
Abstract
Cyanobacteria are recognised for their pivotal roles in aquatic ecosystems, serving as primary producers and major agents in diazotrophic processes. Currently, the primary focus of cyanobacterial research lies in gaining a more detailed understanding of these well-established ecosystem functions. However, their involvement and impact on other crucial biogeochemical cycles remain understudied. This knowledge gap is partially attributed to the challenges associated with culturing cyanobacteria in controlled laboratory conditions and the limited understanding of their specific growth requirements. This can be circumvented partially by the culture-independent methods which can shed light on the genomic potential of cyanobacterial species and answer more profound questions about the evolution of other key biogeochemical functions. In this study, we assembled 83 cyanobacterial genomes from metagenomic data generated from environmental DNA extracted from a brackish water lagoon (Chilika Lake, India). We taxonomically classified these metagenome-assembled genomes (MAGs) and found that about 92.77% of them are novel genomes at the species level. We then annotated these cyanobacterial MAGs for all the encoded functions using KEGG Orthology. Interestingly, we found two previously unreported functions in Cyanobacteria, namely, DNRA (Dissimilatory Nitrate Reduction to Ammonium) and DMSP (Dimethylsulfoniopropionate) synthesis in multiple MAGs using nirBD and dsyB genes as markers. We validated their presence in several publicly available cyanobacterial isolate genomes. Further, we identified incongruities between the evolutionary patterns of species and the marker genes and elucidated the underlying reasons for these discrepancies. This study expands our overall comprehension of the contribution of cyanobacteria to the biogeochemical cycling in coastal brackish ecosystems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the major and significant constituents of aquatic microbial diversity is the members of cyanobacteria. Phylum Cyanobacteria account for 20–30% of Earth’s primary photosynthesis (Pisciotta et al. 2010) and are major primary producers in aquatic ecosystems. They are one of the most important organisms in transforming our planet’s evolutionary history with the capacity for oxygenic photosynthesis. They are the only prokaryotes having this ability hence GOE (Great Oxidation Event) sets the minimum date for oxygenic photosynthesis and diversification of cyanobacteria from other non-photosynthetic members approximately before 2.7 billion years ago (Sanchez-Baracaldo et al. 2021).
Current studies focus on understanding nitrogen fixation, oxygenic photosynthesis and primary production by cyanobacteria with reference to biogeochemical cycling. The sparse representation of cyanobacterial taxa in these genomic databases stems from the tedious and elaborate process of obtaining their axenic cultures as well as their symbiotic growth requirements (Waterbury 2006). Currently, more than 99% of bacteria have not been isolated in pure culture due to our limited understanding of their complex metabolic requirements for their growth and cultivation in laboratory conditions (Yarza et al. 2014). Further, to understand their functional abilities, we need to have their whole genome or RNA sequence information. Notably, we have around 1.8 million genomes of bacteria in the NCBI database (as on 15th December 2023) of which only 0.29% belong to cyanobacteria while 61% of them belong to proteobacteria. Of this, 0.29% (5400 genomes) of available cyanobacterial genomes, approximately 85% are from five of the total nine orders of cyanobacteria. This shows that the genome sequence information in the current databases is skewed and cyanobacteria are severely underrepresented. This “microbial dark matter” can be partially studied through the use of MAGs (Metagenome-Assembled Genomes) obtained from the sequencing of metagenomic DNA directly extracted from an environmental sample. For instance, in a study by Broman et al. (2023) on a brackish water lagoon in the Baltic Sea, MAGs were assembled and genes were predicted. They found that environmental variations played a more prominent role than the taxonomy of bacteria in the composition of functional genes. In another study, the evolutionary link between anaerobic basal thaumarchaeal lineages and mesophilic marine ammonia-oxidising archaea was found by reconstruction of two MAGs from phylum Thaumarchaeota (Reji and Francis 2020). A genome-resolved metagenomic approach was used to recover 37 MAGs from microbial mats of cyanobacteria including a few lineages commonly found in polar region as well as a few less common lineages in 15 Arctic, sub-Antarctic and Antarctic lakes (Pessi et al. 2023).
In this study, we collected water samples and extracted environmental DNA from a brackish water lagoon, Chilika located in the Odisha State on the Eastern coast of India. We assembled the cyanobacteria metagenomes and annotated them for over hundreds of functions. Interestingly, we found two functions which had never been previously attributed to cyanobacteria. One was Dissimilatory nitrate reduction to ammonium (DNRA) encoded by the nirBD gene and another was Dimethylsulfoniopropionate (DMSP) synthase encoded by the dsyB gene. In Dissimilatory nitrate reduction to ammonium (DNRA), nitrate serves as an electron donor and is converted to nitrite and ammonium (NO3− → NO2− → NH4+), providing a bioavailable N source to plants and animals (Lam and Kuypers 2011). DNRA activity has been reported in Proteobacteria namely Beggiatoa alba (Vargas and Strohl 1985) but its presence in cyanobacteria is unclear. A recent study (Tee et al. 2020) indicated the expression of nirBD by Microcoleus in biofilms produced by them. DNRA has been reported to occur in both aerobic (Huang et al. 2020) and anaerobic conditions (Kamp et al. 2011). Further, it was also demonstrated through 15N isotopic studies that nirBD encodes for nitrate reductase and controls the DNRA process in Pseudomonas putida Y-9 (Huang et al. 2020).
The second function of interest was DMSP synthesis. DMSP is an organic tertiary sulfonium compound that is important and extremely abundant (2 pentagram sulphur is produced per annum globally) (Hatton et al. 2012). It is widely distributed in the euphotic zone of aquatic ecosystems and is ampler in marine than in terrestrial ecosystems due to higher sulphur availability (Stefels 2000). DMSP has several physiological and ecological functions. It serves as an osmoprotectant as its chemical structure is similar to an osmolyte glycine betaine (Vairavamurthy et al. 1985). It also can scavenge hydroxyl ions and reactive oxygen species serving as an antioxidant system (Sunda et al. 2002). The significance of DMSP lies not just in its abundance but it is catabolised to DMS (dimethyl sulphide) and hence is the primary precursor of DMS (Lovelock et al. 1972). DMS is a volatile climate-active gas that has a crucial role in forming cloud condensation nuclei and global sulphur cycling (Hatakeyama et al. 1982). DMSP is synthesised from methionine (Met) following the three different pathways across the Tree of Life: first a methylation pathway in Angiosperm (Kocsis et al. 1998), a transamination pathway in heterotrophic bacteria (Curson et al. 2017), marine algae (Gage et al. 1997) and phytoplankton (Curson et al. 2018) and third a decarboxylation pathway in dinoflagellate (Uchida et al. 1996). In the last few years, the molecular pathways and the key enzymes responsible for the synthesis of DMSP in diverse taxa have been elucidated. One of the first DMSP synthesis genes, dsyB was found in marine Alphaproteobacteria (Labrenzia aggregata) which encoded a methyltransferase enzyme and catalysed a committed step of the pathway (Curson et al. 2017). Following this, dsyB homologue was identified in phytoplankton and corals, named DSYB. One report of a cyanobacterial species, Trichodesmium erythraeum, produces DMSP intracellularly and its concentration increased under iron-limiting conditions (Bucciarelli et al. 2013). However, there is negligible work undertaken to comprehend the role of Cyanobacteria in sulphur cycling and DMSP production.
