
Abstract
Snake venom can vary both among and within species. While some groups of New World pitvipers—such as rattlesnakes—have been well studied, very little is known about the venom of montane pitvipers (Cerrophidion) found across the Mesoamerican highlands. Compared to most well-studied rattlesnakes, which are widely distributed, the isolated montane populations of Cerrophidion may facilitate unique evolutionary trajectories and venom differentiation. Here, we describe the venom gland transcriptomes for populations of C. petlalcalensis, C. tzotzilorum, and C. godmani from Mexico, and a single individual of C. sasai from Costa Rica. We explore gene expression variation in Cerrophidion and sequence evolution of toxins within C. godmani specifically. Cerrophidion venom gland transcriptomes are composed primarily of snake venom metalloproteinases, phospholipase A\(_2\)s (PLA\(_2\)s), and snake venom serine proteases. Cerrophidion petlalcalensis shows little intraspecific variation; however, C. godmani and C. tzotzilorum differ significantly between geographically isolated populations. Interestingly, intraspecific variation was mostly attributed to expression variation as we did not detect signals of selection within C. godmani toxins. Additionally, we found PLA\(_2\)-like myotoxins in all species except C. petlalcalensis, and crotoxin-like PLA\(_2\)s in the southern population of C. godmani. Our results demonstrate significant intraspecific venom variation within C. godmani and C. tzotzilorum. The toxins of C. godmani show little evidence of directional selection where variation in toxin sequence is consistent with evolution under a model of mutation–drift equilibrium. Cerrophidion godmani individuals from the southern population may exhibit neurotoxic venom activity given the presence of crotoxin-like PLA\(_2\)s; however, further research is required to confirm this hypothesis.
Resumen
El veneno de las serpientes puede variar entre y dentro de las especies. Mientras algunos grupos de viperidos del Nuevo Mundo—como las cascabeles—han sido bien estudiadas, muy poco se sabe acerca del veneno de las nauyacas de frío (Cerrophidion) que se encuentran en las zonas altas de Mesoamérica. Comparadas con las extensamente estudiadas cascabeles, que estan ampliamente distribuidas, las poblaciones de Cerrophidion, aisladas en montañas, pueden poseer trayectorias evolutivas y diferenciación en su veneno unicos. En el presente trabajo, describimos el transcriptoma de las glándulas de veneno de poblaciones de C. petlalcalensis, C. tzotzilorum, y C. godmani de México, y un individuo de C. sasai de Costa Rica. Exploramos la variación en la expresión de toxinas en Cerrophidion y la evolución en las secuencias geneticas en C. godmani específicamente. El transcriptoma de la glándula de veneno de Cerrophidion esta compuesto principalmente de Metaloproteinasas de Veneno de Serpiente, Fosfolipasas A\(_2\) (PLA\(_2\)s), y Serin Proteasas de Veneno de Serpiente. Cerrophidion petlalcalensis presenta poca variación intraespecífica; sin embargo, los transcriptomas de la glandula de veneno de C. godmani y C. tzotzilorum difieren significativamente entre poblaciones geográficamente aisladas. Curiosamente, la variación intraespecífica estuvo atribuida principalmente a la expresión de las toxinas ya que no encontramos señales de selección en las toxinas de C. godmani. Adicionalmente, encontramos miotoxinas similares a PLA\(_2\) en todas las especies excepto C. petlalcalensis, y PLA\(_2\)s similares a crotoxina en la población sureña de C. godmani. Nuestros resultados demuestran la presencia de variacion intraespecífica presente en el veneno de C. godmani y C. tzotzilorum. Las toxinas de Cerrophidion godmani muestran poca evidencia de selección direccional, y la variación en la secuencias de las toxinas es consistente con evolucion bajo un modelo de equilibrio de mutación-deriva. Algunos individuos de C. godmani de la población del sur potencialmente tienen un veneno neurotóxico dada la presencia de PLA\(_2\)s similares a la crotoxina, sin embargo, se necesita más evidencia para corroborar esta hipótesis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Venoms have emerged as an excellent system for investigating trait evolution given their phenotype–genotype tractability, clear contribution to fitness, and rapid rate of evolution resulting from mutations in protein-coding regions and/or changes in gene expression (Casewell et al. 2011, 2012, 2013; Rokyta et al. 2015b; Aird et al. 2015; Margres et al. 2017). Venom toxins can be linked to specific genes, which facilitates the investigation of how evolutionary processes generate genomic and corresponding phenotypic variation (Brahma et al. 2015; Junqueira-de Azevedo and Ho 2002; Margres et al. 2014b; Rokyta et al. 2015b, 2011). Snake venoms in particular consist of 10–100 secreted toxins, many of which are isoforms from paralogous gene duplications within 10–20 gene families (Casewell et al. 2014; Calvete 2017). Several studies have found evidence for strong, positive directional selection in snake venom toxins (Rokyta et al. 2013; Aird et al. 2015, 2017; Mason et al. 2020; Strickland et al. 2018b); however, others have shown that drift and balancing selection might also have an important role given a lack of differential selection between toxins and nontoxin genes (Ochoa et al. 2020; Rautsaw et al. 2019; Schield et al. 2022) and less selective constraint in toxins than in nontoxins (Aird et al. 2013). These contrasting results may reflect the influence of several key factors, such as the phylogenetic diversity and complex resistance mechanisms of prey (Holding et al. 2021b; Gibbs et al. 2020), the adaptive landscape (Rautsaw et al. 2019), or the demographic history of the species (Ochoa et al. 2020; Margres et al. 2021; Holding et al. 2021a; Rautsaw et al. 2019; Aird et al. 2017). Targeted examinations of specific toxin families have also yielded insights into how genomic variation affects adaptive phenotypes. One example of such gene families are the phospholipase A\(_2\)s (PLA\(_2\)s)—a primary component of many pitviper venoms (Tasoulis and Isbister 2017).
The PLA\(_2\)s in New World pitviper venom evolved via gene duplication and neofunctionalization with gene loss shaping much of the current variation in PLA\(_2\) content (Dowell et al. 2016). For example, certain species possess a heterodimeric PLA\(_2\) \(\beta\)-neurotoxin (herein referred to as “crotoxin-like” PLA\(_2\)) which is formed by an interaction of an acidic and a basic subunit (Rübsamen et al. 1971). Despite an ancient origin of this neurotoxin, the corresponding genes have been lost in several lineages despite being associated with higher lethality in prey (Mackessy 2008; Dowell et al. 2016, 2018; Borja et al. 2018; Rokyta et al. 2015b). Most of the research on PLA\(_2\) variation has been done in rattlesnakes, specifically in the large-bodied, lowland clade, as several species in this group exhibit intraspecific variation in the presence/absence of crotoxin-like PLA\(_2\)s (Strickland et al. 2018b; Dowell et al. 2018; Margres et al. 2021; Zancolli et al. 2019; Neri-Castro et al. 2019). Variation in specific toxins and in venom composition more broadly is expected to be higher among lineages with substantial population genetic structure, with high environmental variation across their distribution, and between populations with low amounts of gene flow (Ochoa et al. 2020; Margres et al. 2017, 2019). These factors are often present in high elevation species, however, these groups are generally not well studied.
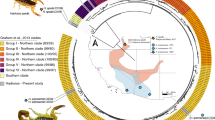
The genus Cerrophidion comprises five species of small montane pitvipers distributed throughout Mesoamerica: C. petlalcalensis and C. tzotzilorum in Mexico; C. godmani from Mexico and Guatemala; C. wilsoni primarily in Honduras, Guatemala, Nicaragua, and El Salvador; and C. sasai in Costa Rica and Panama (Campbell and Lamar 2004; Jadin et al. 2012; Fernández et al. 2017) (Fig. 1). Until recently, C. godmani, C. sasai, and C. wilsoni were thought to be a single, wide-ranging species with substantial morphological variation, however, phylogeographic analyses revealed dynamic biogeographic processes which have resulted in species diversification (Jadin et al. 2012). In particular, montane lineages of pitvipers from Middle America have complex evolutionary histories driven by the geologic events and climatic changes that occurred in the Pliocene and Miocene (Castoe et al. 2009; Daza et al. 2010). Cerrophidion species have likely evolved in response to several vicariant events across well-known biogeographic barriers (Castoe et al. 2009; Daza et al. 2010): C. sasai and C. wilsoni are separated by the Nicaraguan Depression; C. tzotzilorum and C. petlalcalensis are separated by the Tehuantepec Isthmus; and C. godmani is separated from C. sasai and C. wilsoni by the Motagua–Polochic Faults (Castoe et al. 2009; Daza et al. 2010). C. godmani and C. tzotzilorum are the only species that are not separated by a major biogeographic barrier (Fig 1). As a more clear understanding of their evolutionary history emerges, we are now able to further investigate other biological aspects of Cerrophidion.

Cerrophidion distribution in Mesoamerica. Map modified from VenomMaps (Rautsaw et al. 2022), with the localities of samples of each species used herein. Species are represented by different shapes, different outline colors correspond to Northern populations of that species.*Cerrophidion wilsoni was not included in this work. Species tree scaled with IQtree from the inferred Astral tree; support values correspond to the Astral species tree. Node shapes correspond with the populations in the map. Pie charts in the tips show the percentage of expression of the five most abundant toxin families; the remaining toxin families are included in the category “Other.” Photo Credit: Jason M. Jones (C. tzotzilorum)
A thorough understanding of the venom composition of Cerrophidion is lacking; published proteomic data on venom composition are limited to C. sasai and individuals of C. godmani with no associated locality data that may correspond to any one of three distinct species (Gutiérrez et al. 1985; Gené et al. 1989; Arni et al. 1999; Lizano et al. 2000; Chen et al. 2004; Durban et al. 2011; Lomonte et al. 2012, 2014). Cerrophidion sasai venom is composed mostly of snake venom metalloproteinases (SVMPs) and PLA\(_2\)s, inducing intravascular coagulation, hemorrhages, and myonecrosis due to the presence of D49 and K49 type myotoxic PLA\(_2\)s (Lomonte et al. 2012). Our knowledge of C. petlalcalensis venom is restricted to an envenomation report (López-Luna et al. 1999): the venom was described as relatively potent, where localized pain and swelling of the hand and forearm started 15 minutes post bite and lasted for three days despite the administration of polyvalent antivenom (López-Luna et al. 1999). Cerrophidion godmani, C. tzotzilorum, and C. wilsoni venom composition is unknown; however, coagulotoxic effects from their venom have been described (Jones et al. 2022).
