was not possible with Li. sicerae because there is only one complete genome deposited so far. All the phages found in the representatives of this genus belonged to the Siphoviridae family, ranged from 9.9 to 44.5 kb and had a lysogenic trait. The synteny analysis revealed great diversity in the phages inserted in the bacterial chromosome, being unique to each species/strain (Fig. 8). On the other hand, no structures related to transposons and integrons were detected, despite the presence of transposase-coding genes (Fig. 3). Genetic markers commonly detected in mobile elements were hunted. Pathogenicity islands were not detected in any of the bacterial genomes surveyed. Only a few antibiotic resistance genes were found in the genomes of Liquorilactobacillus species (Table 1). Despite the phenotypical similarity of intrinsic resistance to vancomycin, the van operon was incomplete in all strains examined and the genes varied among the species (Table 1). The gene vanT for resistance to glycopeptides was absent in Li. sucicola, while the gene vanH was absent in Li. vini and Li. sicerae. In addition, mdeA encoding a transporter of efflux pumps of the ABC system was found only in Li. hordei and Li. capillatus (Table 1).
Metabolic Diversity of Liquorilactobacillus: Evolution in Action
According to the results above, the metabolism of carbohydrate showed the highest gene diversity among the COG groups and, therefore, appeared as the important source of niche adaptation and speciation (Fig. 9). The presence or absence of the carbohydrate assimilation genes coincided with the metabolic capacity of sugar assimilation checked in the BacDive database (Fig. 9). Li. satsumensis showed the most diverse genes encoding a repertoire of enzymes responsible for the transport and assimilation of different carbohydrates, while Li. cacaonum showed the lowest metabolic (Fig. 9).

Metabolism of sugars among the species of Liquorilactobacillus described in the BacDive database. a Main metabolic routes from the transport by PTS (indicated by the phosphorylated internal form) or by permease (in the case of galactose) to their conversion to glucose 6-phosphate in the central carbon metabolism identified by the presence of the respective genes in the bacterial genomes as described in Fig. 3. b Heat map of the sugar assimilation constructed from the physiological data available in the BacDive database. Shaded areas in red represent the ability of the species in metabolizing each sugar listed
All species had genes for the assimilation of glucose, sucrose, fructose and mannose, ten species had genes for the metabolism of trehalose and mannitol, nine species had genes for the assimilation of cellobiose and seven species had genes for the metabolism of galactose (Fig. 9). The genes for lactose assimilation were only found in the species Li. vini, Li. ghanensis and Li. sicerae that composed clade 1, but not in Li. nageli of this clade (data not shown).
In all species of Liquorilactobacillus, genes for two mechanisms of sucrose assimilation were found: (1) the PTS transport system, for the entry of sucrose in the form of sucrose-6-phosphate for internal hydrolysis, and (2) the dsr gene that encodes dextransucrase that act on synthesis of an extracellular polysaccharide that forms the matrix of biofilms (Fig. 9). An interesting fact is that the genes of the oxidative step of the pentose phosphate pathway were identified in all species of Liquorilactobacillus, but only Li. vini has the genes for both the oxidative and non-oxidative steps of this pathway (data not shown). In addition, none of the species showed genes for the assimilation of pentoses, except Li. vini that contained the gene araA encoding for arabinose isomerase for the assimilation of arabinose (data not shown).
The genes for synthesis and degradation of glycogen were detected only in the four species that composed clade 1 (Li. vini, Li. sicerae, Li. nagelii and Li. ghanensis), as well as in some species of Bacillus, Lacticaseibacillus and Ligilactobacillus (supplementary Figure S3).
As predicted by the results of Figs. 3 and 6, large genetic variation was also identified in the amino acid metabolism as one of the speciation mechanisms in the genus Liquorilactobacillus. In this context, the genomic analyses showed the presence of genes for almost all the biosynthetic pathways of amino acids. The lowest capacity of amino acid biosynthesis was observed for Li. oeni, while Li. ghanensis showed the highest biosynthetic capacity (Fig. 9). All species contained genes for the synthesis of glycine, L-arginine, L-alanine, L-cysteine, L-glutamate and L-histidine. However, the genes for biosynthesis of L-aspartate, L-methionine and L-phenylalanine were restricted to the genome of Li. sucicola (Fig. 10).

Biosynthesis of amino acids in the genus Liquorilactobacillus described in the Biocyc database. a Main metabolic routes for the conversion of the intermediates of the central carbon metabolism to amino acids. The arrows are presenting the direction of the schematic conventional metabolic routes. b Heat map of the sugar assimilation described in the BacDive database. Shaded areas in blue represent the ability of the species in metabolizing each amino acid listed
Species representing clade 1 lost the ability to biosynthesize L-isoleucine (Fig. 9), indicating evolutionary divergence between this clade and the other three clades of the genus. For the remaining amino acids, the biosynthetic capacity identified from the genetic information varied among the species within the clades (Fig. 10), indicating the species-specific differences due to adaptation to the difference ecological niches of these bacteria.
In the context of these minimal genome analysis and genetic divergences related to carbon and nitrogen metabolism, it was possible to identify the metabolic pathways present in the minimal genome of Liquorilactobacillus and which should reflect the evolutionary basis of this group of bacteria such as the lactate fermentation pathway, the synthesis of eight amino acids and the degradation of three of them, lipid and vitamin biosynthesis, stress response mechanisms and a number of transcriptional regulators (Supplementary Figure S4). These seemed the minimal biochemical requirement of these bacteria to live.