We aimed to identify and delineate the evolution of the putative nirBD and dsyB-like genes as a proxy for its potential role in DNRA and DMSP production in cyanobacteria, respectively. We constructed the phylogeny representing the evolution of this gene in cyanobacteria and compared it with their species phylogeny. This highlighted the discrepancies between the evolutionary trajectory of cyanobacterial species and their putative nirBD and dsyB-like genes.
Methods
Study Site and Physiochemical Measurements of Abiotic Factors
Chilika Lagoon (19° 28′ N: 19° 54′ N and 85° 06′ E: 85° 35′ E) is a dynamic brackish coastal ecosystem with an average water spread area of 900 km2 and a catchment area of ~ 4146 km2 (Srichandan et al. 2015). This lagoon is a Ramsar site and is a major biodiversity hotspot in India. Freshwater enters the Chilika Lagoon through 12 major tributaries, while saline water intrudes into the lagoon from the Bay of Bengal, causing significant variability in physicochemical parameters due to the mixing of freshwater and seawater (Tarafdar et al. 2021).
We measured ten physicochemical parameters (water temperature, pH, depth, transparency, Biological Oxygen Demand (BOD), Dissolved Oxygen (DO), silicate. nitrate and phosphate) from water samples across all sampling sites. The water temperature, pH and salinity were measured using water quality Sonde (YSI, Model No. 6600, V2). Depth of water and its transparency were estimated by a Secchi disk. Dissolved Oxygen was quantified using the modified Winkler’s method (Carrit and Carpenter 1996). Biochemical oxygen demand was evaluated after incubation of 5 days at 20 °C. The dissolved inorganic nutrients, namely, nitrate, phosphate and silicate were quantified upon filtering the water through a membrane filter paper of 0.45 μm pore size and a diameter of 47 mm, which was then assessed with autoanalyzer (SKALARSANplus ANALYZER) (Grasshoff et al. 1999).
eDNA Isolation and Sequencing
The water sampling, extraction and sequencing of environmental DNA were carried out as previously described (Manu and Umapathy 2023). Briefly, we filtered water from different locations of Chilika Lake, Odisha, India during the period from December 2019 to December 2020 (Supplementary S1). For this purpose, we used a 0.45 µm mixed-cellulose ester filter (MCE) membrane of 47 mm diameter to trap the extracellular DNA in the water as MCE has been shown to retain the maximum amount of extracellular DNA due to its chemical affinity (Liang and Keely 2013). We collected 16 samples in triplicate from 9 unique locations (Supplementary S1) (minimum 10 km apart) which varied in physiochemical parameters across spatio-temporal ranges of the Chilika lagoon (Muduli and Pattnaik 2020). The timing and sampling sites were chosen based on previous studies (Manu and Umapathy 2023) on bacterial communities (Mohapatra et al. 2020). The above samples were filtered using an eDNA sampler by Smithroot Inc. (Thomas et al. 2018) and filter assemblies were stored at room temperature until eDNA isolation in the laboratory. We used a lysis-free phosphate buffer-based eDNA isolation protocol from the filter membranes (Liang and Keeley 2013 and Taberlet et al. 2012). We randomly fragmented the input DNA into 350 bp regions following which we used the Illumina Truseq DNA PCR-free library preparation method and sequenced for 300 cycles on the Novaseq 6000 platform.
Assembly and Taxonomic and Functional Annotation of MAGs
The raw reads were adapter trimmed and quality filtered with a phred quality threshold of 10 using the BBDUK tool (Bushnell 2014). The quality-filtered reads were in-silico normalised to a target depth of 100x using BBNORM and error-corrected with BBCMS from the BBTOOLS package. The error-corrected reads from all samples were co-assembled using MEGAHIT with the meta-large pre-set. Contigs shorter than 1000 bp were filtered out and the quality-filtered reads from each sample were mapped to the remaining contigs with a minimum of 97% identity using BBMAP (Bushnell 2014). The normalised abundance of contigs in all the samples was calculated using the JGI summarise bam contigs utility and the contigs were binned using METABAT2 (Kang et al. 2019) with default parameters. The quality of bins was assessed with CheckM (Parks et al. 2015) using the lineage workflow and bins with greater than 50% completion and less than 10% contamination were assigned as MAGs and retained for taxonomic classification. We first calculated the mean coverage of each MAG by dividing the number of bases in aligned reads of a sample by the total length of all the contigs in the MAG. The relative abundance of each MAG in a sample was then calculated as the fraction of total coverage of all the MAGs in the sample. The MAGs were taxonomically annotated using the GTDB Toolkit (database v. 207_2) with default parameters and those belonging to the cyanobacteria were selected for functional annotation using KEGG Decoder. Upon functional annotation, we selected two functions which have not been attributed to cyanobacterial species before and further analysed their evolution.
Curation of MAGs
We analysed the sequence properties of binned contigs using uBin v. 1.1 (Bornemann et al. 2023). Briefly, the GC content, read coverage across all the samples and the presence of bacterial single-copy genes were calculated for each contig and visualised through a GC vs. Coverage plot. The contigs were coloured by BIN IDs and checked for clustering pattern of contigs belonging to the same MAG. The outliers in the plot were iteratively excluded and checked for decrease in contamination indicated by the single-copy genes. Further, the GC content and coverage of contigs containing the genes of our interest (nirBD and dsyB) were checked to verify that they fall with the standard deviation of respective MAGs. The gene sequences were translated and searched for the nearest taxa using blastp to verify the taxonomic identity of the best hit matched with the expected taxonomic family of the MAG.
nirBD Gene
Extraction of NirBD Sequences from MAGs and Curation of Sequences of NirBD from the Database
We generated the protein sequences from all cyanobacteria MAGs using Prodigal.v2.6.3 (Hyatt et al. 2010). The predicted ORFs were annotated with KofamKOALA v.2022-01-03 (Aramaki et al. 2020) and searched for DNRA using the definitions from the KeggDecoder tool. The definition for DNRA was nirBD (K00362 + K00363) and/or nrfAH (K03385 + K15876). Using this definition, the ORFs were annotated by KofamKOALA. The e-value cut-off was set to more than 1e-10. These hits were searched using blastp to confirm their identity or to find the nearest match to the existing sequence in database. All the species having a percent identity greater than 50% and having both NirB and NirD proteins were included in the construction of phylogeny. The protein family of the NirBD sequences was classified using InterPro 89.0 (Blum et al. 2021). We considered all the database species of the protein family given that they had both nirB and nirD counterparts and their whole genomes are available in the database for constructing species trees using the ribosomal proteins.
Construction of Species and NirBD Protein Tree
Species Tree
The whole genomes of all representative species were downloaded from the NCBI (National Center for Biotechnology Information) database (Supplementary S2). We used GraftM v0.14.0 (Boyd et al. 2018) to extract 15 single-copy ribosomal protein marker genes (Supplementary S3) from all the representative species from the database as well as from the two MAGs generated as a part of this study. This approach was used because of two main reasons. Firstly, in metagenome data, often 16sRNA genes do not get assembled entirely and cannot be traced back to their respective genome owing to high sequence conservation (Hug et al. 2013). Secondly, species phylogeny constructed using one 16Srna gene yields lower resolution than the phylogeny constructed using many single-copy ribosomal proteins (Teeling and Glöckner 2012). These 15 proteins were then aligned using MEGA11 (Tamura et al. 2021). These were then trimmed using Gblocks 0.91b (Castresana 2000) and concatenated using MEGA11. A maximum-likelihood tree was constructed using an IQ-Tree webserver (Nguyen et al. 2015; Trifinopoulos et al. 2016) using simultaneous model selection. For the construction of the Bayesian tree, PartitionFinder v.2.1.1 (Lanfear et al. 2017) was used to select the model for every partition followed by MrBayes v.3.2.7a (Huelsenbeck and Ronquist 2001).