Given their complex phylogeographic history, montane distribution, and the general lack of information regarding venom composition and variation within Cerrophidion, we aimed to describe their venom gland transcriptomes, determine if there is differential toxin expression among populations, and determine if selection is driving toxin evolution in the venom. To do this, we collected representatives of four of the five described Cerrophidion: six C. godmani, four C. tzotzilorum, three C. petlalcalensis, and one C. sasai (Table 1, Fig. 1). Based on previous proteomic work on C. sasai, we expected to see venoms with a high content of PLA\(_2\)s, SVMPs, and snake venom serine proteases (SVSPs), including myotoxic PLA\(_2\)s (Lomonte et al. 2012). Additionally, we inferred a PLA\(_2\) gene-tree using our newly generated transcriptomic data, and previously published sequence data (Whittington et al. 2018; Neri-Castro et al. 2020b; Mason et al. 2020) to classify Cerrophidion PLA\(_2\) groups and infer their evolutionary history. To investigate potential intraspecific and ontogenetic variation, we tested for differential expression of toxins between populations and body sizes. Last, we tested for signals of selection in toxins, comparing toxins against nontoxins genes. If toxins have a greater mean genetic signal of selection than nontoxins, then an adaptive evolution hypothesis would be supported for Cerrophidion venom. Alternatively, comparable levels of toxin and nontoxin divergence would suggest Cerrophidion venom is largely impacted by drift, possibly influenced by their distribution in isolated mountain ranges and low dispersal capability.
Results
Cerrophidion venom gland transcriptomes were dominated by PLA\(_2\)s and SVMPs, followed by bradykinin-potentiating peptides (BPPs), C-type lectins (CTLs), L-amino acid oxidases (LAAO), and SVSPs. Other toxins families were present in lower percentages (Figs. 2 and 3). Cerrophidion petlalcalensis transcriptomes all had largely similar toxin composition, while C. godmani and C. tzotzilorum both exhibited variation in toxin composition and expression corresponding to the geographic origin of individuals. Toxin composition inferred from the C. sasai venom transcriptome was similar to the results of previous proteomic studies (Fig 2A II; Lomonte et al. 2012). Interestingly, a single individual of C. godmani expressed PLA\(_2\)s that were inferred, based on phylogenetic placement, to be closely related to both subunits of the crotoxin-like PLA\(_2\)s of other pitviper genera, suggesting intraspecific polymorphism for neurotoxic venom.

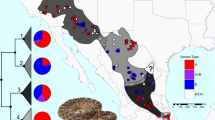
RSEM results for the consensus transcriptomes of A C. sasai, B. C. petlalcalensis, and C. C. tzotzilorum. In A (I) barplot of the log ranked expression of toxin genes, (II) pie charts of the percent expression of each toxin family average of all individuals. In B and C, (I) barplot of the log ranked expression of toxin genes, (II) stacked barplots with the percent expression of each toxin family by sampled individual, (III) pie charts of the percent expression of each toxin family average of all individuals. Photo Credit: A R. Wayne VanDevender (C. sasai), B Carlos E. Montaño-Ruvalcaba (C. petlalcalensis), and C Ramses A. Rosales-García (C. tzotzilorum)

RSEM results for the A average transcriptome of all individual of C. godmani; B average of the northern population; and C average of the southern population. In A (I) barplot of the log ranked expression of toxin genes, (II) stacked barplots with the percent expression of each toxin family by sampled individual, (III) pie chart of the percent expression of each toxin family for individual populations and for all the individuals. In B and C, (I) barplot of the log ranked expression of toxin genes; (II) pie chart of the percent expression of each toxin family for individual populations and for all the individuals. Photo Credit: Carlos E. Montaño-Ruvalcaba (C. godmani)
Our species tree—used to test for selection—did not recover C. godmani as a monophyletic lineage; instead we recovered the northern population as sister to C. tzotzilorum and C. petlalcalensis (Fig. 1). This structure might be a result of possible introgression between the sympatric populations of northern C. godmani and C. tzotzilorum (Fig 1), which future work may help elucidate.
Venom Gland Transcriptome Composition
Toxins were more highly expressed than nontoxins in all species analyzed (\(70.83 - 87.59\%\) of the total expression in transcripts per million (TPM; online resource 2, Table S3, S4, S5). For C. petlalcalensis and C. tzotzilorum, SVMPs were the most highly expressed toxin family followed by PLA\(_2\)s and SVSPs. In comparison, C. godmani and the individual C. sasai had higher expression of PLA\(_2\)s, followed by SVMPs and SVSPs.
Interestingly, C. godmani and C. tzotzilorum both exhibited intraspecific expression variation corresponding to geographic location. For example, PLA\(_2\) expression in the northern population of C. tzotzilorum was higher than in the southern population (36.79% compared to 10.58%, respectively), though SVMPs were the most highly expressed toxin family in both populations (39.07% compared to 61.25%, respectively; Fig. 3C II; online resource 1, Fig. S1). In C. godmani, the southern population exhibited a higher expression of PLA\(_2\)s compared to the northern population (44.06% compared to 26.93%, respectively) and SVMPs were more highly expressed in the northern population (47.61% compared to 31.53%, respectively; Fig. 3 B II and C II)
PLA\(_{2}\) phylogeny
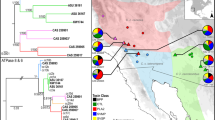
Previous studies on New World pitvipers have characterized five paralogs within PLA\(_2\)s: the K49 myotoxic gK PLA\(_2\)s; two basic PLA\(_2\)s gB1 and gB2 (the basic subunit of crotoxin), and two acidic PLA\(_2\)s gA1 and gA2 (the acidic subunit of crotoxin) (Whittington et al. 2018). We identified PLA\(_2\) sequences similar to the groups gK, gB1 and gA1 in most of the species, with the exception of gK PLA\(_2\)s in C. petlalcalensis, and gB1 in C. sasai (Fig. 4A). Expression of both PLA\(_2\)s related to the crotoxin-like subunits (gA2 and gB2) occurred only in a single individual, a small female from the southern population of C. godmani (CHFCB-0272; Fig. 4). Expression of these PLA\(_2\) subunits suggests that some individuals in the southern population of C. godmani may possess a neurotoxic venom. However, this hypothesis requires isolation of the toxins and confirmation of biological activity from the venom.

A Consensus maximum likelihood tree of the PLA\(_2\)s in Cerrophidion including PLA\(_2\)s used in Whittington et al. (2018), Mason et al. (2020), Neri-Castro et al. (2020b), and from Genbank (accession numbers in online resource 1, Table S1). The Cerrophidion PLA\(_2\)s (names highlighted in blue) are numbered by the toxin’s average expression for each species. Acidic and basic PLA\(_2\)s are identified by red and blue branches, respectively, based on the hypothetical isoelectric point of the amino acid sequences. Nodes with a black dot have \(> 75\) bootstrap support. Cerrophidion crotoxin subunit homologs are identified by a star (*) next to the name. B Species tree scaled with IQtree from the inferred Astral tree; lineages with crotoxin-like subunit homologs are purple; support values correspond to the Astral species tree. C Amino acid alignment of the gA2 clade and the hypothetical homolog from Cerrophidion, dots represent no change from the reference sequence (C._godmani_11). Sites represented with bars match cleavage sites identified in Whittington et al. (2018); a black star (*) at site 5 is the key substitution known in Bothriechis, Crotalus, and Gloydius; a red star (*) is at the alternative cleavage site in C. godmani based on the protein cutter tool from ExPASy server (https://web.expasy.org/peptide_cutter/.; Gasteiger et al. 2005) (Color figure online)
Differential Expression
To assess expression variation within a species, we tested for significant differences in toxin expression between geographically discrete populations using DESeq2 and edgeR R packages (Lov et al. 2014; Robinson et al. 2010). Cerrophidion petlalcalensis and C. sasai were excluded from these analyses due to a lack of population-level sampling. We found 29 toxins and 77 nontoxins differentially expressed between populations of C. godmani (Fig. 5; online resources 2, Table S12), including the toxin families CTL (10), SVMP (10), PLA\(_2\) (5), SVSP (4). The differentially expressed PLA\(_2\)s were similar to the gA1 (C._godmani_1, C._godmani_4) and gK myotoxins PLA\(_2\)s (C._godmani_26, C._godmani_41) based on our tree (Fig. 4). Additionally, we tested whether venom expression varied by body size by using snout-vent length (SVL) as a proxy for discrete “adult” and “juvenile” categorization, as we lacked replicates to directly test for differences between these groups. Four toxins and 38 nontoxins were differentially expressed (Fig. 5), including the toxins families CTL (1), PLA\(_2\) (1), SVMPII (1), SVSP (1). All of the differentially expressed toxins had lower expression in the smaller individual, and the differentially expressed PLA\(_2\) grouped with the gK myotoxins (C._godmani_42). Testing differential expression on the added expression of toxin families, we also found that SVMPIs and SVMPIIs are differentially expressed across SVL, with higher expression in the larger individuals (online resource 1, Fig. S2; online resources 2, Table S13). These data suggest that there are ontogenetic changes in the venom. At the level of entire toxin gene families, no families were differentially expressed by population with either method.

Heatmap showing the log TPM expression of toxins identified as differentially expressed in C. godmani ordered by the average expression. In the left columns (Pop & SVL) the darker colors represent significant differential expression agreement by both DESeq2 and edgeR (\(FDR<0.05\))
For C. tzotzilorum, we found 10 toxins and 42 nontoxins differentially expressed between populations, including the toxin families CTL (3), PLA\(_2\) (1), SVMPIII (2), and SVSP (4) (online resource 1, Fig. S3). The differentially expressed PLA\(_2\) (C._tzotzilorum_1) had the highest average expression, and grouped with the gA1 PLA\(_2\)s in our phylogeny. Only one toxin and 16 nontoxins were differentially expressed across SVL; however, DESeq2 alone indicated that 12 toxins were differentially expressed including the toxin families CTL (3), PLA\(_2\) (2), SVMP (5), SVSP (1), and vascular endothelial growth factor (VEGF; 1) (online resource 2, Table S14). The PLA\(_2\) C._tzotzilorum_1 was differentially expressed for SVL and between populations. Testing for the added expression of toxin families (online resources 1, Fig. S4), we found that PLA\(_2\)s were differentially expressed only by DESeq2, with higher expression in the northern population (Fig. 2C; online resource 2, Table S15). Across SVL, DESeq2 found hyaluronidase (HYAL), SVMPIII, and VEGF as differentially expressed with higher expression of HYAL and SVMPIII in smaller individuals and higher expression of VEGF in larger individuals.