The genes responsible for proteins involved in the regulation of the central carbon and nitrogen metabolisms were also investigated from the synthesis/hydrolysis of the intracellular signalling molecule called alarmone, which regulates the stringent response. It represents a hyperphosphorylated form of the guanosine phosphate derivatives GDP and GTP (Fig. 11a). Genes of two types of proteins RelA and Small Alarmone Synthase (SAS) involved in the alarmone metabolism were mapped (Fig. 10b). The structural analysis of RelA proteins revealed the presence of five functional domains. The hydrolysis (HD) and synthesis domains were identified in the N-terminal region of the proteins, with their respective phosphorylation sites.

Synthesis and degradation of the alarmone molecules ppGpp and pppGpp in Gram-positive bacteria. a Metabolic interconversion of phosphorylated guanosines using ATP as phosphate donor. b Presence or absence of proteins responsible for the synthesis and/or hydrolysis of alarmone in the 4 clades of Liquorilactobacillus, comparing to Bacillus, basal lactobacilli and Ligilactobacillus. c Molecular structures of the enzymes involved in the metabolism of alarmone in different bacteria
In the C-terminal region, the threonyl-tRNA synthase/GTPase/SpoT (TGS) domain, the alpha-helix/conserved cysteine (AH-Ris) domain and the aspartokinase (ACT) domain were recognized (Fig. 11c). In the two SAS proteins, only the alarmone synthesis domain was identified (Fig. 11b). Sequences orthologous to the yjbM and ywaC genes encoding the two types of SAS were maintained from Bacillus to Ligilactobacillus, except in the species of clade 1 of Liquorilactobacillus (Li. vini, Li. sicerae, Li. nagelii and Li. ghanensis) (Fig. 11b). Therefore, RelA is the only protein encoded in the genome of clade 1 species involved in the synthesis/hydrolysis of the alarmone.
Finally, genes were found in the genomes of Liquorilactobacillus species whose proteins may confer some biotechnological potential (Table 2), such as: (1) cellobiase genes, whose encoded protein participates with other cellulose hydrolases in the complete degradation of lignocellulosic material and production of second-generation ethanol, (2) dextran polymer biosynthesis genes useful in the pharmaceutical industry, (3) genes encoding bacteriocins and lactose degraders of interest to the health and food industry, and (4) genes encoding peroxidases useful for the biodegradation of synthetic dyes.
Discussion
A Brief Evolutionary History
The evolution of lactobacilli on earth began 1.8 billion years ago, emerging from a free-living ancestral Bacillus (Makarova et al. 2006; Makarova and Koonin 2007). Hence, the basal lactobacilli represented by the genera Latilactobacillus and Lacticaseibacillus (Fig. 1) appeared before the first eukaryote evolved (Duar et al. 2017). The emergence and diversification of the multicellular eukaryotes created new environments for the lactic acid bacteria to adapt and specialize, like its external (skin of animals and surface of plants) and internal (mucosae) environments. These new ecological niches were exploited by several species from the previously known Lactobacillus salivarius clade, which was composed not only by free-living species but also by vertebrated-associated members (Duar et al. 2017). Recently, Zheng et al. (2020) proposed that this clade should be divided in two genera, Liquorilactobacillus and Ligilactobacillus.
Species of Liquorilactobacillus seemed derived from wide-spread free-living ancestral lactobacilli to a slightly specialized living stiles in aquatic environments, mainly related to human activities of fermentation. On the other hand, a group of plant-associated species, including some ancestral free-living lactobacilli and some representative of liquid-living Liquorilactobacillus, reached the mucosae environment of vertebrates upon the advent of herbivory. Thus, these bacteria turned specialized to compose the set of species in Duar et al. (2017). This proposed evolutionary history was somehow supported in the present work by the phylogenetic analysis that placed the Liquorilactobacillus as ancestral of the Ligilactobacillus (Fig. 1).
Clusterization of Liquorilactobacillus species using the maximum likelihood/maximum parsimony method separated the recognized 13 species of the genus in four clades: clade 1 composed by Li. vini, Li. sicerae, Li. nagelii and Li. ghanensis; clade 2 composed by Li. satsumensis and Li. oeni; clade 3 composed by Li. mali, Li. hordei and Li. cacaonum; and clade 4 composed by Li. aquaticus, Li. capillatus, Li. sucicola and Li. uvarum. According to the tree topology, the clades 2, 3 and 4 derivate from the same branch and share with the Ligilactobacillus a common ancestral, that is not the case for clade 1 that diverged earlier. Thus, the clade 1 would be in a plesiomorphic position in the Ligi-Liquorilactobacillus phylogenetic branch. Together with several evolutionary aspects and its basal position in the group, the clade 1 should assume as the plesiomorphic of the group.
A recent published phylogenetic analysis of the Liquorilactobacillus proposed a different subdivision of this genus taking in account the motility trait (Liu et al. 2023). In that work, the species were separated in the clade A based on the motility phenotype and the presence of motility genes, which includes what was defined herein as clades 1, 2 and 3. Thus, clade B was defined by the absence of these biological markers and included the species of the clade 4 of the present work. Noteworthy, the topology of the phylogenetic tree in that work indicated that the species of the clade 1 were positioned in their own branch separated from the remaining species, clustering in a second and main branch of the tree, like in our tree. Nevertheless, the primitivity of the clade 1 cannot be address in that analysis because of the lack of ancestral genomes to rooting, as we did herein with Bacillus and basal lactobacilli. In addition, the clusters A and B proposed do not resemble the defined tree topology, since the cluster A assembled species of two separated branches and the cluster B includes only part of the species from the main branch. It indicates that the motility trait is not a reliable phylogenetic feature. Moreover, when considering this trait alone, it creates a paraphyletic phylogeny and artificial groups. Since motility is a common feature in free-living bacteria, most likely its genetic information was lost in some species during the speciation. It remains unclear if the motile capability was lost only in the clade 4 or lost and gained several of time among the other Liquorilactobacillus species as consequence of gene exchange produced by, for example, genetic mobile elements. In the present work, we can also separate the species in two distinct groups based on the minimum AAI identity of less than 70% and other unique characteristics that will be discussed below. In this division, the clade 1 would stay alone while the clades 2, 3 and 4 form a large group of species.
