
Abstract
Genes duplicate, mutate, recombine, fuse or fission to produce new genes, or when genes are formed from de novo, novel functions arise during evolution. Researchers have tried to quantify the causes of these molecular diversification processes to know how these genes increase molecular complexity over a period of time, for instance protein domain organization. In contrast to global sequence similarity, protein domain architectures can capture key structural and functional characteristics, making them better proxies for describing functional equivalence. In Prokaryotes and eukaryotes it has proven that, domain designs are retained over significant evolutionary distances. Protein domain architectures are now being utilized to categorize and distinguish evolutionarily related proteins and find homologs among species that are evolutionarily distant from one another. Additionally, structural information stored in domain structures has accelerated homology identification and sequence search methods. Tools for functional protein annotation have been developed to discover, protein domain content, domain order, domain recurrence, and domain position as all these contribute to the prediction of protein functional accuracy. In this review, an attempt is made to summarise facts and speculations regarding the use of protein domain architecture and modularity to identify possible therapeutic targets among cellular activities based on the understanding their linked biological processes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A protein family is typically defined as a collection of protein sequences that have similar evolutionary development, which imply that they have considerable similarity among themselves. Though they might share conserved sections throughout the sequence, the proteins may not necessarily be conserved throughout their entire lengths. These domains or mobile elements normally comprise distinct functional units and are found either alone or in conjunction with other conserved regions. Generally, domains are mobile, free to migrate, and able to pair with other domains, however, domains remain as independent evolutionary elements that typically have their roles conserved over time (Mulder 2010). In a protein, function of a domain may vary, but the overall fold remains the same (Basu et al. 2009). Domain architecture of a protein is defined by the arrangement of these domains within its sequence. As a result of their likely comparable structures, proteins with the same domain architecture maintain the same cellular function (Koonin et al. 2002). It is presumed that, new functions for the organisms might have emerged from the formation of proteins with novel domains and/or domain combinations, and believed to be a key mechanism of evolution (Itoh et al. 2007; Peisajovich et al. 2010). The number of domain combinations can vary greatly, contributing to the dissimilarity of domain repertoires in various organisms. As a result, the diversity of domain structures vary greatly, and influenced by the complexity and way of life of the organisms (Tordai et al. 2005).
In recent years, the avalanche of genomic sequence data has allowed genome-scale comparison analysis to unearth several exciting protein domain architecture (PDA) discoveries (Hsu et al. 2013). It is well known that (a) Even though the PDAs originate from a single ancestral architecture comprising highly conserved two domains, such structures were diversified based on various mechanisms involved indomain duplication, fusion and insertion/deletions. Further, PDAs with multiple origins appear to occur more frequently than mentioned above (Bashton and Chothia 2002; Gough 2005; Kummerfeld and Teichmann 2009; Forslund et al. 2008); (b) gains and losses of the domain in non-repeat PDAs tend to occur at a terminal part rather than within the internal part of the architecture (Bjorklund et al. 2005; Weiner et al. 2006); (c) In conjunction with exponential growth of sequence data, few new single domain architectures are being discovered, concomitantly novel and species-specific multiple-domain architectures are continuing to be identified, leading to the notion that PDA is a major means by which protein functional and species diversity evolves (Levitt 2009), and (d) Some domains have a propensity to promiscuously combine with other domain families to form a variety of PDAs, such as those domains involved in signal transduction (Basu et al. 2008).
Proteins with the same structural fold frequently diverge to very low levels of sequence identity (Rost 1997). These findings imply that the amount of protein sequence divergence is only moderately constrained by the conservation of protein fold, or the general layout and topological linkages of protein secondary structures (Murzin et al. 1995). Unlike protein folds, certain molecular activities are preserved, which might dramatically reduce the long-term divergence of protein orthologs (Konate et al. 2019). However, it is noteworthy that structural similarities could also be the arte fact of convergent evolution contemplated to be a very uncommon phenomenon in terms of protein structure and is mainly limited to examples like a catalytic triad, where three residues might adopt a similar conformation (Gough 2005). Nevertheless, the global structures that enable that arrangement are distinct. The structural classification of proteins (SCOP and CATH) are the two protein structure categorization methods with significant overlap, but some differences capture the similarity (Lo Conte et al. 2000; Knudsen et al. 1994). The other resources like DaliLite (Holm et al. 2019), TM-align (Zhang & Skolnick 2005), MADOKA (Deng et al. 2019), SPalignNS (Brown et al. 2016), and ZEAL (Ljung and Andre 2021). DaliLite and TM-align are conventional, commonly used structure alignment methods, while MADOKA and SPalignNS are more recent methods. Further, a neural network that retrieves proteins with the same fold more precisely than through direct comparison of 3DZDs (3D Zernike descriptors, which is a rotationally invariant, mathematical representation of 3D shape) by using the 3DZDs of the proteins as input. We describe the structure classifications of AlphaFold2 models using 3D-AF-Surfer and talk about the relationship between the models’ degrees of confidence and intrinsically disordered regions (Aderinwale et al. 2022).
These resources offer a masterclass in protein structure space comprehension, while both resources can lead to similar results. Protein families have an apparent evolutionary relationship at the sequence level, whereas super families only have a clear evolutionary relationship based on structural similarity (Gough 2005). Most super families are pretty flexible regarding the spatial position of their functional sites, which is especially true for protein–protein interactions. Catalytic sites are generally found in a few topological locations, albeit limited; the small-ligand binding sites are more diverse than protein–protein interfaces (Dessailly et al. 2013).Each protein family member reuses one or more domains to give an extensive range of functional variation from a presumably small number of domains identified (Weiner et al. 2008). A domain gives a more acceptable level of granularity than fold superfamilies, which are coarse structural measures (Sillitoe et al. 2015). Domains are swapped, added, and taken away to generate a complex repertoire of proteins with various functions as they are compact autonomous folding units considered evolutionary units (Mascarenhas and Gosavi 2017). As an example, consider the P-loop containing nucleoside triphosphate hydrolases superfamily, the most common protein fold that constitutes up to 18% of all gene products in a cell (Daria et al. 2018; Kozlova et al. 2022). According to the superfamily, it appears 1034 times in the human genome. Because they share this domain, all proteins have a similar evolutionary ancestor, yet they serve different purposes (Bourne et al. 2010). Several mechanisms contributed to the evolution of functional differences among these proteins (Sikosek and Chan 2014). An individual domain’s function can alter, but its functional context also influences it. To understand functional evolution of these proteins one must examine the history of their domains (Toll-Rieraand and Albà 2013).
In a multi-domain protein of the superkingdom, two-thirds of prokaryote proteins have two or more domains, whereas of eukaryotes have two or more domains (Teichmann et al. 1998; Gerstein 1998). A study conducted across 40 genomes reveals, a power law was observed for domain combinations, with some two-domain or three-domain combinations known as “supra-domains” (Apic et al. 2001; Vogel et al. 2004a, b). Vogel et al. (2004a, b) evaluated domain combinations in the proteins of 131 complete genomes of species using structural domain assignments. They discovered two-domain and three-domain combinations that frequently occur within various protein settings. These combinations of domains have a specific spatial and functional relationship, hey are referred to as “supra-domains” as they are bigger than individual domains. Over one-third of all structurally assigned multi-domain proteins contain these over-represented supra-domains. Domain combinations appear to be stochastic processes followed by duplication, according to a simulation of domain duplication and combination processes. Gene fusion occurs more frequently than gene fission during domain evolution. According to a recent study, protein domain architecture and the number of protein domains is related to the organism’s complexity, that is measured by the number of cell types that organism contains (Vogel et al. 2004a, b). This information is helpful for systems biology and understanding the dynamic management of networks in which these proteins engage could be aided by knowing about any limitations on protein interactions.
In many human diseases, drug target identification is a rapidly expanding area of research. In the post-genomic era, numerous methods have been developed to find new therapeutic targets for infectious diseases. Exon/ORF shuffling in a genome is frequently discovered by analysis of protein sequences from various organisms. These are known as Rosetta stone sequences and lead to the fusion of proteins or domains, either in the same genome or that of another organism. They provide a bridge between different proteins by describing regional and global interactions among proteomes. Proteins’ functional roles are mostly determined by interactions between their domains, which results in the corresponding signalling pathway. Potential therapeutic targets can be found by re-annotating proteins’ functional roles using domain-based techniques. This innovative method of identifying drug targets using domain-based putative protein re-annotation offers a chance to confirm the suggested therapeutic target’s usefulness as a typical protein targeting both pneumonia-causing species investigated in our recent work (Ramesh et al. 2021).