Signatures of Selection in C. godmani
We tested for signatures of selection within C. godmani given our sample size and geographic coverage. However, we were unable to compare among species as identification of orthologous toxins is difficult due to the high abundance of paralogs in several toxin families.
Sequence Diversity
After filtering by coverage and excluding invariant genes, we had information on 68 toxins and 4,508 nontoxins (Table 2; online resource 2, Table S7). While the number of variants per Kbp was similar between toxins and nontoxins (\(\bar{x}_{toxins}=6.42\pm 3.58\), \(\bar{x}_{nontoxins}=6.77 \pm 4.31\)), nonsynonymous SNPs were proportionally greater in toxins than nontoxins (61 and 26%, respectively; \(\chi ^2 = 230.22\), df = 1, \(p < 0.01\)). The number of variants per Kbp (squared root transformed for normality) was not explained by gene type (\(R^2 < 0.01,\, p = 0.68\), \(b = 0.01\)); however, the number of nonsynonymous variants per Kbp (centered ratio log transformed) was explained by gene type (\(R^2 = 0.01,\, p < 0.01,\, b = 99.1\)) (online resource 1, Fig. S5). We found no significant relationship between nucleotide diversity and gene type (\(R^2 < 0.01,\, p = 0.30,\, b = 0.04\)) (online resource 1, Fig. S7).
Test of Selection
We first used Tajima’s D to test for signatures of selection. The mean Tajima’s D for nontoxins was significantly less than 0 (\(\bar{x} = -0.19 \pm 0.78,\, t= -16.19,\, df= 4507,\, p < 0.01,\, b = 0.48\)), providing evidence for either directional selection of nontoxins or population expansion after a recent bottleneck (Fig. 6A). However, the mean Tajima’s D for toxins is not significantly different from 0 (\(\bar{x} = -0.04 \pm 0.82,\, t = -0.35,\, df = 67,\, p = 0.73\)). This potentially implies that toxins are evolving along a mutation–drift equilibrium or that toxins are under highly variable selection pressures, which neutralizes the signal of the mean value. However, the variance of estimates of Tajima’s D for toxins and nontoxins is not significantly different (\(F = 0.89,\, df_1 = 4507,\, df_2= 67,\, p = 0.46\)). Additionally, toxins and nontoxins are not significantly different in their estimates of Tajima’s D (\(R^2 < 0.01,\, p = 0.11,\, b = 0.10\)), suggesting that there are no differences in selection for toxins and nontoxins.

Selection plots. Top: estimates of selection using A Tajima’s D, B \(F_{ST}\), and C Likelihood Ratio Test (LRT) for the BUSTED model for Toxins and Nontoxins, each with the Nontoxin 95th percentile (dotted lines) to identify outlier toxins. The toxin family and the rank based on highest-to-lowest average expression in the transcriptome is displayed for toxins which fall outside the 95th percentile. Bottom: Linear regressions of the Toxin’s mean expression (Average TPM) and estimates of selection including D Tajima’s D, E \(F_{ST}\), and F LRT of the BUSTED model. For Tajima’s D, dotted lines are regressions considering all the transcripts (center), just positive values (top) and just negative values (bottom)
We also calculated Tajima’s D independently for nonsynonymous and synonymous variants (online resource 1, Fig. S7). For both substitution classes, mean Tajima’s D values for nontoxins and toxins were \(<0\). However, only nontoxins were significantly different from 0 for synonymous substitutions. For both substitution types, toxins and nontoxins did not differ significantly (Synonymous \(R^2 < 0.01,\, p = 0.21,\, b = 0.05\); Nonsynonymous \(\chi ^{2} = 1.84,\, df = 1,\, p = 0.17,\, b = 0\)).
Next, we calculated \(F_{ST}\) between the northern and southern populations of C. godmani (Fig. 6B). The \(F_{ST}\) values were not significantly explained by the gene type (\(R^2 < 0.01,\, p = 0.60,\, b = 0.01\)). Although the difference was nonsignificant, toxin genes had a higher mean \(F_{ST}\) and higher standard deviation than the nontoxin genes(\(\bar{x}_{nontoxins} = 0.19 \pm 0.21, \bar{x}_{toxins} = 0.21 \pm 0.26\)).
We used the Branch-Site Unrestricted Statistical Test for Episodic Diversification (BUSTED) model of HyPhy to detect signals of selection (Murrell et al. 2015). This model is based on the rate of synonymous and nonsynonymous variants (\(\frac{dN}{dS}\)) and tests for evidence that at least one site on at least one test branch has experienced positive selection. To compare toxins against nontoxins, we used a nonparametric Kruskal–Wallis test as the LRT results were not normal (Fig. 6C). We found a significant relationship between the class of genes and the LRT (\(\chi ^2 = 11.79, df = 1,\, p < 0.01,\, b = 0.54\)) with the toxins having a higher mean which indicates that they are more likely to experience selection than nontoxins (\(\bar{x}_{nontoxins} = 0.29 \pm 1.04, \bar{x}_{toxins} = 0.64 \pm 1.46\)). However, only one of the toxins (SVMPIII_39) analyzed was found to be under significant positive selection (LRT \(p < 0.05\)).
Relationship Between Expression and Sequence Evolution in Toxins
To test for a relationship between the expression level in toxins and the sequence divergence, we used the natural log of the average expression of the toxins (TPM). We then evaluated if the differentially expressed genes between populations were under stronger selective pressures or had higher sequence divergence. First, we performed linear regression between the average expression and Tajima’s D. Our models showed that Tajima’s D increased with average expression, however, the relationship was not significant and explained very little of the variation (\(R^2= 0.04,\, p= 0.10\)). Next, we tested if toxins with Tajima’s D \(>0\) or \(<0\) (i.e., those experiencing balancing or positive selection pressures, respectively) had different relationships with expression. In concordance with the full analysis, toxins with Tajima’s D \(<0\) show a positive trend with higher selection pressures being exerted on lowly expressed toxins. This trend was inverted in toxins with Tajima’s D \(>0\); however, neither group was significant (Fig. 6D). Similarly, differential expression did not predict the strength of selection in toxins (\(R^2 < 0.01,\, p = 0.91\)) (online resource, Fig. S8). Expression level also failed to significantly predict \(F_{ST}\), but did show a trend of increased population differentiation in the toxins with higher expression (\(R^2= 0.05,\, p=0.06\)) (Fig. 6E). Differential expression between populations was also not a significant predictor of toxin \(F_{ST}\) (\(R^2 = 0.04,\, p = 0.10\)), which suggests that changes in the expression are not correlated with divergence in the sequences of the genes between populations (online resource, Fig. S8). The nonparametric test of the LRT and the differential expressed toxins was significant (\(\chi ^2 = 4.76,\, p = 0.03\)); however, the LRT was not significantly correlated with the average expression of toxins (\(R^2 = 0.01,\, p = 0.56\)) (Fig. 6F). Nucleotide diversity (\(\pi\)) is not correlated with the differential expression between populations (\(R^2 < 0.01,\, p = 0.72\)), or to the average expression of the genes (log transformed for normality; \(R^2 = 0.03,\, p = 0.14\); online resource 1, Fig. S8). Overall, our results suggest that there is no correlation between expression level and signals of selection in toxins.
Selection on Individual Toxins
We used the distribution of nontoxin values as a reference to identify individual toxins that are outside the 95th percentile of the nontoxins for Tajima’s D, \(F_{ST}\), and BUSTED model LRT (Fig. 6A–C). With Tajima’s D, we identified four toxins, three with values greater than 0 which suggests balancing selection (1 VEGF, 2 SVSPs), and one with a value less than 0 suggesting positive selection (SVMPI). Similarly, we found seven toxins with significantly higher \(F_{ST}\) values (1 VEGF, 3 SVMPIIIs, 3 SVSPs), and eight toxins with a significantly higher LRT value (3 CTLs, HYAL, 2 SVMPIs, SVMPII, SVMPIII, SVSP). From these toxins, only three were found across multiple methodologies (VEGF_30, SVSP_52, SVMPIII_39).
Discussion
Venom Composition and Expression Variation
The venom of Cerrophidion is dominated by five toxin families: SVMPs, PLA\(_2\)s, SVSPs, CTLs, and BPPs (Fig. 2, 3). These toxin families are generally found in high abundance within the venoms of Crotalinae species (Amazonas et al. 2018; Hofmann et al. 2018; Almeida et al. 2021; Strickland et al. 2018a; Tasoulis and Isbister 2017). The venoms of Cerrophidion species likely have proteolytic and hemolytic activity given the high percentages of SVMPs, as well as myotoxic activity due to the presence of PLA\(_2\)s similar to the gK PLA\(_2\)s of Crotalus (e.g., Gutiérrez et al. 1985; Gené et al. 1989; Arni et al. 1999; Lizano et al. 2000; Chen et al. 2004; Durban et al. 2011; Lomonte et al. 2012, 2014).
We found significant intraspecific variation in toxin expression in C. tzotzilorum (Fig. 2C), which could indicate local adaptation between the northern and southern populations (Fraser et al. 2011; Strickland et al. 2018b). This has been observed in other pitviper species, where rapid toxin expression evolution occurred due to local adaptation between mainland and island populations; however, this was restricted to lowly expressed toxins (Margres et al. 2016). Here, we found both lowly and highly expressed toxins, including the most highly expressed toxin, were differentially expressed between populations of C. tzotzilorum (online resource 1, Fig. S3). According to Margres et al. (2016) the observed difference herein might be explained by the difference in divergence time. Cerrophidion lineages are more ancient compared to the intraspecific lineages studied by Margres et al. (2016). Margres et al. (2014a), observed a similar pattern to ours when looking at range-wide geographic variation in toxin expression as opposed to island vs mainland in C. adamanteus.