The Evolution by Genomic Decay: Contraction Towards the Minimal Genomes and Repercussion in COG Composition
In the course of the evolutionary history, it was clear that the species faced a genome retraction mostly related to niche specialization (Figs. 2, 3 and 4). Two evolutionary mechanisms related to genomic reduction involve: 1) horizontal gene transfer by the effects of mobile elements (to be discussed below) and 2) GC content decreasing (Mahajan and Agashe 2022). Our analysis pointed to a drastic reduction in the genome size from the ancestral Bacillus group to basal lactobacilli, and then to Liquorilactobacillus and Ligilactobacillus (Fig. 4). This genomic reshaping affected unevenly each one of the COGs, possibly due to unequally selective pressure to enhance the fitness during the adaptation to new habitats, which overall led to the core genome decay observed (Figs. 3 and 4). The species of Liquorilactobacillus shared more genes with Bacillus than with basal lactobacilli, possibly as a consequence of the maintenance of the generalist lifestyle.
Unlike the motility genetic information, a group of genes were highly conserved among the Liquorilactobacillus, which constituted the minimum genome of the genus. This is composed by 1023 genes (Fig. 6a) distributed in all recognized COGs (Fig. 3). The smallest genome of the genus was identified for Li. cacaonum with 1,893 genes. This species also showed the smallest number of both unique genes (Fig. 6a) and carbohydrate-associated genes (Fig. 9). About one-third of the minimum genome in Liquorilactobacillus, around 380 genes, is related to metabolic activities related to genetic information (replication, transcription and translation), the last two-third is involved with other 19 cellular processes for cell structuring and functioning (Fig. 6). Despite the large portion of the Liquorilactobacillus genome being found in the minimum genome, in median two-third of the gene content are more variable. This suggests that the action of gene gain and loss mechanisms influenced genome evolution as shown in Figs. 2 and 4. Since it diverged from the basal lactobacilli about 398 million of years ago (Fig. 1), and this process somehow involved the loss of transposase genes (Fig. 3); hence, their genomes might be experienced stability and retained more of the Bacillus ancestor genes than the other species of lactobacilli. On the other hand, the species Ligilactobacillus faced a more pronounced decay of the core genome in relation to Bacillus (Fig. 4), accompanied by greater variation in the COG of amino acids (Figs. 3, 4 and 5) in order to adapt to the host-associated lifestyle. Interestingly, COG analysis revealed increasing variation of transposase cluster in Ligilactobacillus genomes, which could be associated with a second phase of genetic instability during this new genomic reshaping speciation. This genomic instability might also affect the capability of the genome to retain the remain ancestral bacilli genes with the outcome the observed core genome decay related to Bacillus genomes in this group.
Genetic Mobile Elements as Managers of Evolutionary Mechanisms
It is well established that in bacterial evolution and speciation, genome reduction and horizontal gene transfer (HGT) played pivotal role in reshaping the genomic information (Haudiquet et al. 2022). Our results indicated the mobile genetic elements such as phages (Fig. 7), together with the presence of transposase-encoding genes in their genomes (Fig. 3), could modulate the genetic content in the Liquorilactobacillus species and mobile genetic elements were important for the evolution of the genus as shown in Fig. 2. HGT mechanisms must have played a large role in the acquisition of genes that became unique or that were shared between few species of the genus (Fig. 5). Promiscuous genetic elements turn feasible the genetic exchange among related species or even species from different genus. An example of this was shown in the work by Mendonça et al. (2016) who reported the transfer of the tn916 transposon-associated tetM gene from the industrial Li. vini to the nosocomial Enterococcus faecalis. Similar events could occur several of times along the species evolution and led not only to gain of genes but also to genes inactivation. However, spatial proximity of the cells involved in those events is critical and allows that species which close niche in the environment shared more genes beyond those found in the minimum genome. In this sense, Popa and Dagan (2011) proposed that the ecology appears to have been the main determinant of gene sharing and genome reshaping along evolution, suggesting that gene transfer occurs to some extent limited by ecological opportunity and occupation of shared habitats.
Phages can also act as an evolutive pressure in the environment, mobilizing genes through transfection and eliminating sensitive competitors in the community. As expected, were identified temperate phages that belong to the Siphoviridae family (Fig. 8) in the analysed genomes. These phages can assist in genetic diversity among these species, such as the production of virulence factors as well as bacteriocins in Li. hordei (Rouse et al. 2008) and in Li. nagelii (Yetiman and Ortakci 2023). The phages identified in the genome of Liquorilactobacillus species have an average size of 30 kb, which gives them the capacity to carry genes of different origins and functions. According to Liu et al. (2023), the species of clade A have two phages per genome, while those of clade B would have up to four phages per genome. However, we only found one complete phage per genome in most of the species, with the exception of Li. mali and Li. hordei which have more than one copy of phage per genome. It may be related to strain variation as function of the habitat of isolation. For example, the bioethanol strain JP789 of Li. vini harbours a phage that is absent in its counterpart wine strain DSM 20605. So, together with HGT events, the phage of the Liquorilactobacillus could also be responsible for both the removal of genes, causing the genomic reduction verified in Fig. 4, and the introduction of genes that became exclusive to one or a few species of the genus. This hypothesis was first proposed by Liu et al. (2023). In the present work, we are showing the consequences of these mobile elements in the reduction/reshaping of bacterial genomes.