NirBD Protein Tree
Blastp of the NirB and NirD ORFs from the two MAGs identified the closest representative species having this protein in the database. These sequences were downloaded and aligned using MEGA v.11, followed by trimming using Gblocks 0.91b and concatenating using MEGA v.11. The same procedure was used to construct the maximum-likelihood and Bayesian tree as described for the species tree.
Reconciling of Species and NirBD Protein Tree
The topology of the phylogenetic tree for both species and the NirBD protein was inspected manually. Since there were several points of discordance between the two, a reconciliation was performed between species and NirBD protein trees by RangerDTL v2.0 (Bansal et al. 2018) to reconcile species and NirBD protein tree using default costs for duplication (2), transfer (3) and loss (1) for 200 reconciliations. These reconciliations were aggregated using RangerAggregator to obtain the most supported events. Individual reconciliation was converted using RecPhyloXML converter (Python script) and visualised using Thinkind (Penel et al. 2022) webserver. The events that show more than 90% consistency of the event, as well as more than 90% mapping consistency, were depicted in the trees.
dsyB Gene
Extraction of dsyB Sequences from MAGs and Curation of Sequences of dsyB from the Database
We used Prodigal.v2.6.3 (Hyatt et al. 2010) to generate protein sequences of the cyanobacterial MAGs. We used HMMER v3.3.2 and HMM profile for dsyB protein as query files to search for them from our target MAGs. The topmost hit was chosen and was used as input for blastP to find the nearest match to the public database sequences. The species that shared more than 50% similarity of sequences from the blastP results were considered to construct phylogenies. Also, the bacterial sequences from Curson et al. 2017 were incorporated to understand the evolutionary link between the different bacterial Phyla.
Construction of Species and DsyB-Like protein Tree
Species Tree
The whole genomes of all the above-mentioned representative species were downloaded from NCBI and details of the same are available in Supplementary S4. The species tree consisted of 91 species from NCBI and 2 of the cyanobacterial MAGs assembled from Chilika Lagoon. The 3 MAGs were not incorporated for constructing the Species Tree as some of their ribosomal proteins could not be retrieved. However, since these three MAGs showed their maximum similarity to Synechococcus sp. hence the sequence of this genus (having a similarity of more than 96% at an amino acid level for DsyB protein) was incorporated from NCBI. We used GraftM v.0.14.0 (Boyd et al. 2018) to extract 15 single-copy ribosomal protein markers (Supplementary S3) from all the genomes. These were then aligned using MEGA v.11 (Tamura et al. 2021) and then trimmed using Gblocks 0.91b (Castresana 2000). These were then concatenated using MEGA v.11. A maximum-likelihood tree was constructed using an IQ-Tree webserver (Nguyen et al. 2015; Trifinopoulos et al. 2016) using simultaneous model selection.
DsyB Protein Tree
The same representative genomes used for constructing Species phylogeny were also used for the dsyB protein tree. All five cyanobacterial MAGs were incorporated in the dsyB protein phylogeny. The sequences were aligned using MEGA11 and then Maximum-likelihood (ML) phylogeny was constructed using IQ-Tree webserver (Nguyen et al. 2015; Trifinopoulos et al. 2016).
Reconciling of Species and DsyB Protein Tree
The Species and DsyB protein tree were manually inspected to infer the points of discordance between them. The two phylogenies were then reconciled to decipher the donor species and the modes by which incongruencies have occurred. The methodology for this analysis was followed as described in section “Reconciling of Species and NirBD Protein Tree”.
Statistical Analyses
We analysed physiochemical parameters data based on the season of sample collection and performed Kruskal–Wallis Test to examine variation among the seasons. As most of the abiotic parameters did not follow a normal distribution, a non-parametric approach (Kruskal–Wallis test) was used as an alternative to one-way ANOVA. This was executed in RStudio using package stats v.4.2.2 and function kruskal.test() and were visualised using ggplot2 v.3.4.2 package.
We performed NMDS analysis to visualise the ordination between the Bray–Curtis dissimilarity of the relative abundances of cyanobacterial MAGs between the sampling sites. For this we used the metaMDS function from vegan v. 2.6-4 package with 999 permutations. We then fitted the environmental variables onto the NMDS ordination using the function envfit from vegan v. 2.6-4 package. This will also highlight which of the environmental variables are significant.
Results
Taxonomic and Functional Annotation
We assembled a total of 83 MAGs from cyanobacteria of which 14 were high quality (more than 90% complete and less than 5% contaminated) and the rest were medium quality (more than or equal to 50% complete and less than 10% contaminated) from Chilika Lagoon.
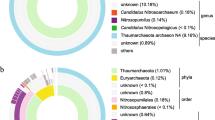
Upon taxonomic annotation of the all 83 cyanobacterial assembled metagenomes from Chilika Lagoon, we found that 92.77% of the MAGs did not match any known representative genome in the GTDB database at the species level implying that these are potentially novel genomes. Upon taxonomic classification of these 83 cyanobacterial MAGs, we found that 71 of them belonged to class Cyanobacteria, 4 to Vampirovibronia and 8 to Sericytochromatia. A large portion of these MAGs from class Cyanobacteria belonged to the family Cyanobiaceae which further had representations from 9 genera (Fig. 1). The other predominant families included Elainellaceae, Microcystaceae, Leptolyngbyaceae, Microcoleaceae, Nostocaceae, Phormidesmiaceae and Prochlorotrichaceae. Also, a few of them belonged to non-photosynthetic cyanobacterial classes of Sericytochromatia and Vampirovibrionia. As of now, few representatives from these non-photosynthetic classes are present in public databases, due to which their functional role in ecology and metabolism remains largely unidentified (Monchamp et al. 2019). A detailed list of taxonomic assignments for all the MAGs are available in Supplementary S5.

Taxonomic annotation of assembled metagenomes from class Cyanobacteria. The other two major classes were Sericytochromatia having 8 MAGs and Vampirovibronia having 4 MAGs
Functional annotation of all 83 MAGs showed that most of them possessed the genes for most amino acid metabolism except alanine and aromatic amino acids like tyrosine and phenylalanine (Supplementary S6). Some of them were devoid of genes for metabolising the amino acids with negatively charged R groups namely aspartate and glutamate. Most of them also possessed genes for carbohydrate metabolism pathways like glycolysis and the TCA cycle. Moreover, 12 MAGs showed a complete absence of photosystem 1, 2 and cytochrome bf6 complex and belonged to either Sericytochromatia or Vampirovibronia which are known to be non-photosynthetic classes of cyanobacteria. A total of 7 MAGs also showed the presence of nitrogen fixation ability. We found that there were certain functions which seemed less predominant and were present only in some of the MAGs. These functions had immense significance in biogeochemical cycles. Two such functions were DNRA and DMSP synthase encoded by nirBD and dsyB genes, respectively. DNRA was present in only two of the MAGs, whereas DMSP synthase was present in five of the MAGs out of total 83.
To verify the quality and contamination of the MAGs, we manually curated them and checked the sequence properties. We visualised the clustering of the MAGs containing NirBD and dsyB genes in GC content v/s coverage plot (Fig. 2). Each MAGs form distinct clusters on the plot verifying their assembly quality. We also checked the NirBD and dsyB protein sequences from the MAGs using blastp and found the best hit corresponds to Cyanobacteria in all the cases. The results of both the analysis are summarised in Supplementary S7.