Lastly, the distribution of C. tzotzilorum is relatively small and discontinuous, which may facilitate either local adaptation or drift in these isolated populations resulting in the differential expression of toxins. Such local adaptation in different geographic regions has been observed in other species with broad distributions, such as Crotalus scutulatus (Strickland et al. 2018b). In contrast, our toxin sequence evolution results for C. godmani suggest that mutation–drift could also be driving evolution in toxin differential expression between populations as it does for sequence evolution.
We similarly found that C. godmani showed significant variation between the southern and northern populations (Fig. 3B and C). In the northern population of C. godmani, the PLA\(_2\)s were expressed less than in the southern population, however, the toxin with the highest expression in the northern population was a PLA\(_2\) not expressed in the southern population. This toxin is an acidic PLA\(_2\) (gA1) related to PLA\(_2\)s from Bothrops and sister to a clade of PLA\(_2\)s from Gloydius (Figs. 4, 5). The southern population of C. godmani, in general, exhibited higher expression of PLA\(_2\)s than the northern population; however, this difference was not significant for total PLA\(_2\) expression. Nonetheless, there were several PLA\(_2\) isoforms differentially expressed between the two populations. Notably, one individual (CHFCB-0272) expressed PLA\(_2\)s similar to both subunits of crotoxin-like toxins of other genera. The remaining individuals showed nearly no expression of these PLA\(_2\)s. The venom of the southern population also showed a large percentage of SVMPs. The venom of CHFCB-0272 was composed of \(21.60\%\) crotoxin subunits, \(25.48\%\) other PLA\(_2\)s, \(26.62\%\) SVMPs, and \(26.30\%\) other toxins families.
Cerrophidion petlalcalensis exhibited little variation in the composition of toxins in its venom (Fig. 2B). Our samples came from populations in Veracruz, Mexico. New populations have recently been discovered in Oaxaca, Mexico, several kilometers south of Veracruz (Torre-Loranca et al. 2019). We lack information regarding the venom of these populations and additional sampling might recover intraspecific venom variation. However, C. petlalcalensis diverged from C. tzotzilorum in the Pliocene, probably by a vicariant event due to the tectonic movement that removed the highlands connections between both sides of the Isthmus of Tehuantepec (Daza et al. 2010; Castoe et al. 2009). It is likely that this lineage underwent a bottleneck post-divergence resulting in a population with low genetic diversity given the short branch lengths between C. petlalcalensis individuals in our species tree. This low genetic diversity may have resulted in largely homogeneous venom phenotypes within this population. Samples from the newly discovered population would facilitate our understanding of the evolutionary history of the species as they would allow us to assess the genetic diversity of the species and test for differential expression of the toxins among populations as we did with C. godmani and C. tzotzilorum.
The sampled individual of C. sasai differed in the percentages of toxin families from what has been previously described for this species using proteomics (Fig. 2A). Here, we found a higher expression of PLA\(_2\)s and lower expression of SVSPs than Lomonte et al. (2012). The differences might be methodological artifacts, given that we investigated the venom gland transcriptome and Lomonte et al. (2012) analyzed the venom directly. Transcriptome and proteome comparisons have variable results, with high or low correlation depending on the methodologies used (Rokyta et al. 2015a; Durban et al. 2011). Another potential source of variation might be that Lomonte et al. (2012) used pooled venom samples from several individuals and therefore profiling within species variation in the expression of toxin families. However, given the isolated distribution of C. sasai and the low genetic variation within the species (Sasa 1997), the venom likely has little variation similar to C. petlalcalensis.
PLA\(_2\)s in Cerrophidion
The evolution of PLA\(_2\)s has been extensively studied, particularly in rattlesnakes (Glenn et al. 1994; Wooldridge et al. 2001; Mackessy 2008; Dowell et al. 2016, 2018; Whittington et al. 2018). Unique combinations of different PLA\(_2\) paralogs have been found in the venom of rattlesnakes both between and within species, and ancestral character reconstruction suggests that the specific arrangement is the result of gene loss (Dowell et al. 2016, 2018). The most recent common ancestor (MRCA) of rattlesnakes likely possessed an expanded array of PLA\(_2\) paralogs, including the two subunits that form the crotoxin-like neurotoxins (Dowell et al. 2016). Cerrophidion species show a high diversity of PLA\(_2\)s with unique paralog compositions based on our phylogeny. Cerrophidion petlalcalensis has gB1 and gA1 PLA\(_2\)s and lacks gK PLA\(_2\)s; C. sasai has gK and gA1 PLA\(_2\)s and lacks gB1 PLA\(_2\)s. Cerrophidion tzotzilorum has copies of gA1, gB1, and gK PLA\(_2\)s in both populations and differs in the expression of the paralogs. Cerrophidion godmani seems to have different PLA\(_2\) gene composition between populations as only the northern population has the gA1 PLA\(_2\) C._godmani_4, while only the southern population has the gA1 PLA\(_2\) C._godmani_1. Additionally, the southern population seems to be polymorphic given the presence of the crotoxin-like subunits in CHFCB-0272, this individual expresses all the other paralogs as well. The composition of PLA\(_2\) genes in Cerrophidion seems to have evolved as in Crotalus, with unique arrangements originating from gene loss. However, there are individuals of C. godmani that express all the PLA\(_2\) groups, as in the hypothetical rattlesnake MRCA proposed by Dowell et al. (2016). This suggests that the genotype with all proposed PLA\(_2\) paralogs was present in the MRCA of Cerrophidion and rattlesnakes, and likely in the MRCA of all New World pitvipers.
The C. godmani acidic PLA\(_2\), similar to the gA2 group, exhibits a proline in position 127 (Fig. 4C at site 5), hypothesized to be the ancestral state of acidic PLA\(_2\)s prior to the evolution of the gA2 group (Whittington et al. 2018). A proline in that position blocks the cleavage recognition residual (phenylalanine) in position 126 (Keil 1992) and cleavage of this site is necessary for the folding of a functional acidic crotoxin subunit (Whittington et al. 2018). A substitution of proline for other amino acids unlocks the cleavage recognition residual specific for chymotrypsin-like SVSPs (Whittington et al. 2018; Keil 1992). Bothriechis, Crotalus, and Gloydius have a serine at position 127 in their homologs of gA2 PLA\(_2\)s and the recently discovered melanurutoxin from Mixcoatlus melanurus has a leucine, demonstrating that different amino acids at position 127 have the potential to unlock this site (Neri-Castro et al. 2020b; Whittington et al. 2018) (Fig. 4C). Whether the toxins present in C. godmani are functional as a chaperone and phospholipase activity agonist for the basic subunit is still unknown (Radvanyi and Bon 1982). If they are neurotoxic, this would mean that a different cleavage recognition residue at site 5 might facilitate cleavage. The homolog of the acidic subunit of C. godmani shows sequence differences upstream of position 127, with a tryptophan and two consecutive phenylalanines at positions 124-126. This may cause local changes in secondary structure or protease recognition that may facilitate an alternative cleavage site in this homolog. According to the peptide cutter tool from the ExPASy server (https://web.expasy.org/peptide_cutter/; Gasteiger et al. 2005) (online resource 1, Fig. S9), tryptophan or phenylalanine residues might act as cleavage recognition sites, although the tryptophan at position 124 has a higher probability of being cleaved (Fig. 4C, red asterisk)). Additionally, this tryptophan residue is highly solvent exposed (site-specific solvent accessible surface area (SASA) 104.31 at position 126, online resource 2, Table S2), which would permit attack by a protease. An alternative hypothesis is that the acidic subunit homologs of C. godmani have functions similar to other acidic PLA\(_2\)s given that the sequence of C. godmani_4 and the gA1s from different taxa in the same group (Gloydius and Bothrops) have tryptophan and two phenylalanines at positions 124–126. The expression levels of the subunits support the former hypothesis for the presence of a crotoxin-like toxin; representing 11.89 and 9.70% of the total venom expression for the acidic and basic subunits, respectively, within CHFCB-0272. Further research on the biological activity and on the proteomics of the venom is needed to discover the function of these PLA\(_2\)s. Clinical records could also help to understand effects of the venom, although snake bite reports from Mexico and Central America usually lack a trustworthy identification of the species involved in the envenomation (Neri-Castro et al. 2020a; Gutiérrez 2014).
Toxin Sequence Variation and Selection
Our results suggest that toxins and nontoxins are evolving under the same evolutionary pressures supporting a main influence of mutation–drift equilibrium. Tajima’s D distributions for toxins and nontoxins were similar with slightly more variation in toxins. However, an F-test showed that the variance of toxins and nontoxins do not differ significantly (\(F = 0.85,\, p = 0.13\)), suggesting it is unlikely that diverse selection pressures are acting on the toxins and therefore masking a true significant difference (Rautsaw et al. 2019). A pattern of toxin evolution by mutation–drift equilibrium has been found in other species including Bothriechis nigroviridis, B. nubestris, Crotalus cerastes, Protobothrops mucrosquamatus, and Sistrurus catenatus (Rautsaw et al. 2019; Aird et al. 2017; Ochoa et al. 2020; Mason et al. 2020).