Metabolic Adaptation as Guide to the Evolutionary Mechanisms
Unlike the transposase cluster in all lactobacilli genera, the highest variation was observed for the of genes in the COG of carbon assimilation, suggesting the influence of the modulation of sugar assimilation in the speciation process of this group of bacteria (Figs. 3, 5 and 9). There seems to be a profusion of events of gain/losses of these genes, possibly induced by HGT and/or mobile genetic elements, with the introduction of unique or little shared genes (Fig. 6). Thus, we propose that sugar metabolism was the main selective pressure of the evolutionary force both to expand the ecological niche, through the metabolization of new resources, and to speciate and specialize to new environments. The definitive settlement to these niches might have been defined by the diversity in the nitrogen metabolism. Interestingly, the genes encoding the enzymes for sucrose, and its monosaccharides, assimilation, the biosynthesis of six (glycine, L-arginine, L-alanine, L-cysteine, L-glutamate and L-histidine) out of 20 amino acids and the genes of basal transcription factors that regulate cell cycle and growth are also conserved. Free-living and plant-associated bacteria, such as of the genus Liquorilactobacillus, evolved with metabolic flexibility for most carbohydrates, such as sucrose, lactose, mannitol, melizitose and cellobiose (Zheng et al. 2015) (Fig. 9). However, it is striking that these bacteria are unable to assimilate pentoses, such as xylose and arabinose, which are abundantly released in the environment by the degradation of decaying plant biomass. The exception is Li. vini with the ability to assimilate and ferment arabinose (Rodas et al. 2006) because of the presence of araA gene in its genome (de Lucena et al. 2012). The fact that the species of this genus have several auxotrophy for amino acids also reveals that these species evolved to live in environments with availability of this nutrient (Fig. 10). This seems to be more pronounced for Ligilactobacillus species, whose genome modifications resulted in the loss of function of carbohydrate and amino acid metabolism genes (Figs. 9 and 10). This kind of evolutive transition narrows the carbohydrate flexibility to align with those found in the diet of the vertebrated host (Makarova et al. 2006; Makarova and Koonin 2007; Sun et al. 2015; Zheng et al. 2020).
Regarding to the nutritional capability, the species of Liquorilactobacillus clade 1 showed some singular features like the genes for glycogen biosynthesis in all species of the clade (Supplementary Figure S3). Altogether, the data suggest that this variability of the gene in COGs related to carbon sources and nitrogen is the outcome of some evolutionary pressure by nutritional availability (Fig. 3).
In line to this carbon- and nitrogen-driven adaptation, the species may fit their metabolism by mechanisms for the regulation of central metabolism, energy metabolism and stress response. An important player in this context is the molecule called alarmone, which is a tetra (ppGpp) or penta (pppGpp) phosphorylated guanidine, involved in the regulatory pathway known as stringent response conserved from Gram-positive and Gram-negative species (Baquero et al. 2021). The alarmone is synthesized from GDP by ATP-dependent Small Alarmone Syntethase (SAS) enzymes (Krishnan and Chatterji 2020). These proteins are classified into 30 families in the different bacterial groups that synthesize ppGpp, with some representatives also synthesizing pppGpp (Gaca et al. 2015). The second type of alarmone is synthesized from GTP by an ATP-dependent pyrophosphosynthase encoded by the relA gene (Atkinson et al. 2011). Unlike SAS proteins, the RelA protein is considered bifunctional because it also performs the pyrophosphohydrolase reaction, working on the balance between synthesis and hydrolysis of alarmone during cell growth (Atkinson et al. 2011) (Fig. 10). RelA is a universal protein, present in almost all types of bacteria, with the exception of obligate intracellular species (Baquero et al. 2021). On the other hand, SAS proteins have variable presence in different bacterial species, with the gene even being duplicated in the bacterial genome, such as yjbM and ywaC in B. subtillis (Nanamiya et al. 2008). Regarding the Gram-negative bacteria, there is the spoT gene that also encodes a bifunctional enzyme like RelA. However, in these bacteria, RelA would have a greater tendency for the synthesis of pppGpp while SpoT would preferentially act on hydrolysis (Atkinson et al. 2011). So far, the accepted theory preconizes that relA and spoT genes evolved from the duplication of an ancestral rel gene found in many groups of bacteria (Mittenhuber 2001). In the present work, the mapping of protein motifs indicated that the yjbM and ywaC genes of Bacillus must have been the result of amplification of spoT or relA, followed by deletion at both ends, with the core structure maintaining the catalytic site for the preferential synthesis of ppGpp (Fig. 11c). In lactobacilli, SAS proteins must have specialized for the production of ppGpp, while RelA specialized in the production of pppGpp and also in the hydrolysis of these two molecules (Fig. 11c). This diversification was observed in the evolutive line of basal lactobacilli to Ligilactobacillus. Once again, the species of Liquorilactobacillus clade 1 differently evolved to present only relA gene involved in the alarmone metabolism.