Plot depicting the GC content v/s the coverage of contigs for the 7 MAGs containing either NirBD or dsyB sequences
nirBD Gene
Cyanobacterial Genomes Having NirBD Signatures
The two cyanobacteria MAGs showing the presence of the nirBD gene belonged to high-quality bins with less than 5% contamination > 90% completeness (Table 1).
Identification of NirB and NirD Protein from MAGs
MAGs showed the presence of a signature for DNRA activity – nirBD (K00362 + K00363) as depicted in Table 2. After classifying the obtained NirB and NirD protein sequences, these belonged to Nitrite reductase [NAD(P)H] large subunit (IPR017121) and Nitrite reductase (NADH) small subunit (IPR017881) protein families, respectively. We considered all the species belonging to these two protein families from cyanobacteria. There were 38 species which had both IPR017121 and IPR017881 sequences available. We checked for genome sequences for all these cyanobacterial species in the NCBI database and finally ended up with 32 genomes. Using the blastp search, we compared the NirB and NirD sequences of the MAGs and identified several proteobacteria with a percentage identity of greater than 65%. The hits from cyanobacteria in the blastp searches were from the same genera that we also obtained from InterPro. We used these 43 sequences from cyanobacteria and proteobacteria and the two of our assembled MAGs for subsequent analysis.
Discordance Between Species and NirBD Protein Tree
We constructed the NirBD protein and species trees (Fig. 3) using both maximum-likelihood and Bayesian methods. In both methods, the topology of the tree is the same but discordance is observed in several branches between the species and the NirBD protein tree. The species tree topology is consistent with the respective taxonomic assignments including the two assembled MAGs from Chilika Lagoon. But in contrast to this, we observe that NirBD protein phylogeny does not follow the pattern as shown by organismal phylogeny. This signifies the evolutionary history of the NirBD protein did not happen concurrently with the evolution of the species. To decipher the reason and the source of these incongruencies, we performed a reconciliation analysis where we embedded the NirBD protein tree into the species tree. Further, this helps to deduce the gene losses, duplications and horizontal gene transfers which could be the probable reasons behind discordance.

A Concatenated NirB and NirD protein phylogeny. The bayesian phylogeny with bootstrap support values is depicted for each node. The NirBD protein extracted from MAGs constructed as a part of this study are represented in red text in this figure. B Species tree. The species phylogeny is constructed using 15 concatenated ribosomal proteins and the bayesian bootstrap values are mentioned for each node. The ribosomal proteins extracted from the MAGs generated as a part of this study are represented in red (Color figure online)
Reconciliation Analysis
The dispersal of DNRA function traced by NirBD protein’s evolution demonstrates cyanobacteria’s intra-phylum dynamics. This protein’s distribution results from a combination of horizontal gene transfer events and speciation among cyanobacteria. Upon reconciling the NirBD protein tree with the species tree (Fig. 3), we observe that the well-supported events with more than 90% consistency happened within the cyanobacterial and not among their closest outgroup members from Proteobacteria. Most of the filamentous cyanobacteria have been responsible for the dissemination of the ability of DNRA. This is evident from the fact that the ancestral donor of NirBD (corresponding to node n22 in Fig. 4) has all of the filamentous species of cyanobacteria. Also, most of the HGT events are mapped to filamentous species of cyanobacteria. Additionally, the deep-branching cyanobacterial species did not contribute to the dispersal of DNRA activity, implying that the event happened later during the evolution. The transfer events were mostly found to be restricted to specific species, indicating a limited ability to perform horizontal gene transfer. Additionally, it appears that most other species gained the NirBD protein through speciation from cyanobacterial ancestors that already possessed the potential for DNRA. The fact that the exact donor could not be identified in most cases suggests that the ancestral species with the potential for DNRA may not have their sequences represented in current databases, highlighting the limitations of an incomplete database.

Reconciliation analysis of Species and NirBD protein tree. Only the events which were more than 90% consistent are shown here. The ones represented in green are filamentous cyanobacteria and the ones in red are MAGs constructed as part of this study. The different coloured circles illustrate the speciation event. For example, the red circle at n28 node of Species tree having six cyanobacterial species speciated during the course of evolution and hence transferred the NirBD protein to ancestral species in m26 node of NirBD protein tree (marked in same colour). The horizontal gene transfer events are marked as dotted lines. Hence both the events could have been responsible for simultaneously disseminating this function (Color figure online)
dsyB-like Gene
Cyanobacterial MAGs Having dsyB-Like Signatures
We found five cyanobacterial MAGs showing the presence of the dsyB-like gene of which 1 was high quality and 4 were medium quality (Table 3).
HMMER Analysis Identification of dysB Protein from MAGs
Upon searching the target MAG sequence with the HMM profile, the topmost hit was used for further analysis. The e-value and the score for the same are tabulated in table 4.
Discordance Between Species and DsyB Protein Tree
The incongruence between species phylogeny and DsyB protein phylogeny was demonstrated. This signified that the evolution of the two phylogenies was distinct.
Figure 5 shows the maximum-likelihood tree for the DsyB protein. The cyanobacterial species do not cluster together but rather are sporadically distributed across the phylogenetic tree. The non-dsyB species (Curson et al. 2017), which have non-functional dsyB and do not produce DMSP, form a separate clade in the DsyB protein tree. On the contrary, Fig. 5 representing the organismal phylogeny depicts two different clusters of species based on their taxonomy. All the Cyanobacterial species form one clade and Proteobacteria form a separate clade as is expected.

A DsyB-like protein phylogeny. The phylogeny is constructed using dsyB amino acid sequence for all the species using maximum-likelihood algorithm. The species marked as non-dsyB refers to non-functional protein which do not produce DMSP. The DsyB-like protein sequences from the five MAGs generated as part of this study are marked in red text. The bootstrap support values are mentioned at the respective node. B Species phylogeny. The phylogeny is constructed using concatenating the 15 single-copy ribosomal protein markers and maximum-likelihood algorithm. The bootstrap values are mentioned at the respective nodes. The two MAGs generated in this study are marked in red text (Color figure online)
Additionally, a few ribosomal proteins could not be extracted from three MAGs to include them in the Species tree. However, the blastp results showed that those three MAGs share the closest similarity to Synechococcus sp. from the public database, which was incorporated in the construction of the phylogenies.
Reconciliation Analysis
The points of incongruence and their reasons were discerned by performing a reconciliation analysis between the two phylogenies. The topology of the two phylogenies was majorly different at most nodes. We found that there were multiple events of lateral and vertical gene transfers during different points. Approximately 95% of the total events occurred with 100% consistency. Approximately 64% of total events were mapped consistently.
The DsyB-like protein tree has 3 major clades of species, marked as m106, m4 and m49 (Fig. 6). The donors for all these 3 clades do not seem to overlap. The donors for m49 consist only of proteobacterial species, whereas those for m106 and m4 consist of donors from cyanobacteria and proteobacteria. The major donor species for node m106 of the dsyB-like protein tree were Thermoleptolyngbya sp., Anabaena elenkinii, Fischerella muscicola from cyanobacteria and Symmachiella dynata and Candidatus Accumulibacter aalborgensis from proteobacteria. The major donor species for node m4 of the DsyB-like protein tree were Moorea producens and members from the Nostocaceae family of cyanobacteria. The prominent donor species from Proteobacteria for this node were Sorangium cellulosum and Parasphingopyxis marina. The dynamics of mapping the exact donor might change upon the addition or deletion of more representatives but it points towards multiple major events of acquiring this gene.