Several factors might influence the evolutionary history of toxins, particularly effective population size and diet (Ochoa et al. 2020; Holding et al. 2021b; Mason et al. 2022). In the case of C. godmani, data regarding population sizes are lacking, though Campbell and Lamar (2004) suggested they are abundant within their range. It is likely that they form isolated populations with low gene flow given that they inhabit high elevation, discontinuous mountain ranges. The mean \(F_{ST}\) (Nontoxins = 0.181, Toxins = 0.182) suggest moderate differentiation between populations, indicating low gene flow at least between the populations sampled herein. Their isolated, patchy distribution might result in smaller effective population sizes, thereby increasing the influence of drift in these populations and potentially masking the effects of selection. Including more populations in future studies might change our current interpretation as the population genetic structure of the species is unknown. Margres et al. (2019) found that species with more generalist diets do not diverge in venom unless there is absence of gene flow. Cerrophidion godmani is considered a diet generalist (Schramer et al. 2018; Campbell and Solórzano 1992), and the mean values of \(F_{ST}\) suggest partial differentiation between southern and northern populations. Both toxin and nontoxin genes show differentiation between populations but were not significantly different from one another, suggesting that toxins are not experiencing directional selection. Instead, balancing selection may play a larger role in toxin differentiation in this species (Margres et al. 2019). The venoms of species with phylogenetically diverse diets evolve under balancing selection that maintains an increased number of toxins (Holding et al. 2021b). Our results partially support this pattern in C. godmani, as its diet is phylogenetically diverse and its venom could be considered complex considering the number of toxins in the consensus transcriptome (a total of 117). However, our results do not suggest a predominant influence of balancing selection on the toxins. Instead, toxins appear to be in mutation–drift equilibrium within C. godmani. These results are limited by the sample size and filtering protocols used here. Specifically, toxins that could be experiencing directional selection in some individuals or are new paralogs of toxin genes might have not been considered as they are not present in most of the individuals and thus not analyzed. A population-wide study would help to further clarify the evolutionary history of these toxins, including genes homologous to crotoxin subunits found in one individual. Only a small number of toxin genes seem to be experiencing directional or balancing selection. According to Holding et al. (2021b), the primary toxin families associated with more complex venom in snakes with phylogenetically diverse diets are PLA\(_2\)s, SVMPs, and SVSPs. Further, Schield et al. (2022) found signals of balancing selection driving sequence evolution in PLA\(_2\)s, SVMPs, and SVSPs within and between Crotalus oreganus and Crotalus viridis. We would expect that these families evolved by balancing selection, assuming a complex venom and a phylogenetically diverse diet. In Cerrophidion godmani, the toxins subjected to balancing selection belong to the toxin families VEGF and SVSP(2), according to Tajima’s D. Therefore, our results do not support the balancing selection hypothesis as more toxins appeared to be in mutation–drift equilibrium. However, our analysis is focused only on intraspecific variation, whereas the study of Holding et al. (2021b) addressed variation on both larger taxonomic and time scales. A more extensive study on Neotropical pitvipers might show agreement with what is known for Nearctic pitvipers, though it is also possible that given the different evolutionary pressures and demographic histories experienced by Nearctic and Neotropical snakes, similar patterns may not be expected to be maintained.
A few toxins—including SVMP, SVSP, and VEGF toxin families (Fig 6A–C)—were regularly found outside of the nontoxin’s \(95^{th}\) percentile distribution in our selection analyses. These toxin families have a wide variety of biological functions; however, they might be most closely associated with hemorrhagic effects in their prey (Fox and Serrano 2005; Yamazaki et al. 2009; Jones et al. 2022). Cerrophidion godmani has a hemorrhagic venom (Jones et al. 2022), which suggests that these few toxins have key functions in prey acquisition by causing massive tissue degradation and internal bleeding.
Expression is not correlated with signals of selection on toxin genes. We found a nonsignificant trend of increasing values of Tajima’s D, \(F_{ST}\) and LRT with higher average expression in the toxins of the venom. This trend suggests that highly expressed toxins are evolving by balancing selection and toxins with low expression have more signals of positive selection. This pattern contrasts with what was found for Crotalus cerastes, where the lower expressed toxins are evolving by balancing selection (Rautsaw et al. 2019). Although expression does not correlate with the signals of sequence selection there is clear expression differentiation between populations in C. godmani given the number of genes that show differential expression (Fig. 5), which might indicate that selection in the venom is acting on levels of expression instead of on sequence evolution.
Conclusions
We found evidence of venom variation between and within species: Cerrophidion petlalcalensis has a homogeneous venom, with little variation between individuals; C. godmani and C. tzotzilorum showed intraspecific variation in the toxins present and in the expression of individual toxins. This variation was observed between geographic populations, as well as by body size, suggesting possible ontogenetic shifts in the venom composition. Overall, sequence variation in toxins and nontoxins in C. godmani was not significantly different across multiple tests for selection. Instead, the evolution of toxin genes seems to be driven by mutation–drift equilibrium with no relationship to toxin expression. The presence of myotoxic PLA\(_2\)s gK seems to be widespread in most Cerrophidion species, except C. petlalcalensis. Additionally, acidic and basic subunits of homologous crotoxin-like PLA\(_2\)s were discovered in one specimen of C. godmani, which suggests that this species might have the heterodimeric crotoxin-like neurotoxin. These PLA\(_2\)s were found in a single, southern population individual, which suggests there is intraspecific variation for the presence of this trait as observed in other pitviper genera such as Crotalus. Overall, our combined results support the hypothesis that drift is potentially the key evolutionary force shaping sequence evolution in highland pitviper species.
Materials and Methods
Sample Collection
We collected six specimens of C. godmani from two populations in Mexico: two from the Central Highlands of Chiapas (Las Margaritas), and four from the Sierra Madre de Chiapas, close to the border with Guatemala (Union Juarez); four specimens of C. tzotzilorum from two populations in the Central Highlands of Chiapas, Mexico (Rayon Mescapala and San Cristobal de las Casas); three specimens of C. petlalcalensis from San Andres Tenejapa (Veracruz, Mexico); and one specimen of C. sasai from Las Nubes Coronado, San Jose, Costa Rica (Fig. 1, Table 1). We extracted venom from the individuals by allowing the snakes to bite a sterile cup covered with parafilm. Four days after the venom extraction, we euthanized the snakes with an injection of sodium pentobarbital (100 \(\frac{mg}{kg}\)) and excised the venom glands in order to maximize mRNA transcription (Rotenberg et al. 1971). We preserved the glands in RNAlater at \(4^{\circ }\)C, then moved the glands to permanent storage at \(-80^{\circ }\)C. All protocols involving live snakes followed ASIH guidelines and were approved by the Clemson University Animal Care and Use Committee (Animal Use Protocol 2017-067).
cDNA Libraries Prep and Sequencing
RNA extraction and sequencing were done following Hofmann et al. (2018). Briefly, we diced the venom glands and placed them in a TRIzol solution (Invitrogen). The homogenized mixture was transferred it to a phase-lock heavy gel tube (5Prime). We waited until the cells were lysed, and then isolated and purified RNA using chloroform followed by isopropyl alcohol and ethanol precipitation. We quantified RNA using a Qubit RNA BroadRange kit, and checked the quality of the RNA using a Bioanalyzer 2100 with an RNA 6000 Pico Kit (Agilent Technologies). The mRNA was isolated with NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB#E7490). We prepared cDNA libraries using a NEB Next Ultra RNA Library Prep Kit for Illumina (NEB#E7530) following the manufacturer’s recommendations. We used a fragmentation time of 13 minutes, 30 seconds to achieve a target mean fragment size of 400 bp, and 14 PCR cycles for amplification of double-stranded cDNA libraries. We quantified library yield and quality with a Bioanalyzer 2100, and determined the total amplifiable concentration of cDNA using KAPA qPCR. We pooled equal concentrations of samples and assessed the final concentration and quality of our pooled libraries with a Bioanalyzer and with KAPA qPCR. The libraries were sequenced on an Illumina HiSeq2500 or NovaSeq 6000 platform at the Translational Science Laboratory in the College of Medicine at Florida State University.
Transcriptome Assembly
We trimmed the adaptors and low-quality sequences of the raw Illumina sequences with TrimGalore! (v0.6.6, https://github.com/FelixKrueger/TrimGalore), set to trim reads with a quality score lower than 5 and a length below 75 bp. We merged the forward and reverse reads with PEAR (v0.9.6) (Zhang et al. 2014). For the subsequent analyses we used GNU parallel 20201122 software for multiprocess computing (Tange 2020). We used Trinity (v2.11.0) (Grabherr et al. 2011), SeqMan Ngen (v14) (using the Lasergene DNAStar software package; Madison, WI, USA: https://www.dnastar.com/t-nextgenseqman-ngen.aspx), and Extender (Rokyta et al. 2012) to de novo assemble contigs following the recommendations of (Holding et al. 2018). We merged the assemblies and ran cd-hit-est (Fu et al. 2012) with a sequence identity threshold of 1 to filter redundancy in our combined assembly. We annotated toxin and nontoxin sequences with ToxCodAn (Nachtigall et al. 2021), a method that uses general Hidden Markov Models (gHMM) to identify the toxin genes. We complemented the output of ToxCodAn with a manual annotation of toxin sequences. Manual annotation was done following the methodology in (Hofmann et al. 2018). Briefly, we ran blastx (v2.10.1) against UniProt animal venom proteins and toxins database (https://www.uniprot.org/program/Toxins), then we used SignalP (Petersen et al. 2011) and cd-hit-est to automatically annotate the genes and extract the coding sequences from the contigs by comparing the contigs to a database of existing pitviper transcripts (Rokyta et al. 2013). The genes that were not automatically annotated were manually annotated using the output of blastx. Specifically, we compared all potential open reading frames (ORF) to the blastx results and annotated the best-matched ORF, with regard to percent identity and sequence length, with the appropriate toxin family. We concatenated the output of ToxCodAn and of the manual annotation and ran cd-hit-est with a threshold of 1 to remove the redundant sequences. We ran ChimeraKiller (https://github.com/masonaj157/ChimeraKiller) to eliminate chimeric sequences. Briefly, reads are mapped to the annotated transcriptome and transcripts with zero coverage at any position are removed. Chimeric transcripts are then reported by searching for a difference \(> 75\%\) (-d 0.75) in the average length of reads on either side of a given site based on the average read size. A final manual revision was done to check for misplaced sequences. We merged the sequences cleaned by ChimeraKiller and ran cd-hit-est with a threshold of 0.99 to keep only one copy of each gene. We made a consensus species transcriptome by concatenating the transcriptomes of all individuals for each species and running cd-hit-est with a threshold of 0.98 to remove any variants between individuals. Additionally, we filtered sequences with internal stop codons, no stop codons, or an incomplete translation frame.