Gene Expression Regulation and Stress Response
In addition to the effects of gene gain and loss events, the adaptation of the species of the genus Liquorilactobacillus took place in the sense of maintaining regulatory elements of the transcription of basal genes and genes related to environmental stress response (Abram et al. 2021). Sigma factors are essential for bacterial gene regulation and alternative sigma factors provide the main line of cellular response, effectively reprogramming the transcription of specific gene sets (Abram et al. 2021). In the genomes of the 13 species of Liquorilactobacillus, genes were identified for the basal sigma 70 (σ70) factor and for the alternative sigma V (σV), 24 (σ24), 28 (σ28) and 54 (σ54) factors. The σ70 factor is related to the transcription of essential genes and cell maintenance and is present in all bacteria (Paget and Helmann 2003). The alternative factors act in a more specific way and their presence is variable in bacterial species. In this study, we identified the genes for σ54 and σ24 factors that are present in all species of Liquorilactobacillus and Ligilactobacillus (Supplementary Figure S4). The first factor acts in the transcription of many different and unrelated genes, ranging from flagellum synthesis to the use of various different sources of carbon and nitrogen (Danson et al. 2019). The genome of all species of these genera contains genes encoding the σ24 factor, the transcription factor HrcA (heat-inducible transcription repressor) and the chaperonins GroESL (Supplementary Figure S4) that are involved in the heat stress response (Rouvière et al. 1995; Hakiem et al. 2020; Stan et al. 2022). The factor σ28 is present in all species of Liquorilactobacillus that have flagellum, being responsible for the regulation of the genes involved in the flagellar structure (Liu et al. 2023). Our results confirmed this finding and complement the information by identifying the σ28 gene in the species of Ligilactobacillus that present flagellum. On the other hand, the σV factor was found only in Li. vini and Li. hordei (Supplementary Figure S3). This transcription factor is responsible for the expression of Bacillus genes involved in the resistance of the cell wall to lysozyme found in tears, saliva and sweat of vertebrate animals and also in plants (Wang et al. 2005). No gene encoding σ32 factor was detected in the genome of Liquorilactobacillus. This factor is one of the proteins responsible for heat shock tolerance (Gourse 2016).
Moreover, the genes of universal stress protein (USP) as well as genes encoding the mechanisms of oxidative and alkaline stress response and the transcriptional regulators ctsR, TetR, HrcA and genes encoding some alternative sigma factors were shared by all members of the genus. The conservation of the genes of the stress response suggests that even without a general stress response known, at least in some level, the Liquorilactobacillus shares some regulatory and responsive mechanisms to handle hazard environmental conditions which had critical role in adaptation and evolution of the group. Therefore, the recent reports of Li. vini stress response involving some of these genes like, uspI to uspV, alternative sigma factor and transcriptional regulators could be employed to understand the adaptation of others Liquorilactobacillus to environment (Mendonça et al. 2019, 2020). All species of Liquorilactobacillus have the five usp genes identified in Li. vini (Supplementary Figure S4). In this species, the uspII gene is the most responsive to different forms of stress and its expression is correlated with the expression of the furR gene (Mendonça et al. 2020). Noteworthy the uspII is the only known gene that behaves like a general stress response gene and acts like the E. coli uspA gene that is part of the general stress response in Gram-negative bacteria. furR encodes one of the transcription factors of the iron uptake regulator family, whose members are also involved in the regulation of several other cellular processes (Troxell and Hassan 2013). The other usp genes seem to play a more stress-specific role, like the uspIII that was up-regulated in Li. vini cells exposed to several stressors, but osmotic and oxidative, with a similar expression pattern with rpoB gene that encodes RNA polymerase beta-subunit (Mendonça et al. 2020). Additionally, the expression of uspV was correlated with the expression of the rpoN gene that encodes σ54 (Mendonça et al. 2020). The genes uspI and uspIV were very responsive to osmotic stress and co-expressed with PspC transcriptional regulator (Mendonça et al. 2020). PspC is the major regulator of the psp regulon involved in the response to cell envelope stress in E. coli, although its function in lactic acid bacteria is still unknown (Bury-Moné et al. 2009). The present analyses revealed that all species of Liquorilactobacillus, as well as Ligilactobacillus and Lacticaseibacillus, have the furR and pspC genes in their genomes together with all five usp genes (data not shown). This indicates that these regulatory modules could represent a common and basal stress response mechanism in these bacteria. Thus, the results on stress response in Li vini reported by Mendonça et al. (Mendonça et al. 2019, 2020) could be considered a model for these groups of bacteria.
Biotechnological Potential of Liquorilactobacillus
Genes with biotechnological potential were found in the genomes of Liquorilactobacillus (Table 2) and the dextransucrase gene for the synthesis of EPS was located in all strains analysed. Many Liquorilactobacillus are known for producing exopolysaccharides (EPS) which have great potential in the food industry. As examples, we can cite Liquorilcatobacillus sicerae CUPV237 that produces a heteropolysaccharide that contained glucose, galactose and rhamnose (Puertas et al. 2023). Liquorilactobacillus satsumensis from water kefir that yields α-glucan polysaccharides with prebiotic and synbiotic qualities (Tan et al. 2022). Already Liquorilactobacillus mali strains, isolated from Algerian food products, are producers of the postbiotic compounds dextran, oligosaccharides and mannitol (Zarour et al. 2024).
Clade 1: Is the Time for Independence Coming?