In this review, an attempt is made to summarise information and hypotheses related to exploiting protein domain architecture and modularity to identify potential therapeutic targets among cellular activities based on understanding their related biological processes.
Promiscuity or Versatility of Domains
A domain can be either a single-domain or multi-domain in a protein component that acts as an evolutionary structural unit (Doolittle and Bork 1993; Doolittle 1995). A domain can function independently or in association with other domains as a multi-domain protein (Vogel et al. 2004a). Even today, the concept of “domain mobility”, which refers to a domain’s capacity to produce a wide range of combinations, is still employed with intron-wise modular recombination (Tordai et al. 2005). Events like protein fusion or the loss of terminal protein fragments are more likely to result in novel combinations than domain transfers from one protein to another, termed “domain versatility” or “domain promiscuity”. Domains differ in their tendency to appear in variable multi-domain contexts, with some being “promiscuous”, i.e., combining with many other domains (Marcotte et al. 1999). Understanding the interactions of domains to form different combinations is crucial for exploring the evolution of proteins (Amoutzias et al. 2004a, b; Przytycka et al. 2006; Amoutzias et al. 2007), genomic comparisons (Ye and Godzik 2004), and even finding out direct medical impact (Lucas et al. 2006).
Existing domain combinations result from selected variables that help them survive in a population and rely on the mechanisms that determine them (Buljan and Bateman 2009). We can comprehend protein evolution as a modular phenomenon by investigating domain structures (Forslund and Sonnhammer 2012). Analysis of domain assignments to genomes has revealed their importance in building the protein repertoire and their role in combinations (Christine et al. 2004). Single-domain features are comparable to those observed in combinations of two or more domains. For example, a few two-domain combinations are adaptable and may be found with various extra domains. Despite this, most two-domain combinations are found in only one or two protein settings. Signal transduction is an important example of the repetition of specific domains and domain combinations (Ersfeld et al. 2005).
Certain domains appear to be proclivein various domain architectures and can be classified as “promiscuous”. These promiscuous domains are frequently found in protein–protein interactions and play an important role in interaction networks, especially those contributing to signal transduction (Basu et al. 2008). Connecting signal transduction network components through specific protein–protein interactions and conveying effectors to the sites of action, chromatin, are two primary modes of action for promiscuous domains.With as many as 654 distinct arrangements in the Swiss Pfam database alone, the SH3–SH2 domain pair, for instance, can be found in a wide range of signal transduction proteins. This recombination flexibility classifies the SH3-SH2 domain pair as a “supra-domain”. Supra-domains are two- or three-domain combinations with various N- and C-terminal neighbors and can be found in diverse domain architecture (Fig. 1). Furthermore, increasing the complexity of the domain structure of proteins will considerably contribute to the development of organism complexity because of the increased capacity for protein–protein interactions and the formation of signal transduction pathways.

An overview of the various features of multidomain protein evolution and the function. a Domain superfamily diversity and b their function in the duplication and recombination-mediated synthesis of multidomain proteins Supra-domains are two- or three-domain combinations that appear in various domain designs with various N–C–terminal neighbours. c The domains contained inside these combinations, including their shape and functional linkages. d Rectangles of the same colour are used to symbolise domains that belong to the same superfamily
Several thousand different combinations of two super families can be found in the proteins of more than 100 different organisms. However, this is significantly fewer (less than 0.5%) than expected, given the total number of super families or multi-domain proteins per proteome (Apic et al. 2001). If membrane proteins are included, this number is expected to decrease further. The small number of domain combinations seen in proteins suggests that all of them have been subjected to intense selection. While a few domain super families are highly versatile and have domains from many super families as neighbours, most super families are not. The number of partner super families per superfamily is distributed according to a power law (Muller et al. 2002; Park et al. 2001). Each domain superfamily has its unique story to answer despite these general rules. Some super families are extremely versatile, while others are highly abundant and yet others are both (Christine et al. 2004).
Several domain promiscuity measures are available, as seen below.
Sl. No. | Domain promiscuity measure | Description | References |
|---|---|---|---|
01 | Co-occurrence (NCO) | The number of domains appears at least once in the same proteins as the selected domain. Using this method, domains that regularly co-occur in the same local context, i.e., have the exact neighbouring domains, demonstrate higher promiscuity. This is because co-occurring domains near the domain of interest differ from co-occurring domains located elsewhere in the protein | Ekman et al. (2005) |
02 | Number of neighbours (NN) | The number of immediate neighbours for a given domain is determined | Apic et al. (2003) |
03 | Number of triplets (NTRP) | There are a lot of distinct combinations X—A—Y for a given domain A, where X and Y are domains or N- or C-termini | Tordai et al. (2005) |
04 | Weighted bigram frequency index \(({\pi }_{i}={\beta }_{i}X log \left(\frac{{\beta }_{i}}{{f}_{i}}\right))\) | The weighted bigram frequency was used to quantify the measure of domain promiscuity. The premise behind this weighting scheme is that multi-domain protein formation is a random process | Basu et al. (2008) |
05 | Domain versatility index (DV I) | In terms of the quantity of occurrences, the Domain Versatility Index (DV I) gauges how closely a domain is related to its neighbours. It is derived using the logarithmic regression of NN over N, with DV I used as the linear coefficient | January Weiner 3rd et al. (2008) |
06 | Poisson distribution | The Poisson distribution is assumed to follow the predicted frequency of domain combinations in a “random” genome. A mixture of two or more Poisson distributions can depict a considerable divergence from a single Poisson distribution. The frequency distributions of domain combinations were analysed using the C.A.MAN program (Bohning et al. 1998) | January Weiner 3rd et al. (2008) |
The promiscuous domain can be estimated using the weighted bigram frequency, which is derived from the Kullback–Leibler information gain formula,
where \({\beta }_{i}\) is the bigram frequency,
where \(t\), is the number of distinct domain types, \({T}_{i}\) is the number of unique domain neighbors of domain \(i\), and \({f}_{i}\) is the frequency of domain \(i\) in the genome, calculated as \(\frac{{n}_{i}}{N}\), where \({n}_{i}\) is the total count of domain \(i\), and \(N\) is the total number of domains detected in the given genome:
Recently, our work used a domain-basedbioinformatics method to re-annotate the proteins using position-specific iterations to discover the probable proteins that are potential therapeutic targets for pneumonia infection (Ramesh et al. 2021). Comparative rosetta stone based domain analysis enabled us to identify the GFO IDH MocA domain in a protein with high abundance and versatility. The protein with accession number EJJ83173 was found to be druggable protein as confirmed by the domain based integrated bioinformatics analysis. Several ligands that are inhibitory to the protein have been evaluated through virtual screening, using the model. These ligands are constituents of commercially available medications, and are also mentioned in traditional Chinese medicine (Ramesh et al. 2021).
Size of the Domain Family’s Distribution
Genome sequencing technology has furnished many proteomes available for thorough analyses of the distributions and evolution of protein domains and domain architectures using various bioinformatics tools. The initial research (Huynen and van Nimwegen 1998; Qian et al. 2001; Luscombe et al. 2002) focused on the total number of copies of a protein family within a species irrespective of kind of domains present. Findings of these early studies appear to hold true for domains, supra-domains and for domain architectures (Apic et al. 2001, 2003; Vogel et al. 2004a, b). Further, these findings show a dominance of the population in a limited number of proteins (Luscombe et al. 2002), i.e., only a small number of domain families occur in most proteins, but with large number of domain families that are present in a majority of proteins (Forslund et al. 2019).
Domain Co-occurrence Networks
Network biology techniques, which focus on biological network structure, dynamics, and functions, are one promising strategy for analyzing the complex connections between biological molecules. Protein domain co-occurrence networks (DCN) were constructed by Wang et al. (2011) to study protein function and interaction at the proteome level. There are numerous methods for determining these interactions and quantifying the degree of co-occurrence. All domains on the same amino acid chain are most typically connected. The co-occurrence of various domains in a given protein is used to construct these networks. Each domain usually serves a different biological function. Domain families/types are groups of domains with similar sequences, structures, and functions. Thousands of domain types are frequently represented in a species’ proteome, which can be found as simple-domain or combined to form multi-domain proteins. Domain combinations are crucial for enhancing a proteome’s complexity in addition to sequential divergence. Instead of inventing new proteins de novo, nature prefers to reuse and recombine existing domains (Fong et al. 2007; Kummerfeld and Teichmann 2009).