Expression Quantification and Differential Expression
We used the consensus transcriptomes generated for each species and the merged reads to calculate the expression of the genes using RSEM (Li and Dewey 2011) with Bowtie2 as the aligner with default settings. We then removed the toxins that were below the 5% percentile of average expression to avoid including contamination from highly expressed toxins from other samples sequenced with our samples. We repeated the RSEM (Li and Dewey 2011) expression analysis with the reduced data set (see online resource 2, Table S3, S4, S5, S6). For visualization, we took the mean transcripts per million reads (TPM) for each toxin in a given species. Results were plotted using (R Core Team 2020) with the plotting script included in ToxCodAn. Names of the toxins in the figures were set as the toxin family and the ranking of average expression of the toxin for the species (see online resource 2, Table S8, S9, S10, S11). We used expected counts from RSEM and R packages DESeq2 (Lov et al. 2014) and edgeR (Robinson et al. 2010) to test for differences in expression between northern and southern populations for C. godmani and for C. tzotzilorum. Cerrophidion petlalcalensis and C. sasai were excluded from differential expression analyses given a lack of population-level sampling and body size variation in the samples. We used SVL to test if body size influences gene expression as a proxy for ontogenetic shifts. Although sexual dimorphism of the venom has been found in some species (Zelanis et al. 2012, 2016), that is not always the case (Saviola et al. 2015; Franco-Servín et al. 2021). We did not add sex as a factor in our experimental design as we lacked of biological replicas of at least one of the sexes in every species. For DESeq2, we used a Wald significance test with a local fit dispersion and used a false-discovery rate (FDR) \(<0.05\) as the threshold for differentially expressed genes. For edgeR, we used a likelihood ratio test (LRT) and, as in DESeq2, we used a FDR \(<0.05\) as the significance threshold. We repeated DESeq and edgeR test for the accumulated expression of each gene family including all nontoxin genes as both packages use all information to fit the dispersion. We created heatmaps of the expression of the toxins in R with package pheatmap (Kolde 2019) to visualize differences in expression and differential expression among populations or across SVL.
PLA\(_2\) Phylogeny and Proteomic Alignment
To reconstruct the PLA\(_2\) phylogeny for Cerrophidion, we combined the PLA\(_2\) sequence data from (Whittington et al. 2018; Neri-Castro et al. 2020b; Mason et al. 2020), and additional PLA\(_2\) sequences available in GenBank (see online resource 2, Table S1). We extracted the annotated PLA\(_2\)s of the consensus transcriptome for each of our species and aligned them with the other sequences using MAFFT v7.475 (Katoh and Standley 2013). We used CIAlign v1.0.10 and trimal v1.4.rev15 to clean and trim the alignment (Tumescheit et al. 2020; Capella-Gutiérrez et al. 2009). As the Python bivittatus PLA\(_2\) was discarded by CIAlign we concatenated the sequence at the end of the cleaning and trimming process and realigned with MAFFT. We used this alignment as input for IQTree using ModelFinder and 1000 ultrafast bootstrap replicates (Kalyaanamoorthy et al. 2017; Hoang et al. 2018; Nguyen et al. 2015). The PLA\(_2\)s we generated were translated with Biopython (Cock et al. 2009) while the PLA\(_2\)s from GenBank were downloaded as amino acid sequences. The theoretical isoelectric points were calculated with biopython (Cock et al. 2009). The translated sequences were aligned with omega clustal algorithm in Geneious Prime 2020.2.4 (https://www.geneious.com/; Sievers and Higgins 2018). The cleavage sites of the sequences similar to the acidic subunit of crotoxin were estimated using the ExPASy Peptide Cutter tool (https://web.expasy.org/peptide_cutter/; Gasteiger et al. 2005). We modeled the structure of the homolog of the gA2 following (Whittington et al. 2018). We used the X-ray crystal structure of the Crotoxin from Crotalus durissus terrificus (pdb:3R0L; Faure et al. 1991) as template for modeling. The model was built using Modeller v 10.2 (Eswar et al. 2006). We built 10 independent structures with the method slow VFTM optimization for 500 iterations, and slow MD refinement. The refinement was repeated 4 times. The models were evaluated using the DOPE-HR. We choose the model with the lower DOPE-HR score to calculate the SASA using GetArea (http://curie.utmb.edu/getarea.html; Fraczkiewicz and Braun 1998).
SNPs Calling and Selection Analysis
We followed the methodology of Rautsaw et al. (2019) for variant calling with our C. godmani data. We excluded the other species for this analysis due to low sample size or lack of population sampling. Briefly, we mapped the merged reads to the consensus transcriptome with BWA-MEM (Li 2013). We used Picard v2.12.1 (http://broadinstitute.github.io/picard/) to sort and index the aligned reads. We followed GATK v3.8.1 and v4.1.9 (software.broadinstitute.org/gatk/) SNP calling pipeline (Auwera et al. 2013); we performed a local realignment of regions with indels, removed reads shorter than 120 nucleotides, called variants, performed joint genotyping, and filtered SNPs. We phased the variants using WhatsHap v0.15 (Martin et al. 2016). We then filtered mapped contigs with a transcript coverage of 0 for more than 5% of total length in order to avoid partial transcripts and poorly mapped reads. We kept genes for analysis if they were present in at least three individuals based on the coverage filter and if they had at least one SNP. We additionally removed toxins that were under the 5% percentile in the first RSEM analysis.
We used Tajima’s D, Weir and Cockerham’s \(F_{ST}\), and Nucleotide Diversity (\(\pi\)) to test if toxins have more selective pressures than the nontoxin genes following (Rautsaw et al. 2019). We used several selection metrics as some of them can potentially be influenced by demographic history and population structure (e.g., Tajima’s D; Nielsen 2005). Additionally, we used the BUSTED model from HyPhy 2.5.31(MP) (Murrell et al. 2015) to test for signals of positive selection in whole genes and used the LRT results to test for significant differences in selection between toxins and nontoxins. First we separated nonsynonymous and synonymous SNPs using SnpEff v.5.0 using our consensus transcriptome as a reference (Cingolani et al. 2012). We calculated the SNPs per kilobase for each gene. We use this value to test for significant differences between toxins and nontoxins using linear regression. We also tested if the type of mutation (i.e., nonsynonymous or synonymous) was significantly associated with the type of gene (toxin or nontoxin) with a \(\chi ^{2}\) test. We used vcftools v0.1.16 (Danecek et al. 2011) to calculate the nucleotide diversity (\(\pi\)), Tajima’s D per gene and per site, and Weir and Cockerham’s \(F_{ST}\) comparing northern and southern populations. We performed all statistical analyses in R software (R Core Team 2020).
Testing for selection with HyPhy 2.5.31(MP) (Murrell et al. 2015) requires a species tree. To generate a phylogeny, we recovered 3,530 single-copy BUSCO loci with BUSCO v5.2.2 (Simão et al. 2015). Next, we aligned each locus with MAFFT v7.475 (Katoh and Standley 2013), and cleaned the alignments with CIAlign v1.0.10 (Tumescheit et al. 2020). We made preliminary gene trees with IQtree using ModelFinder and 1000 ultrafast bootstrap replicates (Kalyaanamoorthy et al. 2017; Hoang et al. 2018; Nguyen et al. 2015). We used the consensus trees and the cleaned alignments to run TreeShrink (Mai and Mirarab 2018). TreeShrink detects and removes outlier long branches in a collections of trees. We recovered the output alignments from TreeShrink and cleaned them again using CIAlign. Finally, we trimmed the alignments with trimAl v1.4.rev15 (Capella-Gutiérrez et al. 2009). We made final gene trees with IQtree using ModelFinder and 1000 ultrafast bootstrap replicates. We concatenated the final 2,122 gene trees with the highest likelihood, and used ASTRAL v5.7.7 (Zhang et al. 2018) to obtain a species tree with default settings (Fig. 1). As the branch lengths from ASTRAL are expressed in coalescent units and all terminal tips have the same length, we constrained the resulting ASTRAL tree topology and re-scaled branch lengths using IQtree. Briefly, we concatenated the BUSCO loci present in all the individuals (306), then used a custom script to concatenate the genes and delimit the partitions of the concatenated alignment to check for the best model for each gene. We used IQtree to select the better evolution model for each partition and scale the tree generated with Astral (Chernomor et al. 2016; Kalyaanamoorthy et al. 2017; Nguyen et al. 2015). We trimmed the branches of C. godmani of the species tree using the ape R package (Paradis and Schliep 2019). We chose one of the two WhatsHap phased alleles at random for each locus and each individual as input for the BUSTED model (HyPhy v2.5.31) using the species tree as the reference phylogeny (Murrell et al. 2015; Pond et al. 2020). The BUSTED models test for evidence that at least one site on at least one test branch has experienced diversifying selection. We analyzed 4,497 nontoxins and 68 toxins with this method; for ten nontoxins the substitution resulted in one internal stop codon for one individual.
For nucleotide diversity, we calculated the mean \(\pi\) (Nei and Li 1979) for each gene and used a linear regression to test for significance in \(\pi\) between toxin and nontoxin genes. To avoid the effect of the sample size driving significant differences regardless of effect size given that the number of nontoxins is higher than the toxins, we performed bootstrapping by taking a random sample of the nontoxins matching the sample size of toxins and repeated the linear regression 1,000 times. We report the proportion of replicates with \(p<0.05\) in the linear regression as bootstrap support. Similarly, we use linear regression and bootstrapping to test for significant differences with Tajima’s D (Tajima 1989), and \(F_{ST}\) (Wright 1949) between toxins and nontoxins. For Tajima’s D, we ran a t-test to evaluate if the mean value from toxins and nontoxins was different from 0; and repeated the linear model using only synonymous SNPs. We performed a nonparametric Kruskal–Wallis test to compare toxins and nontoxins Tajima’s D for nonsynonymous SNPs due to their nonnormal distribution. We used a nonparametric Kruskal–Wallis test to compare the values of the likelihood ratio test (LRT) for the BUSTED model between toxins and nontoxins. As for the linear regression tests, we used bootstrapping for the nonparametric tests with 1000 iterations. To identify specific toxins under selection, we generated a null distribution from the nontoxin transcripts and identified toxins outside the \(95^{th}\) percentile of the distribution for Tajima’s D (two-tail), \(F_{ST}\) (right tail), and LRT of BUSTED model (right tail). To test if low-expression toxins were under stronger positive or balancing selection pressures, we used linear regression with average toxin expression (ln average TPM) as the response variable and Tajima’s D, \(F_{ST}\), and LRT of BUSTED model, we ran a regression for each variable. We tested positive and negative values of Tajima’s D separately to account for potential differences in expression related to alternate selection pressures. We used linear regression and bootstrapping as in the previous steps.