Reconciliation analysis of DsyB protein tree and Species tree – All the events depicted here have occurred with more than 90% probability. The transfer events are marked in dotted lines. The transfer events to node m106 are shown in blue, node m4 are shown in green and node m49 are shown in violet colour (Color figure online)
Another significant inference from the reconciliation analysis was node n2 of the Species tree consisting of all cyanobacterial species mapped as donors for horizontal gene transfer for all the DsyB-like proteins of node m2 consisting of both cyanobacterial and proteobacterial DsyB-like proteins, but not the non-functional DsyB containing species. This event happened with 98% confidence during reconciliation analysis. This meant that cyanobacterial species served as the initial donors for all the cyanobacterial as well as proteobacterial species. This was followed by multiple other events of HGT and speciation which assisted in disseminating this function in the bacterial kingdom.
Spatio-Temporal Variation in Physiochemical Factors and Its Influence on the Distribution of Cyanobacterial MAGs
We found a significant temporal variation in water temperature (p = 0.001), transparency (p = 0.04) and nitrate concentration (p = 0.002) across Chilika Lagoon, while salinity (p = 0.059) was narrowly different (Table 5). However, spatial variations, as assessed between different stations, did not exhibit significance due to constrained sampling efforts stemming from high sequencing costs (Table 5).
Previous research on bacterioplankton communities of Chilika Lagoon has shown strong spatial and seasonal variation in cyanobacterial abundances that depended on salinity gradient (Mohapatra et al. 2020). Cyanobacteria were largly represented by autotrophic picocyanobacteria Synechococcus and Cylindrospermopsis. Synechococcus was more abundant in monsoon oligohaline samples and were negatively correlated with salinity. These were more abundant in ‘riverine’ northern sector of Chilika Lagoon, which experiences low salinity during monsoon.
NMDS analysis highlighted the similarity between the cyanobacterial community among the sampling sites of Chilika Lagoon. The sites sampled during the same season were closer on the plot, signifying a similar cyanobacterial community structure. Upon fitting the environmental variables onto the ordination, water temperature, salinity and nitrate concentration emerge as the most significant variables (Fig. 7). Their significance values were 0.010, 0.015 and 0.054, respectively. This is concordant with the Kruskal–Wallis analysis which also showed significant temporal variations in water temperature, salinity and nitrate concentration.

Spatio-temporal variation in cyanobacterial composition of Chilika Lagoon. Ordination of Bray–Curtis dissimilarity in cyanobacterial MAGs among the spatiotemporal samples of the Chilika lagoon. The sampling sites are represented by station number (S1, S6, S14, S17, S26, S27, S28, S29, S30) followed by season of sampling (MS = Monsoon and WN = Winter). The physiochemical parameters are represented by blue arrows (W.Temp = water temperature, Trans = transparency, salinity, depth, DO =Dissolved Oxygen, BOD = Biological Oxygen demand, pH, N = nitrate, P = phosphate, Si = silicate) (Color figure online)
Discussion
In this study, we assembled 83 MAGs from cyanobacteria of Chilika Lagoon. We functionally and taxonomically annotated them and found DNRA and DMSP synthesis functions which were not previously attributed to cyanobacteria. We also deciphered the evolution of these functions in cyanobacteria. Further, we found temporal changes in physiochemical parameters of Chilika Lagoon drive the cyanobacterial composition. We will see these findings in detail.
nirBD Gene: Proxy for DNRA Function
DNRA is the conversion of nitrate to ammonium and its marker gene NirBD is reported in some bacterial species, but there is minimal information in cyanobacteria many of which are diazotrophic. Hence, finding it in our assembled genomes from Chilika Lagoon was interesting but posed new questions. Upon subsequent analysis of cyanobacterial whole genomes from public databases (NCBI) and finding the NirBD gene in them suggested that the NirBD gene in our cyanobacterial MAGs was not any artefact and represents an unexplored area.
We used Kyoto Encyclopedia of Genes and Genomes (KEGG) modules to identify the MAGs having NirBD gene. This is because KEGG is a resource that systematically connects genomic data to functional insights and presents as a curated collection. Within the KEGG modules, the genes associated with nitrate assimilation (M00531) and DNRA (M00530) are catalogued. Specifically, nirB and nirD feature within the DNRA module, whereas they are notably absent from the nitrate assimilation module. Hence, in our study, we have used the presence of NirBD in the genome as a proxy for DNRA but we acknowledge that there are studies which have demonstrated that NirBD controls both DNRA and nitrate assimilation processes (Huang et al. 2020), which is difficult to ascertain from only genome information.
Some cyanobacteria are well-known diazotrophs and are broadly classified into heterocystous and non-heterocystous forms. The enzyme nitrogenase, which is vital for nitrogen fixation, is extremely sensitive to oxygen. Hence, heterocystous nitrogen-fixing cyanobacteria have spatially parted the oxygenic photosynthesis from nitrogen fixation by use of specialised cells called heterocyst. These specialised cells are devoid of oxygenic photosystem and possess a glycolipid cell wall aiding to minimise the oxygen concentration for nitrogen fixation to happen efficiently in these organisms (Stal and Zehr 2008). In non-heterocystous nitrogen fixers, this is accomplished by temporal partitioning where nitrogen fixation happens during the dark when these species are grown under alternating light–dark cycles (Stal et al. 1994; Bergman et al. 1997). Interestingly, our analysis showed that many of the cyanobacterial genera which are non-heterocystous nitrogen fixers also had NirBD (a marker of DNRA function). Some of these genera were Lyngbya, Phormidium, Oscillatoria, Microcoleus and Limnothrix. Both the MAGs also showed the presence of nitrogen-fixing function based on the KEGG decoder hence showing the ability to perform Biological Nitrogen Fixation (BNF) besides DNRA. This hints towards the existence of multiple strategies in cyanobacteria for acquiring bioavailable nitrogen sources (ammonium) and could be beneficial under certain stress conditions such as prolonged N limitation.
Discordance Between the Cyanobacterial Species Tree and NirBD Protein Tree
One of our assembled genomes (MAG827261_f_Nostocaceae) forms a separate clade in the NirBD protein tree demonstrating its amino acid sequence is quite divergent from the ones from the database. This MAG in the Species tree forms a sister clade with members of the Calothrix genus which also belongs to the family Nostocaceae (according to Genome Taxonomy Database, GTBD). This discordance in the phylogeny of the NirBD protein and the species (MAG827261_f_Nostocaceae) is due to their different evolutionary paths. Further, this also shows inadequate genome information in databases to fathom the evolution of these functional genes. This further leaves us with the resonant question about the indefinite diversity of such functions in ecosystems emanating from inadequate genome sequence information.