Domain co-occurrence networks (DCNs) can capture the intense, permanent and definite relationship between domains. A DCN is a graph that depicts all species as nodes and protein domain types as edges. When the two domain types (nodes) coexist in the same protein, and an edge connects them (Wuchty and Almaas 2005). The DCN has the following advantages over the protein–protein interaction network:
Accurate and Reliable
Sequential analysis-based domain co-existence relationships are almost 100% accurate, significantly more so than experimental methods like the yeast-two-hybrid system, which predict protein interactions.
Higher Coverage
A DCN constructed using homologous sequence analysis can frequently recall around 70% of the domain’s co-existence associations, in contrast to the limited coverage of protein interaction networks in most species. DCN-based assessments are frequently more reliable (Ekman et al. 2005).
Easy to Construct
It is significantly easier to create a genome’s DCN by comparing the protein sequences of well-known protein domain databases such as Pfam (Sonnhamma et al. 1998) and ProDoM (Servant et al. 2002) than it is to do so by utilizing experimental or computational approaches to generate protein interaction networks.
Additionally, rather than representing a sequence, each node in a DCN represents a domain type. DCNs, which share a lot of similar domain types, make it simpler to compare or match them between different species than sequence-based networks like PPI networks. Additionally, a DCN edge represents a permanent combination relationship stronger than the relationship in the PPI network.
Domain Versatility in Regulatory Networks
Domains have an important role in mediating protein–protein interactions, as previously stated. Developing higher-level organizational units like complexes and the evolution of organisms generally requires domain versatility. Protein–protein interactions are primarily responsible for these complexes in transcription and signalling networks. The increasing multicellular organisms’ complexity cannot be explained just by accumulating more genes. The increasing complexity of regulatory networks and network rewiring is the primary reason. For example, the proportion of transcription factors in an organism increases as the organism’s organization increases. Individual genes may be regulated by multiple transcription factors, individually or collectively, and each transcription factor can control many genes (Levine and Tjian 2003). The average number of transcription factors that regulate a gene is higher in more complex organisms. Modularly restructuring the factor’s components, such as proteins with domains that interact with DNA or other proteins, is required to rewire transcriptional circuits (Nimwegen 2006).
According to the lineage, a small, old core set of DNA binding domains that have been reused in different combinations and frequencies is assumed to have given rise to the DNA binding domains of many organisms (Babu and Teichmann 2003; Perez-Rueda and Collado-Vides 2000; Aravind and Koonin 1999). PPIs, such as those found in transcription factor complexes, develop and are dependent on domain combinatorial rearrangements. According to a structural study, the number of domain interactions between proteins, like domain combinations inside proteins, is limited (Park et al. 2001; Russell et al. 2004).The interaction network has been examined and merged with the investigation of three families of eukaryotic transcription factors, including bHLH, NR, and bZIP proteins (Amoutzias et al. 2004a, b). Single-gene duplications were thought to have evolved from homo-dimerizing to hetero-dimerizing proteins, conserving homodimeric contacts while accumulating homologous heterodimeric interactions to the bHLH interaction network (Amoutzias et al. 2004a, b).
A hub-like (star-shaped) network arose due to this interaction, as demonstrated in Fig. 2. Furthermore, networks are primarily isolated because most contacts occur within one family. This family-specific interaction pattern is linked to alternate dimerization domains unique to each family. The modular domain rearrangement has direct consequences for creating new networks, at least in this context such configurations have repeatedly been proven to have minor but critical structural influences on dimerization specificities. The nuclear receptor (NR) and bZIP protein families have similar results (Bornberg-Bauer et al. 2005).

Network of interactions amongst the basic helix-loop-helix (bHLH) group transcription factors. Proteins that can only homodimerize are shown by ovals; proteins that can only heterodimerize by boxes; and proteins that already interact with one another are represented by edges. Different bHLH families are represented by different colours. Under the respective family name, the domain arrangements of the various bHLH families are demonstrated (Courtesy from Amoutzias et al. 2004a, b)
Mechanisms for Domain Architecture Evolution
The propensities of different protein-domain families to combine with other domains vary; some produce a small number of combinations, while others form a considerable number (Apic et al. 2001). Given that a protein domain typically has a length of 120 amino acids, there are significant alterations in domain architecture at the gene level (Buljan and Bateman 2009). Understanding protein evolution requires empathy for how multi-domain proteins gain, lose and rearrange domains. In recent years, with the availability of many fully sequenced genomes significant progress has been made in understanding the evolution of domain architectures in multi-domain proteins(Marsh and Teichmann 2010; Forslund et al. 2019; Thomas et al. 2020).Molecular mechanisms such as gene fusion and fission, exon shuffling (van Rijk and Bloemendal 2003; Liu and Grigoriev 2004)via intron recombination, alternative gene splicing, and retro positioning all these direct the domain rearrangement process. It has been elucidated that, the composition of protein domains in prokaryotes has been shown to shift due to gene fusion and fission (Liu and Grigoriev 2004). Yet, over time, a protein domain’s function is frequently conserved and can even be changed or modified. If a domain sequence with a divergent structure and/or function has resulted through mutations, deletions, and insertions, the new domain will have different properties from the original. However, even minor alterations in the domain sequence, such as point mutations would lead to significant impact on a protein domain’s activity and, protein’s overall function may get altered (Bhattacharya et al. 2017).
For example, amino acids are generally highly conserved in the active site of an enzyme, and mutations in them can ultimately degenerate the enzyme’s activity. It has been discovered that changes in the active-site residues lead to catalytically inactive versions or with the potential for novel activities which sometimes may be other than the catalysis itself. Enzymes have evolved to catalyse numerous processes on the same structural scaffold thanks to mutations in their catalytic domains, which can adapt the enzyme’s specificity to a different substrate (Buljan and Bateman 2009). The proteins with similar domain architectures were assumed to be homologous, and discrepancies in domain composition were sought. One of the major discoveries is that most evolutionary changes to multi-domain protein structures at the amino and carboxyl termini can be attributed to new domain insertions, domain repetitions, and domain deletions. All observed alterations can be classified as domain deletions because they occur more frequently than domain insertions.
However, domain design changes tend to occur more frequently after gene duplication. Due to the likelihood that domain insertion and deletion at any point in a protein would affect the structure of the present domain, this demonstrates the bias towards protein termini. The key molecular processes that contribute to domain gain and loss occur at the termini, such as gene fusion and fission, as well as the insertion of new start and stop codons. These processes also dictate the location of domain gain and loss. Multi-domain protein synthesis in bacteria is facilitated by gene fusion. However, eukaryotes’ domain gain processes are more varied due to their complex exon–intron gene architectures. Gene fusion is also necessary for eukaryotes, but it typically does not entail the direct assembly of exons from nearby genes. Instead, due to altered splicing patterns, a fused gene is produced from the still-separated exons (Fig. 3a). Dohmen et al. 2020 used maximum parsimony to show that the rate of gene fusion in archaebacteria, and eukaryotes appears to be 4 times greater than the rate of gene fission. A unique domain gain method extends an exon into a non-coding region, as seen in Fig. 3b (Buljan et al. 2010). Further, Dohmen et al. (2020) demonstrated that domain modularity is important for evolutionary innovation. The recombination of existing units to form larger complexes with new functionalities exempts the need to create novel elements from scratch. It has also been demonstrated that domain rearrangements are based on a canonical set of mutational events with rates within a relatively narrow and consistent range (Dohmen et al. 2020).

Protein domain gains could be caused in a variety of ways. Exons are represented by colored blocks, with blue, orange, and green denoting exons coding for various domains. Introns are shown by solid black lines, and intergenic regions are shown by red lines. a Fusion of genes. In order for the first gene’s exons to be spliced into the second gene, the non-coding region between the two genes is altered. b Exon expansion. Following an exon, a non-coding region is incorporated into the exon and codes for a new domain. Exon recombination (c). Exons from two genes are linked together directly. Intron recombination is (d). A gene’s intron is altered by the insertion of an exon from another gene. e Repositioning. An mRNA intermediate is used by a retrotransposon sequence (RT, purple) to mediate the copying of both itself and a nearby gene region, which is then followed by insertion into another gene (Color figure online)
Recombination is another technique for creating protein domains. For instance, there may be direct connections through sequence exchange or sharing between the exons of two distinct genes as illustrated in Fig. 3c (Patthy 1999). It is possible to introduce exons from one gene into an intron and vice versa (Fig. 3d). In complex eukaryotes, one of the key drivers of domain architectural variety is intronic recombination, often known as exon shuffling (Patthy 1999).Strong correlations between domain-exon borders and the fact that symmetric phase introns flank most domain-corresponding exons indicate that intron recombination contributes significantly to domain rearrangements (that is, introns are inserted at the same positions concerning codon triplets) (Liu and Grigoriev 2004).