Data Availability
The raw data-sets generated and analyzed during the current study are available in the National Center for Biotechnology Information (NCBI) repository under the accession numbers SRR19227107, SRR19227106, SRR19227101, SRR19227100, SRR19227099, SRR19227098, SRR19227097, SRR19227096, SRR19227095, SRR19227094, SRR19227105, SRR19227104, SRR19227103, and SRR19227102. The consensus transcriptomes generated and analyzed during the current study are available at the github repository https://github.com/RamsesRosales/Cerrophidion_Selection, a similar version is available at the DDBJ/EMBL/GenBank under the accession numbers: C. sasai GJZX00000000, the version described in this paper is the first version, GJZX01000000; C. petlalcalensis GKAV00000000, the version described in this paper is the first version, GKAV01000000; C. godmani GKCA00000000, the version described in this paper is the first version, GKCA01000000. C. tzotzilorum GKIB00000000, the version described in this paper is the first version, GKIB01000000. Scripts utilized in this work can be found in the online resources 3 and 4, and in the github repositories: https://github.com/RamsesRosales/Cerrophidion_Selection, https://github.com/RamsesRosales/ModelCrotA.
References
Aird SD, Aggarwal S, Villar-Briones A, Tin MMY, Terada K, Mikheyev AS (2015) Snake venoms are integrated systems, but abundant venom proteins evolve more rapidly. BMC Genom 16:1–20. https://doi.org/10.1186/s12864-015-1832-6
Aird SD, Arora J, Barua A, Qiu L, Terada K, Mikheyev AS (2017) Population genomic analysis of a pitviper reveals microevolutionary forces underlying venom chemistry. Genome Biol Evol 9:2640–2649
Aird SD, Watanabe Y, Villar-briones A, Roy MC, Terada K, Mikheyev AS (2013) Quantitative high-throughput profiling of snake venom gland transcriptomes and proteomes (Ovophis okinavensis and Protobothrops flavoviridis). BMC Genom 14:1–26
Almeida DD, Viala VL, Nachtigall PG, Broe M, Gibbs HL, Serrano SMdT, Moura-da Silva AM, Ho PL, Nishiyama-Jr MY, Junqueira-de Azevedo ILM (2021) Tracking the recruitment and evolution of snake toxins using the evolutionary context provided by the Bothrops jararaca genome. Proc Natl Acad Sci 118:e2015159118. https://doi.org/10.1073/pnas.2015159118
Amazonas DR, Portes-Junior JA, Nishiyama-Jr MY, Nicolau CA, Chalkidis HM, Mourão RH, Grazziotin FG, Rokyta DR, Gibbs HL, Valente RH, Junqueira-de Azevedo IL, Moura-da Silva AM (2018) Molecular mechanisms underlying intraspecific variation in snake venom. J Proteom 181:60–72. https://doi.org/10.1016/j.jprot.2018.03.032
Arni RK, Fontes MR, Barberato C, Gutiérrez JM, Díaz C, Ward RJ (1999) Crystal structure of myotoxin II, a monomeric Lys49-phospholipase A\(_2\) homologue isolated from the venom of Cerrophidion (Bothrops) godmani. Arch Biochem Biophys 366:177–182
Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA (2013) From fastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protocols Bioinfo 43:1–33. https://doi.org/10.1002/0471250953.bi1110s43
Junqueira-de Azevedo IDL, Ho PL (2002) A survey of gene expression and diversity in the venom glands of the pitviper snake Bothrops insularis through the generation of expressed sequence tags (ESTs). Gene 299:279–291
Borja M, Neri-Castro E, Castañeda-Gaytán G, Strickland JL, Parkinson CL, Castañeda-Gaytán J, Ponce-López R, Lomonte B, Olvera-Rodríguez A, Alagón A, Pérez-Morales R (2018) Biological and proteolytic variation in the venom of Crotalus scutulatus scutulatus from Mexico. Toxins 10:1–19
Brahma RK, McCleary RJ, Kini RM, Doley R (2015) Venom gland transcriptomics for identifying, cataloging, and characterizing venom proteins in snakes. Toxicon 93:1–10. https://doi.org/10.1016/j.toxicon.2014.10.022
Calvete JJ (2017) Venomics: integrative venom proteomics and beyond. Biochem J 474:611–634
Campbell JA, Lamar WW (2004) Venomous reptiles of the western hemisphere, vol 1. Cornell University Press, Ithaca
Campbell JA, Solórzano A (1992) The distribution, variation, and natural history of the middle American montane pitviper Porthidium godmani. In: Campbell JA, Brodie ED (eds) Biology of the pitvipers. Selva, Tyler, pp 223–250
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) Trimal: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. https://doi.org/10.1093/bioinformatics/btp348
Casewell NR, Huttley GA, Wüster W (2012) Dynamic evolution of venom proteins in squamate reptiles. Nat Commun 3:1066
Casewell NR, Wagstaff SC, Harrison RA, Renjifo C, Wuster W (2011) Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol Biol Evol 28:2637–2649. https://doi.org/10.1093/molbev/msr091
Casewell NR, Wagstaff SC, Wüster W, Cook DA, Bolton FM, King SI, Pla D, Sanz L, Calvete JJ, Harrison RA (2014) Medically important differences in snake venom composition are dictated by distinct postgenomic mechanisms. Proc Natl Acad Sci USA 111:9205–9210. https://doi.org/10.1073/pnas.1405484111
Casewell NR, Wüster W, Vonk FJ, Harrison RA, Fry BG (2013) Complex cocktails: the evolutionary novelty of venoms. Trends Ecol Evol 28:219–229
Castoe TA, Daza JM, Smith EN, Sasa MM, Kuch U, Campbell JA, Chippindale PT, Parkinson CL (2009) Comparative phylogeography of pitvipers suggests a consensus of ancient Middle American highland biogeography. J Biogeogr 36:88–103. https://doi.org/10.1111/j.1365-2699.2008.01991.x
Chen YH, Wang YM, Hseu MJ, Tsai IH (2004) Molecular evolution and structure-function relationships of crotoxin-like and asparagine-6-containing phospholipases A\(_2\) in pit viper venoms. Biochem J 381:25–34
Chernomor O, von Haeseler A, Minh BQ (2016) Terrace aware data structure for phylogenomic inference from supermatrices. System Biol 65:997–1008. https://doi.org/10.1093/sysbio/syw037
Cingolani P, Platts A, Coon M, Nguyen T, Wang L, Land S, Lu X, Ruden D (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain \(W^{118}\); iso-2; iso-3. Fly 6:80–92.
Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B et al (2009) Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 25:1422–1423. https://doi.org/10.1093/bioinformatics/btp163
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R (2011) The variant call format and VCF tools. Bioinformatics. https://doi.org/10.1093/bioinformatics/btr330
Daza JM, Castoe TA, Parkinson CL (2010) Using regional comparative phylogeographic data from snake lineages to infer historical processes in Middle America. Ecography 33:343–354. https://doi.org/10.1111/j.1600-0587.2010.06281.x
Dowell NL, Giorgianni MW, Griffin S, Kassner VA, Selegue JE, Sanchez EE, Carroll SB (2018) Extremely divergent haplotypes in two toxin gene complexes encode alternative venom types within rattlesnake species. Curr Biol 28:1016–1026
Dowell NL, Giorgianni MW, Kassner VA, Selegue JE, Sanchez EE, Carroll SB (2016) The deep origin and recent loss of venom toxin genes in rattlesnakes. Curr Biol 26:2434–2445
Durban J, Juárez P, Angulo Y, Lomonte B, Flores-Diaz M, Alape-Girón A, Sasa M, Sanz L, Gutiérrez JM, Dopazo J, Conesa A, Calvete JJ (2011) Profiling the venom gland transcriptomes of Costa Rican snakes by 454 pyrosequencing. BMC Genom. https://doi.org/10.1186/1471-2164-12-259
Eswar N, Webb B, Marti-Renom MA, Madhusudhan M, Eramian D, My Shen, Pieper U, Sali A (2006) Comparative protein structure modeling using modeller. Curr Protocols Bioinfo 15:1–30. https://doi.org/10.1002/0471250953.bi0506s15
Faure G, Guillaume JL, Camoin L, Saliou B, Bon C (1991) Multiplicity of acidic subunit isoforms of crotoxin, the phospholipase A\(_2\) neurotoxin from Crotalus durissus terrificus venom, results from posttranslational modifications. Biochemistry 30:8074–8083
Fernández M, Martínez-Fonseca J, Salazar-Saavedra M, Gutiérrez L, Loza J, Sunyer J (2017) First verified record of Cerrophidion wilsoni (Reptilia: Squamata: Viperidae) from Nicaragua. Mesoamerican Herpetol 4:481–484
Fox JW, Serrano SM (2005) Structural considerations of the snake venom metalloproteinases, key members of the M12 reprolysin family of metalloproteinases. Toxicon 45:969–985
Fraczkiewicz R, Braun W (1998) Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem 19:319–333
Franco-Servín C, Neri-Castro E, Bénard-Valle M, Alagón A, Rosales-García RA, Guerrero-Alba R, Poblano-Sánchez JE, Silva-Briano M, Guerrero-Barrera AL, Sigala-Rodríguez JJ (2021) Biological and biochemical characterization of Coronado Island Rattlesnake (Crotalus helleri caliginis) venom and antivenom neutralization. Toxins 13:582
Fraser DJ, Weir LK, Bernatchez L, Hansen MM, Taylor EB (2011) Extent and scale of local adaptation in salmonid fishes: review and meta-analysis. Heredity 106:404–420
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152. https://doi.org/10.1093/bioinformatics/bts565
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The proteomics protocols handbook. Humana Press, Totowa, pp 571–607. https://doi.org/10.1385/1-59259-890-0:571
Gené J, Roy A, Rojas G, Gutiérrez J, Cerdas L (1989) Comparative study on coagulant, defibrinating, fibrinolytic and fibrinogenolytic activities of Costa Rican crotaline snake venoms and their neutralization by a polyvalent antivenom. Toxicon 27:841–848