Phylogenetic Reconciliation and Evolution of NirBD in Cyanobacteria
The genome of bacteria consists of core gene sets and flexible gene sets. The core gene sets are highly conserved in their amino acid sequence and are resistant to Horizontal Gene Transfers (HGT) and hence represent robust organismal phylogeny. However, flexible gene sets are variable and allow changes in their amino acid sequences according to selection pressure and can be transferred among organisms. In cyanobacteria genomes, genes encoding photosynthetic and ribosomal proteins form the densest component of core gene sets (Shi and Falkowski 2008). Thus, the single-copy conserved ribosomal proteins are used to construct Species trees in bacteria since they are a reflection of the species evolutionary history (Hug et al. 2016) and (Moore et al. 2019). To understand the source of conflict between the phylogenies of a target gene tree and a Species tree, we need to comprehend their respective evolution. The discrepancies between a target gene tree with a species tree stem from the concept of independent evolution of species and the genes encoded by its genome. Embedding the gene tree into a species tree called phylogenetic reconciliation can help in discerning the reason behind the discordance of the gene and organismal phylogeny (Waglechner et al. 2019) and (Shang et al. 2022). Analysing the phylogenetic reconciliation of our dataset illustrated that the deep-diverging nodes of the organismal phylogeny were not mapped as the donor for the NirBD protein. This meant that acquiring and disseminating NirBD protein happened later during evolution. Also, the Acaryochloris marina does not possess genes for nitrogen fixation as well (Pfreundt et al. 2012). Overall, we uncovered the genomic capacity and evolutionary dynamics of DNRA function within the Cyanobacteria Phylum.
dsyB-Like Gene: Proxy for DMSP Synthesis
DMSP production using the transamination pathway has a committed step catalysed by a methyltransferase enzyme. It is reported in heterotrophic bacteria, marine algae and phytoplankton but has no reports from phototrophic bacteria like cyanobacteria. The dsyB gene and its eukaryotic homolog, DSYB, are used as reporters for DMSP production (Curson et al 2017). The evolution of such a significant function of suphur biogeochemical cycling is ambiguous. There can be multiple explanations of why a product is synthesised using a certain pathway in a taxon but possessing the genes to undergo that pathway, serves as a prerequisite. Sharing a similar pathway across different taxa stems from either their common ancestry or gene duplication and its divergence or convergent evolution of the pathway under similar environmental conditions. Evolution is a complex process influenced by multiple factors and discerning the exact reason can be enigmatic.
dsyB-Like Gene
The production of DMSP by cyanobacteria has not been extensively investigated and there are no reported studies on delineation of involved molecular pathways. Therefore, the identification of the putative dsyB-like gene in our cyanobacterial MAGs, generated as part of this study, was intriguing. Four of the MAGs that possessed dsyB-like genes showed similarity of only 70–80% at the amino acid level. This meant that there were no sequences in the public databases that were entirely similar and could only indicate the closely related representative from the already-sequenced genomes. The dsyB-like genes from three of the MAGs were closest to Synechococcus sp. Cyanobacteria have 20 orders and 49 families according to Genome Taxonomy Database (GTBD) but of these, only approximately 14 families had shown to have a dsyB-like gene. This could be due to skewed genome sequence information from only a few of the cyanobacterial families in these public databases, probably due to well-standardised culturing methods for them. This study also highlights the probability that many more cyanobacterial species have the dsyB-like gene but their whole genome sequence is not available to date. Further, this also illustrates inadequate genome information in databases which consequently provides an incomplete representation to fathom the evolution of these genes having significant functions in the ecosystem.
Discordance Between DsyB Protein and Species Phylogeny
The Cyanobacterial species having DsyB-like protein do not cluster together in the phylogeny signifying that this gene did not undergo only vertical transfer but also horizontal gene transfer. The evolution of the gene is hence not congruent to the evolution of the Species ensuing sporadic pattern. Additionally, the species having confirmed reports of inability to produce DMSP and non-functional DsyB protein formed a separate clade.
Phylogenetic Reconciliation and the Evolution of dsyB-Like Gene in Cyanobacteria
After performing reconciliation analysis, we made an intriguing observation: the DsyB-like proteins from both heterotrophic and phototrophic bacteria were mapped to the cyanobacterial clade in the species tree. Based on this evidence, we hypothesise that the dsyB-like gene was initially acquired by a cyanobacterial ancestor and subsequently transferred to other bacterial groups. This could explain the presence of a similar transamination-based pathway in these bacteria, albeit with potential differences in substrates or co-factors due to sequence divergence over time. Furthermore, the DsyB-like protein phylogeny exhibited three distinct clades, and the species acting as donors for these clades did not overlap. This indicates that multiple vertical and lateral gene transfer events occurred, leading to the widespread dissemination of dsyB-like genes across the bacterial kingdom.
Spatio-Temporal Variations in Physiochemical Factors and Its Impact on Cyanobacteria
This shows the scope for further research on critical ecological functions and the adaptability of microbes to various environmental conditions. Environmental conditions play a vital role by favouring species of cyanobacteria to proliferate under varied conditions as observed in the NMDS analysis (Fig. 7). The samples from the same season are closer on the NMDS ordination plot than samples from different seasons, signifying variation in cyanobacterial community temporally. There are studies showing that besides deterministic environmental factors, bacterioplankton including cyanobacteria have been shown to be governed by biotic interactions (positive and negative species relationships) with phytoplankton and bacterial communities of Chilika Lagoon (Tarafdar et al. 2021; Mohapatra et al. 2023). For instance, Synechococcus showed a positive correlation with Spartobacteria, suggesting its significance as the primary source of carbohydrate substrates and, consequently, its crucial role in carbon cycling (Mohapatra et al. 2020).
Significance of Cyanobacteria in Evolution and Global Biogeochemical Cycling in Aquatic Ecosystems
The identification of DNRA and DMSP synthesis functions in cyanobacteria in Chilika Lagoon will have short- and long-term implications on ecology of brackish water lagoon. DNRA function in Cyanobacteria might be involved in nitrogen biogeochemical cycling under prolonged N-limitation and thus assists as alternate strategies for nitrogen acquisition. Furthur, it also possibly helps to proliferate during oxygen-deficient conditions (Kamp et al. 2011), which helps maintaining recycling of nutrients in the lagoon. DMSP is a crucial component of sulphur cycling which is synthesised by Proteobacteria, algae and diatoms. But there is no information available about cyanobacteria’s contribution to DMSP synthesis even though cyanobacteria are a major proportion of aquatic bacterial communities. Our findings reveal the new capability of Cyanobacteria in DMSP synthesis which might be helpful for nutrient cycling, thus supporting the stability of brackish lagoons.
Cyanobacteria are commonly found in the photic zones of aquatic ecosystems and often engage in symbiotic relationships with other microbes. These microorganisms play pivotal roles in both aquatic ecology and Earth’s evolutionary history. They have a crucial role in transitioning Earth from an anoxygenic to an oxygenic environment, fundamentally altering the course of evolution (Blankenship 2010). Furthermore, Cyanobacteria contributed significantly to the evolution of eukaryotes by endosymbiosis, providing them with the ability for oxygenic photosynthesis (Archibald 2015). Another noteworthy role is their ability to convert atmospheric nitrogen (N2) into bioavailable ammonia (NH3) or ammonium ions (NH4+). While nitrogen fixation is found in various bacterial phyla, Cyanobacteria in the ocean surface are presumed to make a significant contribution (Zehr 2011). Additionally, Cyanobacteria are key contributors to primary production in aquatic ecosystems. Globally, they have estimated global biomass in the order of 3 × 1014 g , or 1015 g wet biomass (Garcia-Pichel et al. 2009).
Conclusion
Our study revealed the presence of putative nirBD and dsyB-like genes in Cyanobacterial genomes, shedding light on their potential to engage in DNRA function and DMSP synthesis, respectively. While some Cyanobacterial species are known for Biological Nitrogen Fixation (BNF), research on other alternate pathways is limited. Certain Cyanobacterial species possess these alternate pathways for creating bioavailable ammonium in the ecosystem, which might be conferred with a competitive advantage. Additionally, we investigated the evolutionary history of these two genes, uncovering several significant implications. Notably, both vertical and horizontal gene transfers played a role in disseminating this function among Cyanobacteria in both cases, but certain species were frequently identified as donors in these events. Moreover, it seems that the dsyB-like gene was initially acquired by a Cyanobacterial ancestor and later transferred to other bacterial Phyla, but this needs further validation by incorporating more genomes from various bacterial taxa in analysis. These studies open new avenues for the research community to further define the overall contribution of such functions in biogeochemical cycling and other important processes facilitated by microorganisms and when are they chosen by them. This further reciprocates the roles of these microscopic creatures which go unnoticed but might be extremely helpful for ecosystem services. Future research should be directed to apply molecular assays to test the function of the target gene for laboratory validation. Furthermore, the integration of omics (meta-transcriptomics) along with evolutionary studies can further facilitate a more detailed understanding of these important functions.