Retrotransposons, which can duplicate and insert themselves into different chromosomal sites, are another probable source of protein domain gain (Fig. 3e). An inserted region will be absent of the gene’s introns because an mRNA intermediate achieves retroposition. By using the maximum parsimony method, it was possible to identify the evolutionary scenario that would result in the fewest gain or loss events and estimate the domain composition of ancestral proteins (Marija Buljan et al. 2009). It also shows that domain gains and losses follow the same pattern of alterations, with changes occurring at the protein’s termini rather than in the centre. According to experts, the pattern results from a complicated interaction between processes that add and remove domains from protein termini and selective forces that favour gains and losses of domains within a protein, rather than being solely explained by the causal mechanisms. It is reasonable to assume that adding or removing domains at protein termini will have a less significant effect on the rest of the structure because these regions are frequently charged, flexible, and located on the surface of proteins, especially if the domains in question are structurally independent.
Animal genes have been reported to acquire domains in the extracellular or cellular control mechanisms, such as signal transduction or DNA binding. The first domains include the EGF (epidermal growth factor), immunoglobulin superfamily, and the CUB (complement protein subcomponents, in addition to the leucine-rich repeat protein family, other domains include the C1r/C1s, urchin embryonic growth factor, and BMP-1. The PH (pleckstrin homology) domain, leucine-rich repeat, SH3 (Src homology 3), and zinc finger (C2H2 type) domains are examples of domains in the second group (Buljan and Bateman 2009; Marsh and Teichmann 2010). Several approaches through mutations have been used to investigate de novo domain formation (Forslund et al. 2019). These strategies provide a simple framework and explain most protein domain gains. There is still much to learn about the various genetic processes at molecular level, that would certainly lead to reconnoitre the diversity of multi-domain protein structures in complex eukaryotes.
Assessing How Different Domain Gain Strategies Contribute
Various molecular genetic pathways have contributed to the current understanding diversity of multi-domain protein structures in complex eukaryotes, over the amino acid level to facilitate multi-domain protein evolution (Buljan et al. 2010). The vital idea is to analyse the relative contributions of various domain-gain strategies to ensure they leave various genomic traces. A domain produced from a retrotransposon should only have one exon since the retrotransposon replicates through an intermediate translated mRNA. Therefore, it is improbable that retroposition led to the emergence of domains with many exons. For instance, exon recombination and gene fusion are considerably more likely to occur near protein termini than intron recombination, that occurs only in the middle of a protein. The location of the obtained domain therefore could be utilized to ascertain which mechanisms are most likely would lead to domain gain. Homologs within the genomes have been searched to identify potential ‘donor’ genes for all gained domains (Marsh and Teichmann 2010). This provides information on whether gene duplication preceded domain gains and can identify potential source genes for retroposition (Buljan et al. 2010). Most domain gains, or 71% of the total, were discovered to occur at the amino- or carboxyl termini of proteins. Several exons were implicated in the majority of these additions. Gene fusion is the only explanation for the 32% increase at termini that covers several exons. In addition, gene fusion is probably responsible for most of the remaining 39% increase at termini; however, additional pathways cannot be ruled out in these conditions. These facts imply that gene fusion is required for mammals to acquire domains (Buljan et al. 2010). However according to Gough (2005), most domain increases in animal proteins appear to be driven directly by gene fusion rather than duplication and recombination. Retroposition and intron recombination seem to have played a minor part in recent evolutionary history. In the near future, the number of fully sequenced genomes may drastically rise due to major recent advances in next-generation sequencing technologies (Marsh and Teichmann 2010).
The domain-oriented approach allows the detection of discordant domain orthologs. Cases where different domains on the same protein have different evolutionary histories. In addition to domain-level analysis, protein level orthology based on the fraction of domains that are orthologous can be inferred. Domainoid orthology assignments were compared to those yielded by the conventional full-length approach InParanoid, and were validated in a standard benchmark (Persson et al. 2019).
Convergent Evolution of Domain
Understanding the dynamics of domain architectural evolution is crucial to exploring how proteins function. Rearrangement and duplication events have produced complex architectures throughout evolution. It is fascinating to consider how often a particular design has been created, is it a form of convergent evolution or domain architectural reinvention? (Forslund et al. 2008). According to Gough (2005), convergent evolution refers to a sequence of independent evolutionary processes (recombination) that produce the same domain organization in numerous genomes.
A novel domain tree-based method that uses maximum parsimony to identify ancestor protein architectures was used to examine the incidence of domain architecture reinvention in 96 genomes. The creation of domain architectures was done using Pfam. Forslund et al. (2008) employed bootstrapping trees to ensure robustness and only selected results with solid statistical support. Various aspects of domain architecture evolution like gene fusion, fission and circular permutations were examined by several studies (Björklund et al. 2005; Bornberg-Bauer et al. 2005). The occurrence of numerous independent domain architectural invention events, on the other hand, has received little attention.
To assess the degree to which the domain architectures observed in the genomes result from functional necessity or evolutionary descent. The evolution of domain architectures across 62 genomes of known phylogeny, including all kingdoms of life, was observed for the possibility of convergent evolution. Numerous approaches, including phylogenetic grouping, sequence similarity/alignment, mutation rates, and comparative genomics, were used to approach this challenging topic from various angles (Gough 2005). Convergent evolution appears to result from random events with no apparent functional or structural preferences. Even though the sample size is modest, variations in the number of tandem repeat domains happen more frequently than variations in domain composition.
Domain Associations Conservation and Variation
The number of distinct neighbours for each domain family is distributed according to a power law. Only one N-to-C-terminal order is present for each domain pair; however, between 5 and 10% of domain pairings do so. In addition, domain combination and duplication is important because it increases the likelihood that new domain combinations will arise from duplications of already-existing combinations rather than recombination. As a result, domain combinations are conserved at numerous levels in proteins. There are fewer potential forms of domain pairs than predicted from the random shuffling of domains. Various domain combinations can be present in multiple proteins within a genome. There are two possible reasons: numerous proteins with identical domain designs exist, or proteins with diverse domain architectures share two or more sequential domains. Proteins produced by gene duplication are more likely to have the same domain architecture or domains arranged in the same order from the polypeptide chain’s N-terminus to its C-terminus. This is supported by sequence and structural evidence. The same data suggests that the evolutionary conservation of the nearby domains explains why two proteins share a fraction of those domains (Apic et al. 2003; Bashton & Chothia 2002).
P-loop nucleotide triphosphate hydrolase domains that are close to translation protein domains in 26 proteins in the human genome provide evidence for this idea. In addition to the P-loop and translation protein domains, these primary translation factors feature domains from four different families and eight distinct domain designs. This example demonstrates the preservation of a domain combination with specific functional attributes and the flexibility in developing new domain structures (Vogel et al. 2004a, b). The preservation of domain combinations within the protein repertoire, caused by gene duplication and gene conservation, is demonstrated by the fact that only around 1% of all possible domain combinations are observed in multi-domain proteins with assigned domain architecture. Despite 150 fully sequenced genomes having over 9114 SCOP domain combinations, there is significant evolutionary conservation of domain combinations because of duplication and domain change due to sequence divergence. This also explains why domain order is conserved from N to C: the same domain pair recurs as a single unit through duplication and divergence rather than independent recombination processes leading to domain order inversion (Bornberg-Bauer et al. 2005).
Also, orthologous proteins are predicted to maintain function more commonly than other homologs and are typically used for functional annotation transfer between species. A large-scale analysis of such events between humans and 40 other species spanning the evolutionary spectrum was carried out to determine the level of domain architectural conservation among orthologs. A metric for assessing domain architecture similarity was developed, and it was used to investigate differences in domain architecture conservation across orthologs and paralogs compared to primary sequence conservation. Unique domain switching occurrences between pairs of orthologs and paralogs was statistically analysed and determined (Kristoffer Forslund et al. 2011). The findings show that orthologous homologs exhibit better domain architectural conservation than paralogous homologs, showing that orthologs are under more selection pressure than paralogs to preserve the domain architecture necessary for proteins to perform a specific function. Even at enormous evolutionary distances, the domain structures of orthologs and their closest paralogous homologs are highly similar. The insertion or deletion of new domains was the most frequent domain architectural alteration seen in ortholog and paralog pairs, but domain shuffles and segment duplication/deletion were infrequent. Analysing the lineage-based protein domain architecture content in 14 full green plant genomes was used to investigate the evolutionary dynamics of domain architecture in the plant kingdom (Zhang and Skolnick 2005).