Even though all stages of speciation of the species studied here are still unknown, it is a fact that Liquorilactobacillus have at least two distinct groups within the genus separated with a minimum AAI lower than 70%. The first group herein classified as clade 1 showed the basic characteristics to constitute a phylogenetic independent branch since their constituting species have a common taxonomic, genetic and ecological history as well as the isolation from other populations in time and space (Van Noordwijk 1988). Their fours species (Li. vini, Li. sicerae, Li. nagelii and Li. ghanensis) are separated by 141 million years from the other nine species of the genus and have unique genetic and metabolic characteristics that distinguish them from the other nine species of the genus. For example, these species are the only with the capacity to synthesize and degrade glycogen and to assimilate lactose. Also distinct from species from the other three clades, these species collectively present the complete pentose phosphate pathway and have lost the ability to biosynthesize L-isoleucine. In its singular evolutionary trajectory, the species of clade 1 have lost the genes encoding SAS proteins, keeping only the relA gene for a bifunctional RelA protein for the synthesis and degradation of alarmone. All these modifications took part during the evolution of this group the resulted in four species that present less than 70% of similarity in the AAI with the other species of the genus. Therefore, the present work reveals that evolution tends to separate them as a new genus positioned between basal lactobacilli and the genus Liquorilactobacillus. To support this hypothesis, it is necessary to aggregate additional information based on other bioanalytical tools, such as the analysis of Conserved Signature Indels (CSI) that was robustly used by Gupta et al. (2020) in the reorganization of the genus Bacillus and definition of novel genera and species within this large heterogeneous group.
Conclusions
The phylogenetic analysis produced in this work has enriched the understanding of the evolutionary history of the genus Liquorilactobacillus. After the general definitions of what characterizes a Liquorilactobacillus, carried out in this study, it will be possible to understand the variations of the biological types and isolates of the genus not yet known. The applied evolutionary pluralism allowed us to discuss different processes that show the changes in gene and genotypic frequencies unifying the biology of the genus. Some of the variations are more highlighted by chance (phages and mobile genetic elements) and fixed by the action of natural selection (use of carbohydrates and amino acids, and the stress response). In addition, gene flow may result in the great diversity of the genus, and this diversity might very soon separate them into distinct two groups within Liquorilactobacillus, one of them potentially representing a new genus. This shows that the taxonomy of the group is not yet completed. Everything depends on how time and evolutionary factors will shape these species.
References
Abram F, Arcari T, Guerreiro D, O’Byrne CP (2021) Evolutionary trade-offs between growth and survival: the delicate balance between reproductive success and longevity in bacteria. pp 133–162
Atkinson GC, Tenson T, Hauryliuk V (2011) The RelA/SpoT homolog (RSH) superfamily: distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLoS ONE 6:e23479. https://doi.org/10.1371/journal.pone.0023479
Baquero F, Coque TM, Galán JC, Martinez JL (2021) The origin of niches and species in the bacterial world. Front Microbiol. https://doi.org/10.3389/fmicb.2021.657986
Bertelli C, Laird MR, Williams KP et al (2017) IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res 45:W30–W35. https://doi.org/10.1093/nar/gkx343
Bhunchoth A, Blanc-Mathieu R, Mihara T et al (2016) Two asian jumbo phages, ϕRSL2 and ϕRSF1, infect Ralstonia solanacearum and show common features of ϕKZ-related phages. Virology 494:56–66. https://doi.org/10.1016/j.virol.2016.03.028
Bury-Moné S, Nomane Y, Reymond N et al (2009) Global analysis of extracytoplasmic stress signaling in Escherichia coli. PLoS Genet 5:e1000651. https://doi.org/10.1371/journal.pgen.1000651
Camacho C, Coulouris G, Avagyan V et al (2009) BLAST+: architecture and applications. BMC Bioinform 10:421. https://doi.org/10.1186/1471-2105-10-421
Carr JG, Davies PA (1970) Homofermentative Lactobacilli of Ciders including Lactobacillus mali nov. spec. J Appl Bacteriol 33:768–774. https://doi.org/10.1111/j.1365-2672.1970.tb02261.x
Chen K, Durand D, Farach-Colton M (2000) NOTUNG: a program for dating gene duplications and optimizing gene family trees. J Comput Biol 7:429–447. https://doi.org/10.1089/106652700750050871
da Silva PKNPKN, Mendonça AAAA, De Miranda ARAR et al (2019) Nutritional requirements for Lactobacillus vini growth in sugarcane derivative substrate of ethanol fermentation. FEMS Microbiol Lett 366:1–9. https://doi.org/10.1093/femsle/fnz202
Danson AE, Jovanovic M, Buck M, Zhang X (2019) Mechanisms of σ54-dependent transcription initiation and regulation. J Mol Biol 431:3960–3974. https://doi.org/10.1016/j.jmb.2019.04.022
Darling ACE, Mau B, Blattner FR, Perna NT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. https://doi.org/10.1101/gr.2289704