Data Availability
The raw reads from the metagenomic sequencing are available in NCBI SRA under the BioProject accession PRJNA691704. The NCBI accession numbers for the cyanobacterial MAGs generated as a part of this study can be found in supplementary material S5.
References
Aramaki T, Blanc-Mathieu R, Endo H et al (2020) KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36:2251–2252. https://doi.org/10.1093/bioinformatics/btz859
Archibald JM (2015) Endosymbiosis and eukaryotic cell evolution. Curr Biol 25:R911–R921. https://doi.org/10.1016/j.cub.2015.07.055
Bansal MS, Kellis M, Kordi M, Kundu S (2018) RANGER-DTL 2.0: rigorous reconstruction of gene-family evolution by duplication, transfer and loss. Bioinformatics 34:3214–3216. https://doi.org/10.1093/bioinformatics/bty314
Bergman B, Gallon JR, Rai AN, Stal LJ (1997) N2 fixation by non-heterocystous cyanobacteria. FEMS Microbiol Rev 19:139–185. https://doi.org/10.1111/j.1574-6976.1997.tb0096.x
Blankenship RE (2010) Early Evolution of Photosynthesis. Plant Physiol 154:434–438. https://doi.org/10.1104/pp.110.161687
Blum M, Chang HY, Chuguransky S et al (2021) The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49:D344–D354. https://doi.org/10.1093/nar/gkaa977
Bornemann TLV, Esser SP, Stach TL et al (2023) uBin: A manual refining tool for genomes from metagenomes. Environ Microbiol 25:1077–1083. https://doi.org/10.1111/1462-2920.16351
Boyd JA, Woodcroft BJ, Tyson GW (2018) GraftM: a tool for scalable, phylogenetically informed classification of genes within metagenomes. Nucleic Acids Res 46:E59. https://doi.org/10.1093/NAR/GKY174
Bucciarelli E, Ridame C, Sunda WG et al (2013) Increased intracellular concentrations of DMSP and DMSO in iron-limited oceanic phytoplankton Thalassiosira oceanica and Trichodesmium erythraeum. Limnol Oceanogr 58:1667–1679. https://doi.org/10.4319/lo.2013.58.5.1667
Bushnell B (2014) BBTools software package. https://sourceforge.net/projects/bbmap
Carrit DE, Carpenter JH (1996) Recommendation procedure for Winkler analyses of sea water for dissolved oxygen. J Mar Res 24:313–318
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552. https://doi.org/10.1093/oxfordjournals.molbev.a026334
Curson ARJ, Liu J, Bermejo Martínez A et al (2017) Dimethylsulfoniopropionate biosynthesis in marine bacteria and identification of the key gene in this process. Nat Microbiol. https://doi.org/10.1038/nmicrobiol.2017.9
Curson ARJ, Williams BT, Pinchbeck BJ et al (2018) DSYB catalyses the key step of dimethylsulfoniopropionate biosynthesis in many phytoplankton. Nat Microbiol 3:430–439. https://doi.org/10.1038/s41564-018-0119-5
Gage DA, Rhodes D, Nolte KD, Hicks WA, Leustek T, Cooper AJ, Hanson AD (1997) A new route for synthesis of dimethylsulphoniopropionate in marine algae. Nature 387(6636):891–894. https://doi.org/10.1038/43160
Garcia-Pichel F, Belnap J, Neuer S, Schanz F (2009) Estimates of global cyanobacterial biomass and its distribution. Arch Hydrobiol Suppl Algol Stud 109:213–227. https://doi.org/10.1127/1864-1318/2003/0109-0213
Grasshoff K, Kremling K, Ehrhardt M (1999) Methods of seawater analysis. Wiley, New York, p 600
Hatakeyama S, Okuda M, Akimoto H (1982) Formation of sulfur dioxide and methanesulfonic acid in the photooxidation of dimethyl sulfide in the air. Geophys Res Lett 9(5):583–586. https://doi.org/10.1029/GL009i005p00583
Hatton AD, Shenoy DM, Hart MC et al (2012) Metabolism of DMSP, DMS and DMSO by the cultivable bacterial community associated with the DMSP-producing dinoflagellate Scrippsiella trochoidea. Biogeochemistry 110:131–146. https://doi.org/10.1007/s10533-012-9702-7
Huang X, Weisener CG, Ni J et al (2020) Nitrate assimilation, dissimilatory nitrate reduction to ammonium, and denitrification coexist in Pseudomonas putida Y-9 under aerobic conditions. Bioresour Technol 312:123597. https://doi.org/10.1016/j.biortech.2020.123597
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755. https://doi.org/10.1093/bioinformatics/17.8.754
Hug LA, Castelle CJ, Wrighton KC et al (2013) Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1:1–17. https://doi.org/10.1186/2049-2618-1-22
Hug LA, Baker BJ, Anantharaman K et al (2016) A new view of the tree of life. Nat Microbiol 1:1–6. https://doi.org/10.1038/nmicrobiol.2016.48
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ (2010) Integrated nr database in protein annotation system and its localization. Nat Commun 6:1–8
Kamp A, De Beer D, Nitsch JL et al (2011) Diatoms respire nitrate to survive dark and anoxic conditions. Proc Natl Acad Sci USA 108:5649–5654. https://doi.org/10.1073/pnas.1015744108
Kang DD, Li F, Kirton E et al (2019) MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019:1–13. https://doi.org/10.7717/peerj.7359
Kocsis MG, Nolte KD, Rhodes D et al (1998) Dimethylsulfoniopropionate biosynthesis in Spartina alterniflora: evidence that S-methylmethionine and dimethylsulfoniopropylamine are intermediates. Plant Physiol 117:273–281. https://doi.org/10.1104/pp.117.1.273
Lam P, Kuypers (2011) Microbial nitrogen cycling processes in oxygen minimum zones. Ann Rev Mar Sci 3:317–345. https://doi.org/10.1146/annurev-marine-120709-142814
Lanfear R, Frandsen PB, Wright AM et al (2017) Partitionfinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34:772–773. https://doi.org/10.1093/molbev/msw260
Liang Z, Keeley A (2013) Filtration recovery of extracellular DNA from environmental water samples. Environ Sci Technol 47:9324–9331. https://doi.org/10.1021/es401342b
Lovelock JE, Maggs RJ, Rasmussen RA (1972) Atmospheric dimethyl sulphide and the natural sulphur cycle. Nature 237(5356):452–453. https://doi.org/10.1038/237452a0
Manu S, Umapathy G (2023) Deep sequencing of extracellular eDNA enables total biodiversity assessment of ecosystems. Ecol Indic 156:111171. https://doi.org/10.1016/j.ecolind.2023.111171
Mohapatra M, Behera P, Kim JY, Rastogi G (2020) Seasonal and spatial dynamics of bacterioplankton communities in a brackish water coastal lagoon. Sci Total Environ 705:134729. https://doi.org/10.1016/j.scitotenv.2019.134729
Mohapatra M, Manu S, Kim JY, Rastogi G (2023) Distinct community assembly processes and habitat specialization driving the biogeographic patterns of abundant and rare bacterioplankton in a brackish coastal lagoon. Sci Total Environ 879:163109. https://doi.org/10.1016/j.scitotenv.2023.163109
Monchamp ME, Spaak P, Pomati F (2019) Long term diversity and distribution of non-photosynthetic cyanobacteria in peri-alpine lakes. Front Microbiol. https://doi.org/10.3389/fmicb.2018.03344