Every domain family can be represented as a node in a graph, and families can be connected close to one another or appear to be a component of the same protein, as a convenient method of displaying the group of nearby domains in a genome or across numerous genomes. The domain graph \(G\) is given by \(G=(V,E)\), where each node or vertices is a node or a vertice. Each of the vertices or nodes \(Vi\) is a family of domains, such as the Rossmann or Protein Kinase domains. An edge regardless of their order, the term \({E}_{i,j}\) refers to the adjacency or coexistence of domains within a protein that comprises both domains \(i\) and \(j\). Two different graphs are generated, whether the edges represent contiguous domains or merely domain co-occurrence (Bornberg-Bauer et al. 2005). Overall, the evidence is consistent with the hypothesis that, in contrast to primary sequence conservation, ortholog function conservation necessitates stronger domain architectural conservation than other types of homologs. This bolsters the idea that homologs with similar evolutionary distances are more functionally identical than other homologs.
Implications: Heat Shock Proteins (Hsp’s) as Emerging Therapeutic Targets
Two ATP-dependent molecular chaperones that fold and restructure proteins are Hsp90 and Hsp70. Both play an essential role in nearly every cellular activity, including protein transport, signal transduction, and receptor development. They also regulate protein homeostasis. Hsps are produced in response to cellular stress. Heat shock, for example, is an example of a rapid shift in the cellular environment to which the cell is unprepared to adapt. Practically all forms of cellular stress induce Hsp. Hsp’s are commonly referred to as stress proteins due to the widespread nature of this phenomenon. The theory underlying this phenomenon is that following a stressful event, the necessity for Hsp’s chaperone function increases, triggering their induction. The increased amount of damaged proteins, the inhibition of their removal via the proteasome, and the damage to the chaperones themselves all contribute to this need. Hsp’s induction may aid in the renature of chaperones and hence may result in a ‘cascading amplification’ of available chaperone activity (Genest et al. 2019).
However, chaperone activity modulators are required since chaperone function has been connected to the emergence of several diseases. Both Hsp90 and Hsp70 collaborate with different Hsp90 and Hsp70 co-chaperones in eukaryotes. On the other hand, Hsp90 and Hsp70 in bacteria are less complicated; Hsp90 works without co-chaperones assistance, while Hsp70 uses two co-chaperones. Additionally, some Hsp90 clients are carcinogenic proteins, and stabilizing these proteins by Hsp90 can aid in cancer development. This makes Hsp90 a promising therapeutic target (Genest et al. 2019; Soti et al. 2005).
Hsp90 and Hsp70 Protomer Domain Arrangements
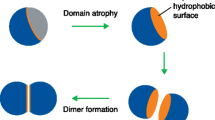
Humans and E. coli share 50% of the sequence of the highly conserved protein known as Hsp90. In each protomer of Hsp90, there are three domains: an N-terminal domain (NTD), a middle domain (MD), and a C-terminal domain (CTD) (Fig. 4A and B).The NTD binds ATP, and the NTD and MD work together to hydrolyze ATP (Schopf et al. 2017; Prodromou 2016; Röhl et al. 2013). In E. coli and yeast, a portion of the MD interacts directly with Hsp70, and higher eukaryotes may also (Genest et al. 2015; Kravats et al. 2018). The dimerization area is found in the CTD (Harris et al. 2004). Most clients interact with residues in the MD and CTD; however, residues in the NTD also bind some particular clients (Radli and Rüdiger 2018). Although all Hsp90 proteins have the same three-domain structure, there are two key differences between eukaryotic and bacterial Hsp90 (Fig. 4A). A lengthy, flexible, charged linker connects the NTD and the MD in cytosolic eukaryotic Hsp90 and endoplasmic reticulum Hsp90, Grp94, but not in bacterial Hsp90 or mitochondrial Hsp90, TRAP1 (Lavery et al. 2014; Shiau et al. 2006). This charged linker is hypothesized to influence co-chaperone binding and alterations in Hsp90 conformation (Hainzl et al. 2009; Jahn et al. 2018). Several co-chaperones containing tetratricopeptide repeats (TPR) domains, including Hop, PP5, CHIP, Tpr2, Sgt1, and Tom70, which can interact with the MEEVD motif, which is present in cytosolic eukaryotic Hsp90 proteins (Huck et al. 2017), is the second distinguishing characteristic (Fig. 4A). In multiple nucleotide-bound states for the Hsp90 homodimer, the structures of Hsp90 alone and in combination with various co-chaperones and clients have been determined. The apo, AMP-PNP and ADP-bound forms of full-length E. coli Hsp90 were seen in three different distinct conformations. Hsp90 connected to AMP-PNP adopted a more closed conformation with dimerizing N-domains, whereas Apo-Hsp90 generally inhabited an open V-shaped conformation (Fig. 4B). The ADP-bound Hsp90 was found to exist in a less compact form by X-ray crystallography. In contrast, a twisted compact form was discovered using negative stain EM (Fig. 4C).

In Hsp90 homologs, the domain arrangement is preserved. A Hsp90 protomers are made up of a CTD (blue) that contains the dimerization area and is engaged in client binding, an NTD (green) that is involved in ATP binding and hydrolysis as well as interaction with some clients, and an MD (yellow) that is directly involved in interaction with Hsp70. B model of the E. coli Hsp90 dimer’s crystal structure in the apo form (PDB code 2IOQ) (PDB code 1SF8). C E. coli Hsp90 dimer structure in the ADP-bound configuration (PDB code 2IOP) (Color figure online)
The DnaK family of proteins, which includes human and bacterial Hsp70, is highly conserved. The Hsp70 NBD and C-terminal substrate-binding domain (SBD) are joined by a flexible linker (Fig. 5A). The four subdomains that make up the nucleotide-binding domain (NBD) form a deep cleft where nucleotides bind. The substrate-binding site is located in a-sheet subdomain of the SBD, while the lid is a helical subdomain of the SBD (Mayer 2018; Mayer and Kityk 2015). The β-sheet subdomain is separated from the helical lid in the ATP-bound state. The substrate-binding site is left open for rapid, low-affinity interactions due to the attachment of both subdomains to various NBD regions. The interdomain linker docks into a crease that is made visible in the NBD when the helical lid of the SBD rotates to connect with the NBD in the ATP-bound conformation (Zhu et al. 1996; Mayer 2013).

Chaperone Hsp70/DnaK. E. coliHsp70, DnaK, structures in the closed (A) ADP-bound conformation (PDB code 2KHO) and the open (B) ATP-bound conformation (PDB code 4B9Q), which is stabilized in an oxidized triple mutant (E47C, T199A, and F529C) when ATP is present. The NBD (grey) of the DnaK protomer is joined to the SBD (teal), which is composed of two subdomains: the “lid” (α-helical subdomain), and the substrate-binding site (β-sheet subdomain), by a flexible linker (blue) (purple). B, a model of ATP as a CPK is displayed in black (Color figure online)
Hsp90 serves as a “holdase” and is an ATP-dependent molecular chaperone in cooperation with Hsp70. The capacity of Hsp90 to bind client proteins and inhibit their aggregation is called holdase activity. Aggregation can occur spontaneously, as it does with synuclein, a protein implicated in Parkinson’s disease pathogenesis, or with heat treatment or chemical denaturation of model substrates (Nakamoto et al. 2014; Daturpalli et al. 2013). Hsp90’s activity is unaffected by ATP binding or hydrolysis (Young, and Hartl 2000). It also requires neither Hsp70 nor Hsp90 co-chaperones in bacteria or eukaryotes. Client released from Hsp90 has been demonstrated independent of ATP in specific experiments (Fig. 5B). Several clients have been demonstrated to fold back into their active conformation after being released from Hsp90, indicating that the Hsp90-client complexes are transient and reversible (Garcie et al. 2016).