de Lucena BTL, Silva GGZ, Manoel Dos Santos B et al (2012) Genome sequences of the ethanol-tolerant Lactobacillus vini strains LMG 23202T and JP7.8.9. J Bacteriol 194:3018. https://doi.org/10.1128/JB.00446-12
Dieckmann MA, Beyvers S, Nkouamedjo-Fankep RC et al (2021) EDGAR3.0: comparative genomics and phylogenomics on a scalable infrastructure. Nucleic Acids Res 49:W185–W192. https://doi.org/10.1093/nar/gkab341
Duar RM, Lin XB, Zheng J et al (2017) Lifestyles in transition: evolution and natural history of the genus Lactobacillus. FEMS Microbiol Rev 41:S27–S48. https://doi.org/10.1093/femsre/fux030
Farris JS (1972) Estimating phylogenetic trees from distance matrices. Am Nat 106:645–668. https://doi.org/10.1086/282802
Fels L, Jakob F, Vogel RF, Wefers D (2018) Structural characterization of the exopolysaccharides from water kefir. Carbohydr Polym 189:296–303. https://doi.org/10.1016/j.carbpol.2018.02.037
Gaca AO, Kudrin P, Colomer-Winter C et al (2015) From (p)ppGpp to (pp)pGpp: characterization of regulatory effects of pGpp synthesized by the small alarmone synthetase of enterococcus faecalis. J Bacteriol 197:2908–2919. https://doi.org/10.1128/JB.00324-15
Goris T, Schubert T, Gadkari J et al (2014) Insights into organohalide respiration and the versatile catabolism of S ulfurospirillum multivorans gained from comparative genomics and physiological studies. Environ Microbiol 16:3562–3580. https://doi.org/10.1111/1462-2920.12589
Gourse RL (2016) Classic spotlight: the heat shock response and the discovery of alternative sigma factors in Escherichia coli. J Bacteriol 198:2550–2550. https://doi.org/10.1128/JB.00576-16
Gupta RS, Patel S, Saini N, Chen S (2020) Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int J Syst Evol Microbiol 70:5753–5798. https://doi.org/10.1099/ijsem.0.004475
Hakiem OR, Parijat P, Tripathi P, Batra JK (2020) Mechanism of HrcA function in heat shock regulation in Mycobacterium tuberculosis. Biochimie 168:285–296. https://doi.org/10.1016/j.biochi.2019.11.012
Haudiquet M, de Sousa JM, Touchon M, Rocha EPC (2022) Selfish, promiscuous and sometimes useful: how mobile genetic elements drive horizontal gene transfer in microbial populations. Philos Trans R Soc B Biol Sci. https://doi.org/10.1098/rstb.2021.0234
Kanehisa M (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. https://doi.org/10.1093/nar/28.1.27
Krishnan S, Chatterji D (2020) Pleiotropic effects of bacterial small alarmone synthetases: underscoring the dual-domain small alarmone synthetases in mycobacterium smegmatis. Front Microbiol. https://doi.org/10.3389/fmicb.2020.594024
Kumar S, Suleski M, Craig JM et al (2022) TimeTree 5: an expanded resource for species divergence times. Mol Biol Evol. https://doi.org/10.1093/molbev/msac174
Lefort V, Desper R, Gascuel O (2015) FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program: Table 1. Mol Biol Evol 32:2798–2800. https://doi.org/10.1093/molbev/msv150
Letunic I, Bork P (2024) Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. https://doi.org/10.1093/nar/gkae268
Liu X, Li W, Sun Z et al (2023) Phylogenomics of the Liquorilactobacillus Genus. Curr Microbiol 80:274. https://doi.org/10.1007/s00284-023-03336-7
Lucena BT, Dos Santos BM, Moreira JLS et al (2010) Diversity of lactic acid bacteria of the bioethanol process. BMC Microbiol 10:298. https://doi.org/10.1186/1471-2180-10-298
Mahajan S, Agashe D (2022) Evolutionary jumps in bacterial GC content. G3 Genes|Genomes|Genetics. https://doi.org/10.1093/g3journal/jkac108
Makarova K, Slesarev A, Wolf Y et al (2006) Comparative genomics of the lactic acid bacteria. PNAS 103:15611–15616
Makarova KS, Koonin EV (2007) Evolutionary genomics of lactic acid bacteria. J Bacteriol 189:1199–1208. https://doi.org/10.1128/JB.01351-06
McArthur AG, Waglechner N, Nizam F et al (2013) The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57:3348–3357. https://doi.org/10.1128/AAC.00419-13
Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform 14:60. https://doi.org/10.1186/1471-2105-14-60
Meier-Kolthoff JP, Carbasse JS, Peinado-Olarte RL, Göker M (2022) TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res 50:D801–D807. https://doi.org/10.1093/nar/gkab902
Meier-Kolthoff JP, Göker M (2019) TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun 10:2182. https://doi.org/10.1038/s41467-019-10210-3
Mendonça AA, da Silva PKN, Calazans TLS et al (2020) Gene regulation of the Lactobacillus vini in response to industrial stress in the fuel ethanol production. Microbiol Res 236:126450. https://doi.org/10.1016/j.micres.2020.126450
Mendonça AA, da Silva PKN, Calazans TLS et al (2019) Lactobacillus vini: Mechanistic response to stress by medium acidification. Microbiology (united Kingdom) 165:26–36. https://doi.org/10.1099/mic.0.000738
Mendonça AA, De Lucena BTL, de Morais MAMMCMA, de Morais MAMMCMA (2016) First identification of Tn916-like element in industrial strains of Lactobacillus vini that spread the tet-M resistance gene. FEMS Microbiol Lett 363:fnv240. https://doi.org/10.1093/femsle/fnv240
Mittenhuber G (2001) Comparative genomics and evolution of genes encoding bacterial (p)ppGpp synthetases/hydrolases (the Rel, RelA and SpoT proteins). J Mol Microbiol Biotechnol 3:585–600
Nanamiya H, Kasai K, Nozawa A et al (2008) Identification and functional analysis of novel (p)ppGpp synthetase genes in Bacillus subtilis. Mol Microbiol 67:291–304. https://doi.org/10.1111/j.1365-2958.2007.06018.x