Moore KR, Magnabosco C, Momper L et al (2019) An expanded ribosomal phylogeny of cyanobacteria supports a deep placement of plastids. Front Microbiol 10:1–14. https://doi.org/10.3389/fmicb.2019.01612
Muduli PR, Pattnaik AK (2020) Spatio-temporal variation in physicochemical parameters of water in the Chilika Lagoon. In: Finlayson C, Rastogi G, Mishra D, Pattnaik A (eds) Ecology, conservation, and restoration of Chilika Lagoon, India. Wetlands: ecology, conservation and management, vol 6. Springer, Cham
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Parks DH, Imelfort M, Skennerton CT et al (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. https://doi.org/10.1101/gr.186072.114
Pessi IS, Popin RV, Durieu B et al (2023) Novel diversity of polar Cyanobacteria revealed by genome-resolved metagenomics. Microb Genomics 9:1–14. https://doi.org/10.1099/mgen.0.001056
Pfreundt U, Stal LJ, Voß B, Hess WR (2012) Dinitrogen fixation in a unicellular chlorophyll d-containing cyanobacterium. ISME J 6:1367–1377. https://doi.org/10.1038/ismej.2011.199
Pisciotta JM, Zou Y, Baskakov IV (2010) Light-dependent electrogenic activity of cyanobacteria. PLoS ONE. https://doi.org/10.1371/journal.pone.0010821
Reji L, Francis CA (2020) Metagenome-assembled genomes reveal unique metabolic adaptations of a basal marine Thaumarchaeota lineage. ISME J 14:2105–2115. https://doi.org/10.1038/s41396-020-0675-6
Sanchez-Baracaldo P, Bianchini G, Wilson J, Knoll A (2021) Cyanobacteria and biogeochemical cycles through Earth history. Trends Microbiol. https://doi.org/10.1016/j.tim.2021.05.008
Shang H, Rothman DH, Fournier GP (2022) Oxidative metabolisms catalyzed Earth’s oxygenation. Nat Commun. https://doi.org/10.1038/s41467-022-28996-0
Shi T, Falkowski PG (2008) Genome evolution in cyanobacteria: the stable core and the variable shell. Proc Natl Acad Sci USA 105:2510–2515. https://doi.org/10.1073/pnas.0711165105
Srichandan S, Kim JY, Kumar A et al (2015) Interannual and cyclone-driven variability in phytoplankton communities of a tropical coastal lagoon. Mar Pollut Bull 101:39–52. https://doi.org/10.1016/j.marpolbul.2015.11.030
Stal LJ, Paerl HW, Bebout B, Villbrandt M (1994) Heterocystous versus non-heterocystous cyanobacteria in microbial mats. Microb Mats. https://doi.org/10.1007/978-3-642-78991-5_41
Stefels J (2000) Physiological aspects of the production and conversion of DMSP in marine algae and higher plants. J Sea Res 43:183–197. https://doi.org/10.1016/S1385-1101(00)00030-7
Sunda W, Kieber DJ, Kiene RP, Huntsman S (2002) An antioxidant function for DMSP and DMS in marine algae. Nature 418(6895):317–320. https://doi.org/10.1038/nature00851
Taberlet P, Coissac E, Pompanon F et al (2012) Towards next-generation biodiversity assessment using DNA metabarcoding-TABERLET-2012-Molecular Ecology-Wiley Online Library. Mol Ecol 21:2045–2050
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38:3022–3027. https://doi.org/10.1093/molbev/msab120
Tarafdar L, Kim JY, Srichandan S et al (2021) Responses of phytoplankton community structure and association to variability in environmental drivers in a tropical coastal lagoon. Sci Total Environ 783:146873. https://doi.org/10.1016/j.scitotenv.2021.146873
Tee HS, Waite D, Payne L et al (2020) Tools for successful proliferation: diverse strategies of nutrient acquisition by a benthic cyanobacterium. ISME J 14:2164–2178. https://doi.org/10.1038/s41396-020-0676-5
Teeling H, Glöckner FO (2012) Current opportunities and challenges in microbial metagenome analysis-a bioinformatic perspective. Brief Bioinform 13:728–742. https://doi.org/10.1093/bib/bbs039
Thomas AC, Howard J, Nguyen PL et al (2018) ANDeTM: a fully integrated environmental DNA sampling system. Methods Ecol Evol 9:1379–1385. https://doi.org/10.1111/2041-210X.12994
Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ (2016) W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res 44:W232–W235. https://doi.org/10.1093/NAR/GKW256
Uchida A, Ooguri T, Ishida T, Kitaguchi H, Ishida Y (1996) Biosynthesis of dimethylsulfopropionate in Crypthecodinium cohnii (Dinophyceae). Biological and Environmental Chemistry of DMSP and related Sulfonium Compounds. Springer, Boston, MA, p 97–107
Vairavamurthy A, Andreae MO, Iverson RL (1985) Biosynthesis of dimethylsulfide and dimethylpropiothetin by Hymenomonas carterae in relation to sulfur source and salinity variations. Limnol Oceanogr 30:59–70. https://doi.org/10.4319/lo.1985.30.1.0059
Vargas A, Strohl WR (1985) Utilization of nitrate by Beggiatoa alba. Arch Microbiol 142:279–284. https://doi.org/10.1007/BF00693404
Waglechner N, McArthur AG, Wright GD (2019) Phylogenetic reconciliation reveals the natural history of glycopeptide antibiotic biosynthesis and resistance. Nat Microbiol 4:1862–1871. https://doi.org/10.1038/s41564-019-0531-5
Waterbury JB (2006) The Cyanobacteria—Isolation, purification and identification. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (eds), The Prokaryotes. Springer, New York. https://doi.org/10.1007/0-387-30744-3_38
Yarza P, Yilmaz P, Pruesse E et al (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635–645. https://doi.org/10.1038/nrmicro3330
Zehr JP (2011) Nitrogen fixation by marine cyanobacteria. Trends Microbiol 19:162–173. https://doi.org/10.1016/j.tim.2010.12.004
Acknowledgements
We acknowledge the support received by G.U. from the Department of Biotechnology (DBT), Govt. of India, vide Grant No. BT/PR29032/FCB/125/4/2018 for this study which also included funding of PhD fellowship for M.R. S.M. was supported by a BINC fellowship from DBT, Govt. of India. We thank Chilika Development Authority, Balugaon, Odisha for providing logistics for sample collection and laboratory facility for initial processing of sample. We also acknowledge Dr. Mihir Trivedi for his constructive suggestions during phylogenetic analysis.
Funding
Funding was provided by Department of Biotechnology, Ministry of Science and Technology, India (Grant No. BT/PR29032/FCB/125/4/2018).
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: John Bracht.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ray, M., Manu, S., Rastogi, G. et al. Cyanobacterial Genomes from a Brackish Coastal Lagoon Reveal Potential for Novel Biogeochemical Functions and Their Evolution. J Mol Evol 92, 121–137 (2024). https://doi.org/10.1007/s00239-024-10159-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-024-10159-y