Hsp90 Inhibition as an Effective Anticancer Therapeutic Strategy
Hsp90’s N-terminal domain contains a unique ATP-binding site called the Bergerat-fold, which is only seen in a small number of bacterial gyrases, topoisomerases, and histidine-kinases (Prodromou 2016). Specialized Hsp90 inhibitors have been made because of the specific ATP-binding site. The recent discovery of a second nucleotide-binding site in the C-terminal domain of Hsp90 may provide new opportunities to inhibit this chaperone. Raf, Akt, and cyclin-dependent serine kinases, as well as numerous essential participants in the malignant transformation, such as the ErbB2, Src, Abl, or Met tyrosine kinases, are among the numerous kinases that Hsp90 interacts with and stabilizes. Raf, Akt, and cyclin-dependent serine kinases (Sõti et al. 2005). Instead of interacting with effector proteins, chaperone-based inhibitors stop the associated chaperone(s) from retaining their activation-competent conformation. Chaperone inhibitors act like regular multitarget medications, more effective in many applications than highly selective single-target therapies. Geldanamycin, its less toxic analog, 17-allylamino-17-dimethoxy-geldanamycin (17AAG), radicicol, and its more stable oxime derivatives show a greater affinity for Hsp90 than geldanamycin. Purine-scaffold inhibitors, the third class of tumor-specific Hsp90 inhibitors, and new geldanamycin analogs have recently been developed, and attempts are currently being made to produce even more Hsp90-interacting therapeutic options (Soti et al. 2005).
Inhibiting the 90 kDa heat shock protein (Hsp90), according to Soti et al. has recently gained attention as a highly effective method of treating many cancers. On the other hand, it has been demonstrated that inducing the 70 kDa Hsp70 is highly efficient assistance in the recovery from a variety of diseases, for instance, ischemic heart disease, diabetes, and neurodegeneration. There may be significant therapeutic advantages in developing drugs that interact with membranes to alter certain membrane domains and control the heat shock response. Hsp is produced when the heat shock factor (HSF)-1 is activated. HSF-1 was found bound by Hsp90 and kept dormant in resting cells, along with other chaperones. Because misfolded proteins pick up these repressive chaperones, the cytoplasmic chaperone/HSF-1 complex separates under stress. The dissociation of HSF-1 from Hsp90 reveals the nuclear localization signal of this transcription factor, allowing for its translocation to the cell nucleus. HSF-1 is trimerized and phosphorylated (Morimoto 2002).
Even though the exact sequence of these processes is still unknown, a study has implicated the polo-like kinase-1 as a significant participant in the phosphorylation and subsequent nuclear translocation of HSF-1 at the Ser-419 site (Kim et al. 2005). However, other studies found that Ser-326, not Ser-419, was a critical site for phosphorylating HSF-1 in association with activation (Guettouche et al. 2005). A component of the nuclear HSF-1 is contained in heat shock granules, which may alter the chromatin structure (Jolly et al. 2004). When HSF-1 attaches to the heat shock elements of the heat shock-inducible genes, the RNA polymerase in most of these genes stops (or “pauses”) after transcribing the first segment of the mRNA. HSF-1 is liberated from the DNA by a nuclear Hsp90 complex and is most likely transported back to the cytoplasm. There is still much to understand about how Hsp90 enters the nucleus in response to stress, how their complex develops there, and how they are then drawn to the DNA-bound HSF-1. The HSF-1 activation and downregulation cascade’s additional proteins, including the Ralbinding protein-1 and tubulin in the cytoplasmic HSF-1/Hsp90 complex and p23 in the nuclear HSF-1/Hsp90 complex, are highly intriguing as prospective therapeutic targets (Fig. 6; Soti et al. 2005).

Potential pharmacological targets for HSF-1 activation components. The main components of the activation of HSF-1, the main transcription factor causing the stimulation of Hsp production, are shown in the picture. The following are some potential pharmacological targets: HSF-1 is distributed intracellularly, moves from the cytoplasm to the nucleus, forms a complex with Hsp90 in the nucleus, and moves back from the nucleus to the cytoplasm. RalBP-1, also known as p23, is a cochaperone of Hsp90
Conclusion
The role of domain combinations in forming the protein repertoire has been outlined in this review. According to the relatively extensive domain assignments available for completely sequenced genomes most proteins are multi-domain, even in simple genomes. Many domain acquisitions and rearrangements are presumably the consequence of a mix of processes due to the complexity of evolution. Despite the fact that the observed domain combinations only make up a small portion of all possible combinations in the repertoire of protein families, the emergence of new combinations is linked to speciation and particular phylogenetic groups. The creation of increasingly complicated animals is aided by domain modulations/alterations, domain super families expansions, and other methods. To understand the molecular subtleties of domain combinations’ functions, the three-dimensional structure of the domain architecture is essential. Structural genomic programs might discover new domain modulations/alterations and the structures of individual domains. The number of proteins that contain a given domain combination and their responsiveness to various domain functional architectures may be used to target particular domain modulations/alterations. Regional disparities between paralogs or (partial) overlap between interaction partners have also been discovered by large-scale expression investigations and evolutionary relationships between gene expression levels and timings. We may be able to comprehend the adaptable power of evolution that moulded the protein cosmos and life on earth if we can grasp the spatiotemporal characteristics of inter- and intramolecular interactions. Finally, domain architecture analysis leads to identifying several possible pharmacological targets. This was looked into in the Hsp70 and Hsp90 chaperon families, which have been proved to help people recover from various disorders, including ischemic heart disease, diabetes, and neurodegeneration. Hsp90 inhibition as an anticancer treatment is also being explored.
Abbreviations
- PDA:
-
Protein domain architecture
- DCN:
-
Protein domain co-occurrence networks
- SCOP:
-
Structural classification of proteins
- CATH:
-
Protein structural classification database; class (C), architecture (A), topology (T) and homologous superfamily (H)
- Pfam:
-
The protein families database ProDoM
- EGF:
-
Epidermal growth factor
- CUB domain:
-
Complement protein subcomponents C1r/C1s, urchin embryonic growth factor, and bone morphogenetic protein 1
- HSP:
-
Heat shock protein
References
Aderinwale T, Bharadwaj V, Christoffer C, Terashi G, Zhang Z, Jahandideh R, Kagaya Y, Kihara D (2022) Real-time structure search and structure classification for AlphaFold protein models. Commun Biol 5(1):316
Amoutzias GD, Robertson DL, Oliver SG, Bornberg-Bauer E (2004a) Convergent evolution of gene networks by single-gene duplications in higher eukaryotes. EMBO Rep 5(3):274–279
Amoutzias GD, Robertson DL, Bornberg-Bauer E (2004) The evolution of protein interaction networks in regulatory proteins. Comp Funct Genomics 5(1):79–84
Amoutzias GD, Veron AS, Weiner J 3rd, Robinson-Rechavi M, Bornberg-Bauer E, Oliver SG, Robertson DL (2007) One billion years of bZIP transcription factor evolution: conservation and change in dimerization and DNA-binding site specificity. Mol Biol Evol 24(3):827–835
Apic G, Gough J, Teichmann SA (2001) Domain combinations in archaeal, eubacterial and eukaryotic proteomes. J Mol Biol 310(2):311–25
Apic G, Huber W, Teichmann SA (2003) Multi-domain protein families and domain pairs: comparison with known structures and a random model of domain recombination. J Struct Funct Genomics 4(2):67–78
Aravind L, Koonin EV (1999) DNA-binding proteins and evolution of transcription regulation in the archaea. Nucleic Acids Res 27(23):4658–4670. https://doi.org/10.1093/nar/27.23.4658