Paget MS, Helmann JD (2003) The σ70 family of sigma factors. Genome Biol 4:203. https://doi.org/10.1186/gb-2003-4-1-203
Passoth V, Blomqvist J, Schnürer J (2007) Dekkera bruxellensis and Lactobacillus vini form a stable ethanol-producing consortium in a commercial alcohol production process. Appl Environ Microbiol 73:4354–4356. https://doi.org/10.1128/AEM.00437-07
Popa O, Dagan T (2011) Trends and barriers to lateral gene transfer in prokaryotes. Curr Opin Microbiol 14:615–623. https://doi.org/10.1016/j.mib.2011.07.027
Potter SC, Luciani A, Eddy SR et al (2018) HMMER web server: 2018 update. Nucleic Acids Res 46:W200–W204. https://doi.org/10.1093/nar/gky448
Puertas AI, Arahal DR, Ibarburu I et al (2014) Lactobacillus sicerae sp. nov., a lactic acid bacterium isolated from Spanish natural cider. Int J Syst Evol Microbiol 64:2949–2955. https://doi.org/10.1099/ijs.0.059980-0
Puertas AI, Llamas-Arriba MG, Etxebeste O et al (2023) Characterization of the heteropolysaccharides produced by Liquorilactobacillus sicerae CUPV261 and Secundilactobacillus collinoides CUPV237 isolated from cider. Int J Food Microbiol 397:110199. https://doi.org/10.1016/j.ijfoodmicro.2023.110199
Rodas AM, Chenoll E, Macián MC et al (2006) Lactobacillus vini sp. nov., a wine lactic acid bacterium homofermentative for pentoses. Int J Syst Evol Microbiol 56:513–517. https://doi.org/10.1099/ijs.0.63877-0
Rouse S, Canchaya C, van Sinderen D (2008) Lactobacillus hordei sp. nov., a bacteriocinogenic strain isolated from malted barley. Int J Syst Evol Microbiol 58:2013–2017. https://doi.org/10.1099/ijs.0.65584-0
Rouvière PE, De Las PA, Mecsas J et al (1995) rpoE, the gene encoding the second heat-shock sigma factor, sigma E, in Escherichia coli. EMBO J 14:1032–1042. https://doi.org/10.1002/j.1460-2075.1995.tb07084.x
Schwengers O, Jelonek L, Dieckmann MA et al (2021) Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb Genom 7:685. https://doi.org/10.1099/mgen.0.000685
Stan G, Lorimer GH, Thirumalai D (2022) Friends in need: how chaperonins recognize and remodel proteins that require folding assistance. Front Mol Biosci. https://doi.org/10.3389/fmolb.2022.1071168
Starikova EV, Tikhonova PO, Prianichnikov NA et al (2020) Phigaro: high-throughput prophage sequence annotation. Bioinformatics 36:3882–3884. https://doi.org/10.1093/bioinformatics/btaa250
Sun Z, Harris HMB, McCann A et al (2015) Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat Commun 6:8322. https://doi.org/10.1038/ncomms9322
Tan LL, Ngiam JJ, Sim ESZ et al (2022) Liquorilactobacillus satsumensis from water kefir yields α-glucan polysaccharides with prebiotic and synbiotic qualities. Carbohydr Polym 290:119515. https://doi.org/10.1016/j.carbpol.2022.119515
Troxell B, Hassan HM (2013) Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front Cell Infect Microbiol 3:1–13. https://doi.org/10.3389/fcimb.2013.00059
Van Noordwijk AJ (1988) Futuyma, D. J. 1986. Evolutionary Biology 2nd edition, Sinauer Associates Inc. Sunderland, Mass. J Evol Biol 1:89–90. https://doi.org/10.1046/j.1420-9101.1988.1010089.x
Wang S, Ng TB, Chen T et al (2005) First report of a novel plant lysozyme with both antifungal and antibacterial activities. Biochem Biophys Res Commun 327:820–827. https://doi.org/10.1016/j.bbrc.2004.12.077
Yetiman AE, Ortakci F (2023) Genomic, probiotic, and metabolic potentials of Liquorilactobacillus nagelii AGA58, a novel bacteriocinogenic motile strain isolated from lactic acid-fermented shalgam. J Biosci Bioeng 135:34–43. https://doi.org/10.1016/j.jbiosc.2022.10.008
Zarour K, Zeid AF, Mohedano ML et al (2024) Leuconostoc mesenteroides and Liquorilactobacillus mali strains, isolated from Algerian food products, are producers of the postbiotic compounds dextran, oligosaccharides and mannitol. World J Microbiol Biotechnol 40:114. https://doi.org/10.1007/s11274-024-03913-3
Zavistanaviciute P, Lele V, Antanaitis R et al (2020) Separate and synergic effects of Lactobacillus uvarum LUHSS245 and arabinogalactan on the in vitro antimicrobial properties as well as on the fecal and metabolic profile of newborn calves. Animals 10:593. https://doi.org/10.3390/ani10040593
Zheng J, Ruan L, Sun M, Gänzle M (2015) A genomic view of Lactobacilli and Pediococci demonstrates that phylogeny matches ecology and physiology. Appl Environ Microbiol 81:7233–7243. https://doi.org/10.1128/AEM.02116-15
Zheng J, Wittouck S, Salvetti E et al (2020) A taxonomic note on the genus Lactobacillus: description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int J Syst Evol Microbiol 70:2782–2858. https://doi.org/10.1099/ijsem.0.004107
Acknowledgements
D.S.S. thanks the Brazilian agency FACEPE for granting the doctoral scholarship. The authors thank UFPE's “researcher support” program for the financial support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
None.
Additional information
Handling editor: Greg Lang.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
da Silva Santos, D., Freitas, N.S.A., de Morais, M. et al. Liquorilactobacillus: A Context of the Evolutionary History and Metabolic Adaptation of a Bacterial Genus from Fermentation Liquid Environments. J Mol Evol 92, 467–487 (2024). https://doi.org/10.1007/s00239-024-10189-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-024-10189-6