Bashton M, Chothia C (2002) The geometry of domain combination in proteins. J Mol Biol 315(4):927–939
Basu MK, Carmel L, Rogozin IB, Koonin EV (2008) Evolution of protein domain promiscuity in eukaryotes. Genome Res 18(3):449–461
Basu MK, Poliakov E, Rogozin IB (2009) Domain mobility in proteins: functional and evolutionary implications. Brief Bioinform 10(3):205–216. https://doi.org/10.1093/bib/bbn057
Bhattacharya R, Rose PW, Burley SK, Prlić A (2017) Impact of genetic variation on three dimensional structure and function of proteins. PLoS ONE 12(3):e0171355
Björklund ÅK, Ekman D, Light S, Frey-Skött J, Elofsson A (2005) Domain rearrangements in protein evolution. J Mol Biol 353(4):911–923
Böhning D, Dietz E, Schlattmann P (1998) Recent developments in computer-assisted analysis of mixtures. Biometrics 54(2):525–36
Bornberg-Bauer E, Beaussart F, Kummerfeld SK, Teichmann SA, Weiner J (2005) The evolution of domain arrangements in proteins and interaction networks. Cell Mol Life Sci CMLS 62(4):435–445
Bourne PE, Briedis K, Dupont C, Valas R, Yang S (2010) Genome evolution studied through protein structure. Evolutionary genomics and systems biology. Wiley, pp 153–164
Brown P, Pullan W, Yang Y, Zhou Y (2016) Fast and accurate non-sequential protein structure alignment using a new asymmetric linear sum assignment heuristic. Bioinformatics 32:370–377
Buljan M, Bateman A (2009) The evolution of protein domain families. Biochem Soc Trans 37(4):751–755
Buljan M, Frankish A, Bateman A (2010) Quantifying the mechanisms of domain gain in animal proteins. Genome Biol 11(7):1–15
Daria N, ShalaevaDmitry A, CherepanovMichael Y, GalperinAndrey V, GolovinArmen Y, Mulkidjanian (2018) Evolution of cation binding in the active sites of P-loop nucleoside triphosphatases in relation to the basic catalytic mechanism. eLife 7:e37373
Daturpalli S, Waudby CA, Meehan S, Jackson SE (2013) Hsp90 inhibits α-synuclein aggregation by interacting with soluble oligomers. J Mol Biol 425(22):4614–4628
Deng L, Zhong G, Liu C, Luo J, Liu H (2019) MADOKA: an ultra-fast approach for large-scale protein structure similarity searching. BMC Bioinform 20:662
Dessailly BH, Dawson NL, Mizuguchi K, Orengo CA (2013) Functional site plasticity in domain superfamilies. Biochim Biophys Acta 1834(5):874–889
Dohmen E, Klasberg S, Bornberg-Bauer E, Perrey S, Kemena C (2020) The modular nature of protein evolution: domain rearrangement rates across eukaryotic life. BMC Evol Biol 20(1):30. https://doi.org/10.1186/s12862-020-1591-0
Doolittle RF (1995) The origins and evolution of eukaryotic proteins. Philosophical transactions of the royal society of London. Ser B: Biol Sci 349(1329):235–240
Doolittle RF, Bork P (1993) Evolutionarily mobile modules in proteins. Sci Am 269(4):50–56
Ekman D, Björklund ÅK, Frey-Skött J, Elofsson A (2005) Multi-domain proteins in the three kingdoms of life: orphan domains and other unassigned regions. J Mol Biol 348(1):231–243
Ersfeld K, Barraclough H, Gull K (2005) Evolutionary relationships and protein domain architecture in an expanded calpain superfamily in kinetoplastid parasites. J Mol Evol 61(6):742–757
Fong JH, Geer LY, Panchenko AR, Bryant SH (2007) Modeling the evolution of protein domain architectures using maximum parsimony. J Mol Biol 366(1):307–315
Forslund K, Sonnhammer EL (2012) Evolution of protein domain architectures. Evolutionary genomics. Humana Press, Totowa, pp 187–216
Forslund K, Henricson A, Hollich V, Sonnhammer EL (2008) Domain tree-based analysis of protein architecture evolution. Mol Biol Evol 25(2):254–264
Forslund K, Pekkari I, Sonnhammer EL (2011) Domain architecture conservation in orthologs. BMC Bioinform 12(1):1–14
Forslund SK, Kaduk M, Sonnhammer EL (2019) Evolution of protein domain architectures. Evolutionary genomics. Springer, New York, pp 469–504
Garcie C, Tronnet S, Garénaux A, McCarthy AJ, Brachmann AO, Pénary M, Martin P (2016) The bacterial stress-responsive Hsp90 chaperone (HtpG) is required for the production of the genotoxin colibactin and the siderophore yersiniabactin in Escherichia coli. J Infect Dis 214(6):916–924
Genest O, Wickner DSM (2019) Hsp90 and Hsp70 chaperones: collaborators in protein remodeling. J Biol Chem 294(6):2109–2120
Genest O, Hoskins JR, Kravats AN, Doyle SM, Wickner S (2015) Hsp70 and Hsp90 of E. coli directly interact for collaboration in protein remodeling. J Mol Biol 427(24):3877–3889
Gerstein M (1998) How representative are the known structures of the proteins in a complete genome? A comprehensive structural census. Fold Des 3:497–512
Gough J (2005) Convergent evolution of domain architectures (is rare). Bioinformatics 21(8):1464–71. https://doi.org/10.1093/bioinformatics/bti204
Guettouche T, Boellmann F, Lane WS, Voellmy R (2005) Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem 6(1):1–14
Hainzl O, Lapina MC, Buchner J, Richter K (2009) The charged linker region is an important regulator of Hsp90 function. J Biol Chem 284(34):22559–22567
Harris SF, Shiau AK, Agard DA (2004) The crystal structure of the carboxy-terminal dimerization domain of htpG the Escherichia coli Hsp90 reveals a potential substrate binding site. Structure 12(6):1087–1097
Holm L (2019) Benchmarking fold detection by DaliLite vol 5. Bioinformatics 35:5326–5327
Hsu CH, Chen CK, Hwang MJ (2013) The architectural design of networks of protein domain architectures. Biol Let 9(4):20130268
Huck JD, Que NL, Hong F, Li Z, Gewirth DT (2017) Structural and functional analysis of GRP94 in the closed state reveals an essential role for the pre-N domain and a potential client-binding site. Cell Rep 20(12):2800–2809
Huynen MA, van Nimwegen E (1998) The frequency distribution of gene family sizes in complete genomes. Mol Biol Evol 15(5):583–589
Itoh M, Nacher JC, Kuma K, Goto S, Kanehisa M (2007) Evolutionary history and functional implications of protein domains and their combinations in eukaryotes. Genome Biol 8:R121
Jahn M, Tych K, Girstmair H, Steinmaßl M, Hugel T, Buchner J, Rief M (2018) Folding and domain interactions of three orthologs of Hsp90 studied by single-molecule force spectroscopy. Structure 26(1):96–105
Jolly C, Metz A, Govin J, Vigneron M, Turner BM, Khochbin S, Vourc’h C (2004) Stress-induced transcription of satellite III repeats. J Cell Biol 164(1):25–33
Kim SA, Yoon JH, Lee SH, Ahn SG (2005) Polo-like kinase 1 phosphorylates heat shock transcription factor 1 and mediates its nuclear translocation during heat stress. J Biol Chem 280(13):12653–12657
Knudsen LM, Hippe E, Hjorth M, Holmberg E, Westin J (1994) Renal function in newly diagnosed multiple myeloma—a demographic study of 1353 patients. Eur J Haematol 53(4):207–212
Konaté MM, Plata G, Park J, Usmanova DR, Wang H, Vitkup D (2019) Molecular function limits divergent protein evolution on planetary timescales. Elife 8:e39705. https://doi.org/10.7554/eLife.39705
Koonin EV, Wolf YI, Karev GP (2002) The structure of the protein universe and genome evolution. Nature 420:218–223
Kozlova MI, Shalaeva DN, Dibrova DV, Mulkidjanian AY (2022) Common patterns of hydrolysis initiation in P-loop fold nucleoside triphosphatases. Biomolecules 12(10):1345. https://doi.org/10.3390/biom12101345
Kravats AN, Hoskins JR, Reidy M, Johnson JL, Doyle SM, Genest O, Wickner S (2018) Functional and physical interaction between yeast Hsp90 and Hsp70. Proc Natl Acad Sci USA 115(10):E2210–E2219
Kummerfeld SK, Teichmann SA (2009) Protein domain organisation: adding order. BMC Bioinform 10:39
Lavery LA, Partridge JR, Ramelot TA, Elnatan D, Kennedy MA, Agard DA (2014) Structural asymmetry in the closed state of mitochondrial Hsp90 (TRAP1) supports a two-step ATP hydrolysis mechanism. Mol Cell 53(2):330–343
Levine M, Tjian R (2003) Transcription regulation and animal diversity. Nature 424(6945):147–151
Levitt M (2009) Nature of the protein universe. Proc Natl Acad Sci 106(27):11079–11084
Liu M, Grigoriev A (2004) Protein domains correlate strongly with exons in multiple eukaryotic genomes–evidence of exon shuffling? Trends Genet 20(9):399–403
Ljung F, Andre I (2021) ZEAL: protein structure alignment based on shape similarity. Bioinformatics 37(18):2874–2881. https://doi.org/10.1093/bioinformatics/btab205
Lo Conte L, Ailey B, Hubbard TJ, Brenner SE, Murzin AG, Chothia C (2000) SCOP: a structural classification of proteins database. Nucleic Acids Res 28(1):257–259
Lucas JI, Arnau V, Marin I (2006) Comparative genomics and protein domain graph analyses link ubiquitination and RNA metabolism. J Mol Biol 357:9
Luscombe NM, Qian J, Zhang Z, Johnson T, Gerstein M (2002) The dominance of the population by a selected few: power-law behaviour applies to a wide variety of genomic properties. Genome Biol 3(8):00401
Madan Babu M, Teichmann SA (2003) Evolution of transcription factors and the gene regulatory network in Escherichia coli. Nucleic Acids Res 31(4):1234–1244
Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D (1999) Detecting protein function and protein–protein interactions from genome sequences. Science 285:751–753
Marsh JA, Teichmann SA (2010) How do proteins gain new domains? Genome Biol 11(7):1–4
Mascarenhas NM, Gosavi S (2017) Understanding protein domain-swapping using structure-based models of protein folding. Prog Biophys Mol Biol 128:113–120. https://doi.org/10.1016/j.pbiomolbio.2016.09.013
Mayer MP (2013) Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem Sci 38(10):507–514
Mayer MP (2018) Intra-molecular pathways of allosteric control in Hsp70s. Philos Trans R Soc B: Biol Sci 373(1749):20170183
Mayer MP, Kityk R (2015) Insights into the molecular mechanism of allostery in Hsp70s. Front Mol Biosci 2:58
Morimoto RI (2002) Dynamic remodeling of transcription complexes by molecular. Cell 110(3):281–284
Mulder NJ (2010) Protein domain architectures. Methods Mol Biol 609:83–95. https://doi.org/10.1007/978-1-60327-241-4_5
Müller A, MacCallum RM, Sternberg MJ (2002) Structural characterization of the human proteome. Genome Res 12(11):1625–1641
Murzin AG, Brenner SE, Hubbard T, Chothia C (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247:536–540. https://doi.org/10.1016/S0022-2836(05)80134-2
Nakamoto H, Fujita K, Ohtaki A, Watanabe S, Narumi S, Maruyama YH (2014) Physical interaction between bacterial heat shock protein (Hsp) 90 and Hsp70 chaperones mediates their cooperative action to refold denatured proteins. J Biol Chem 289(9):6110–6119
Nimwegen EV (2006) Scaling laws in the functional content of genomes. Power laws, scale-free networks and genome biology. Springer, pp 236–253
Park J, Lappe M, Teichmann SA (2001) Mapping protein family interactions: intramolecular and intermolecular protein family interaction repertoires in the PDB and yeast. J Mol Biol 307(3):929–938
Patthy L (1999) Genome evolution and the evolution of exon-shuffling–a review. Gene 238(1):103–114
Peisajovich SG, Garbarino JE, Wei P, Lim WA (2010) Rapid diversification of cell signaling phenotypes by modular domain recombination. Science 328:368–372
Pérez-Rueda E, Collado-Vides J (2000) The repertoire of DNA-binding transcriptional regulators in Escherichia coli K-12. Nucleic Acids Res 28(8):1838–1847
Persson E, Kaduk M, Forslund SK, Sonnhammer ELL (2019) Domainoid: domain-oriented orthology inference. BMC Bioinform 20(1):523. https://doi.org/10.1186/s12859-019-3137-2
Prodromou C (2016) Mechanisms of Hsp90 regulation. Biochem J 473(16):2439–2452
Przytycka T, Davis G, Song N, Durand D (2006) Graph theoretical insights into evolution of multidomain proteins. J Comput Biol 13(2):351–363
Qian J, Luscombe NM, Gerstein M (2001) Protein family and fold occurrence in genomes: power-law behaviour and evolutionary model11Edited by. J Thornton J Mol Biol 313(4):673–681
Radli M, Rüdiger SG (2018) Dancing with the diva: Hsp90–client interactions. J Mol Biol 430(18):3029–3040
Ramesh P, Nagendrappa JH, Shivashankara SKH (2021) Comparative analysis of Rosetta stone events in Klebsiella pneumoniae and Streptococcus pneumoniae for drug target identification. Beni-Suef Univer J Basic Appl Sci 10(1):1–11
Röhl A, Rohrberg J, Buchner J (2013) The chaperone Hsp90: changing partners for demanding clients. Trends Biochem Sci 38(5):253–262
Rost B (1997) Protein structures sustain evolutionary drift. Fold Des 2:S19–S24. https://doi.org/10.1016/S1359-0278(97)00059-X
Russell RB, Alber F, Aloy P, Davis FP, Korkin D, Pichaud M, Sali A (2004) A structural perspective on protein–protein interactions. Curr Opin Struct Biol 14(3):313–324
Schopf FH, Biebl MM, Buchner J (2017) The HSP90 chaperone machinery. Nat Rev Mol Cell Biol 18(6):345–360
Servant F, Bru C, Carrere S, Courcelle E, Gouzy J, Peyruc D, Kahn D (2002) ProDom: automated clustering of homologous domains. Brief Bioinform 3(3):246–251
Shiau AK, Harris SF, Southworth DR, Agard DA (2006) Structural analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 127(2):329–340
Sikosek T, Chan HS (2014) Biophysics of protein evolution and evolutionary protein biophysics. J R Soc Interface 11(100):20140419. https://doi.org/10.1098/rsif.2014.0419
Sillitoe I, Dawson N, Thornton J, Orengo C (2015) The history of the CATH structural classification of protein domains. Biochimie 119:209–217. https://doi.org/10.1016/j.biochi.2015.08.004
Sonnhammer EL, Eddy SR, Birney E, Bateman A, Durbin R (1998) Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res 26(1):320–322
Sõti C, Nagy E, Giricz Z, Vígh L, Csermely P, Ferdinandy P (2005) Heat shock proteins as emerging therapeutic targets. Br J Pharmacol 146(6):769–780
Teichmann SA, Park J, Chothia C (1998) Structural assignments to the Mycoplasma genitalium proteins show extensive gene duplications and domain rearrangements. Proc Natl Acad Sci USA 95:14658–14663
Thomas GWC, Dohmen E, Hughes DST, Murali SC, Poelchau M, Glastad K, Anstead CA, Ayoub NA, Batterham P, Bellair M, Binford GJ, Chao H, Chen YH, Childers C, Dinh H, Doddapaneni HV, Duan JJ, Dugan S, Esposito LA, Friedrich M (2020) Gene content evolution in the arthropods. Genome Biol 21:15. https://doi.org/10.1186/s13059-019-1925-7
Toll-Riera M, Albà MM (2013) Emergence of novel domains in proteins. BMC Evol Biol 13:47. https://doi.org/10.1186/1471-2148-13-47
Tordai H, Nagy A, Farkas K, Bányai L, Patthy L (2005) Modules, multidomain proteins and organismic complexity. FEBS J 272:5064–5078
van Rijk A, Bloemendal H (2003) Molecular mechanisms of exon shuffling: illegitimate recombination. Genetica 118(2):245–249
Vogel C, Bashton M, Kerrison ND, Chothia C, Teichmann SA (2004a) Structure, function and evolution of multidomain proteins. Curr Opin Struct Biol 14(2):208–216. https://doi.org/10.1016/j.sbi.2004.03.011
Vogel C, Berzuini C, Bashton M, Gough J, Teichmann SA (2004b) Supra-domains: evolutionary units larger than single protein domains. J Mol Biol 336(3):809–823. https://doi.org/10.1016/j.jmb.2003.12.026
Wang Z, Zhang XC, Le MH, Xu D, Stacey G, Cheng J (2011) A protein domain co-occurrence network approach for predicting protein function and inferring species phylogeny. PLoS ONE 6(3):e17906
Weiner J 3rd, Beaussart F, Bornberg-Bauer E (2006) Domain deletions and substitutions in the modular protein evolution. FEBS J 273(9):2037–2047
Weiner J, Moore AD, Bornberg-Bauer E (2008) Just how versatile are domains? BMC Evol Biol 8(1):1–14
Wuchty S, Almaas E (2005) Evolutionary cores of domain co-occurrence networks. BMC Evol Biol 5:24. https://doi.org/10.1186/1471-2148-5-24
Ye Y, Godzik A (2004) Comparative analysis of protein domain organization. Genome Res 14(3):343–353
Young JC, Hartl FU (2000) Polypeptide release by Hsp90 involves ATP hydrolysis and is enhanced by the co-chaperone p23. EMBO J 19(21):5930–5940
Zhang Y, Skolnick J (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33:2302–2309
Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272(5268):1606–1614
Acknowledgements
We thank Dr. Riaz Mahmood, Professor, Department of Biotechnology, Kuvempu University, Shankaraghatta, India, for assisting in language editing.
Funding
No funding was available for the present work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflict of interest.
Additional information
Handling editor: Erich Bornberg-Bauer.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gollapalli, P., Rudrappa, S., Kumar, V. et al. Domain Architecture Based Methods for Comparative Functional Genomics Toward Therapeutic Drug Target Discovery. J Mol Evol 91, 598–615 (2023). https://doi.org/10.1007/s00239-023-10129-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-023-10129-w