Gibbs HL, Sanz L, Pérez A, Ochoa A, Hassinger AT, Holding ML, Calvete JJ (2020) The molecular basis of venom resistance in a rattlesnake-squirrel predator-prey system. Mol Ecol 29:2871–2888. https://doi.org/10.1111/mec.15529
Glenn J, Straight R, Wolt T (1994) Regional variation in the presence of canebrake toxin in Crotalus horridus venom. Compar Biochem Physiol C 107:337–346
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Gutiérrez J (2014) Current challenges for confronting the public health problem of snakebite envenoming in Central America. J Venom Anim Toxins Including Trop Dis 20:7. https://doi.org/10.1186/1678-9199-20-7
Gutiérrez JM, Gené JA, Rojas G, Cerdas L (1985) Neutralization of proteolytic and hemorrhagic activities of Costa Rican snake venoms by a polyvalent antivenom. Toxicon 23(6):887–893
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS (2018) UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol 35:518–522
Hofmann EP, Rautsaw RM, Strickland JL, Holding ML, Hogan MP, Mason AJ, Rokyta DR, Parkinson CL (2018) Comparative venom-gland transcriptomics and venom proteomics of four Sidewinder Rattlesnake (Crotalus cerastes) lineages reveal little differential expression despite individual variation. Sci Rep 8:15534. https://doi.org/10.1038/s41598-018-33943-5
Holding ML, Margres MJ, Mason AJ, Parkinson CL, Rokyta DR (2018) Evaluating the performance of de novo assembly methods for venom-gland transcriptomics. Toxins 10:1–21
Holding ML, Sovic MG, Colston TJ, Gibbs HL (2021) The scales of coevolution: comparative phylogeography and genetic demography of a locally adapted venomous predator and its prey. Biol J Linnean Soc 132:297–317
Holding ML, Strickland JL, Rautsaw RM, Hofmann EP, Mason AJ, Hogan MP, Nystrom GS, Ellsworth SA, Colston TJ, Borja M, Castañeda-Gaytán G, Grünwald CI, Jones JM, Freitas-de Sousa LA, Viala VL, Margres MJ, Hingst-Zaher E, Junqueira-de Azevedo ILM, Moura-da Silva AM, Grazziotin FG, Gibbs HL, Rokyta DR, Parkinson CL (2021) Phylogenetically diverse diets favor more complex venoms in North American pitvipers. Proc Natl Acad Sci 118:e2015579118. https://doi.org/10.1073/pnas.2015579118
Jadin RC, Townsend JH, Castoe TA, Campbell JA (2012) Cryptic diversity in disjunct populations of Middle American montane pitvipers: a systematic reassessment of Cerrophidion godmani. Zoologica Scripta 41:455–470
Jones L, Youngman NJ, Neri-Castro E, Guadarrama-Martínez A, Lewin MR, Carter R, Frank N, Fry BG (2022) Differential antivenom and small-molecule inhibition of novel Coagulotoxic variations in atropoides, Cerrophidion, Metlapilcoatlus, and Porthidium American viperid snake venoms. Toxins 14. https://www.mdpi.com/2072-6651/14/8/511
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. https://doi.org/10.1093/molbev/mst010
Keil B (1992) Specificity of proteolysis. Springer, Berlin, Heidelberg
Kolde R (2019) pheatmap: pretty heatmaps. https://CRAN.R-project.org/package=pheatmap. R package version 1.0.12
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinfo 12:323
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 00:1–3. arXiv:1303.3997
Lizano S, Angulo Y, Lomonte B, Fox JW, Lambeau G, Lazdunski M, Gutiérrez JM (2000) Two phospholipase A\(_2\) inhibitors from the plasma of Cerrophidion (Bothrops) godmani which selectively inhibit two different group-II phospholipase A\(_2\) myotoxins from its own venom: isolation, molecular cloning and biological properties. Biochem J 346:631
Lomonte B, Fernández J, Sanz L, Angulo Y, Sasa M, Gutiérrez JM, Calvete JJ (2014) Venomous snakes of Costa Rica: biological and medical implications of their venom proteomic profiles analyzed through the strategy of snake venomics. J Proteom 105:323–339. https://doi.org/10.1016/j.jprot.2014.02.020
Lomonte B, Rey-Suárez P, Tsai WC, Angulo Y, Sasa M, Gutiérrez JM, Calvete JJ (2012) Snake venomics of the pit vipers Porthidium nasutum, Porthidium ophryomegas, and Cerrophidion godmani from Costa Rica: Toxicological and taxonomical insights. J Proteom 75:1675–1689
López-Luna MA, Vogt RC, De la Torre-Loranca MA (1999) A new species of montane pitviper from Veracruz, México. Herpetologica 55:382–389
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with deseq2. Genome Biol 15:550
Mackessy SP (2008) Venom composition in rattlesnakes: trends and biological significance. In: Hayes WK, Beaman KR, Cardwell MD, Bush SP (eds) The biology of rattlesnakes. Loma Linda University Press, Loma Linda, pp 495–510
Mai U, Mirarab S (2018) Treeshrink: fast and accurate detection of outlier long branches in collections of phylogenetic trees. BMC Genom 19. https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-4620-2
Margres MJ, McGivern JJ, Seavy M, Wray KP, Facente J, Rokyta DR (2014) Contrasting modes and tempos of venom expression evolution in two snake species. Genetics 199:165–176. https://doi.org/10.1534/genetics.114.172437
Margres MJ, McGivern JJ, Wray KP, Seavy M, Calvin K, Rokyta DR (2014) Linking the transcriptome and proteome to characterize the venom of the eastern diamondback rattlesnake (Crotalus adamanteus). J Proteom 96:145–158. https://doi.org/10.1016/j.jprot.2013.11.001
Margres MJ, Patton A, Wray KP, Hassinger ATB, Ward MJ, Lemmon EM, Lemmon AR, Rokyta DR (2019) Tipping the scales: the migration-selection balance leans toward selection in snake venoms. Mol Biol Evol 36:271–282
Margres MJ, Wray KP, Hassinger ATB, Ward MJ, McGivern JJ, Moriarty Lemmon E, Lemmon AR, Rokyta DR (2017) Quantity, not quality: rapid adaptation in a polygenic trait proceeded exclusively through expression differentiation. Mol Biol Evol 34:3099–3110
Margres MJ, Wray KP, Sanader D, McDonald PJ, Trumbull LM, Patton AH, Rokyta DR (2021) Varying intensities of introgression obscure incipient venom-associated speciation in the timber rattlesnake (Crotalus horridus). Toxins 13:782
Margres MJ, Wray KP, Seavy M, McGivern JJ, Herrera ND, Rokyta DR (2016) Expression differentiation is constrained to low-expression proteins over ecological timescales. Genetics 202:273–283
Martin M, Patterson M, Garg S, Fischer SO, Pisanti N, Klau G, Schöenhuth A, Marschall T (2016) Whatshap: fast and accurate read-based phasing. bioRxiv P. 085050. https://www.biorxiv.org/content/10.1101/085050v2
Mason AJ, Holding ML, Rautsaw RM, Rokyta DR, Parkinson CL, Gibbs HL (2022) Venom gene sequence diversity and expression jointly shape diet adaptation in pitvipers. Mol Biol Evol 39:1–14
Mason AJ, Margres MJ, Strickland JL, Rokyta DR, Sasa M, Parkinson CL (2020) Trait differentiation and modular toxin expression in palm-pitvipers. BMC Genom 21:147
Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, Eren K, Pollner T, Martin DP, Smith DM, Scheffler K, Pond SLK (2015) Gene-wide identification of episodic selection. Mol Biol Evol 32:1365–1371
Nachtigall PG, Rautsaw RM, Ellsworth SA, Mason AJ, Rokyta DR, Parkinson CL, de Azevedo ILMJ (2021) Toxcodan: a new toxin annotator and guide to venom gland transcriptomics. Brief Bioinfo. https://doi.org/10.1093/bib/bbab095/6235957
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci 76:5269–5273
Neri-Castro E, Bénard-Valle M, Gil G, Borja M, de León JL, Alagón A (2020) Venomous snakes in Mexico: a review of the study of venoms, antivenom and epidemiology. Revista Latinoamericana de Herpetologia 3:05–22
Neri-Castro E, Hernández-Dávila A, Olvera-Rodríguez A, Cardoso-Torres H, Bénard-Valle M, Bastiaans E, López-Gutierrez O, Alagón A (2019) Detection and quantification of a \(\beta\)-neurotoxin (crotoxin homologs) in the venom of the rattlesnakes Crotalus simus, C. culminatus and C. tzabcan from Mexico. Toxicon: X 2:100007. https://doi.org/10.1016/j.toxcx.2019.100007
Neri-Castro E, Sanz L, Olvera-Rodríguez A, Bénard-Valle M, Alagón A, Calvete JJ (2020) Venomics and biochemical analysis of the black-tailed horned pitviper, Mixcoatlus melanurus, and characterization of Melanurutoxin, a novel crotoxin homolog. J Proteom 225:103865
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
Nielsen R (2005) Molecular signatures of natural selection. Annu Rev Genet 39:197–218
Ochoa A, Broe M, Moriarty Lemmon E, Lemmon AR, Rokyta DR, Gibbs HL (2020) Drift, selection and adaptive variation in small populations of a threatened rattlesnake. Mol Ecol 29:2612–2625
Paradis E, Schliep K (2019) ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35:526–528
Petersen TN, Brunak S, Von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Pond SLK, Poon AFY, Velazquez R, Weaver S, Hepler NL, Murrell B, Shank SD, Magalis BR, Bouvier D, Nekrutenko A, Wisotsky S, Spielman SJ, Frost SDW, Muse SV (2020) Hyphy 2.5–a customizable platform for evolutionary hypothesis testing using phylogenies. Mol Biol Evol 37:295–299
R Core Team (2020) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Radvanyi FR, Bon C (1982) Catalytic activity and reactivity with p-bromophenacyl bromide of the phospholipase subunit of crotoxin. influence of dimerization and association with the noncatalytic subunit. J Biol Chem 257:12616–12623
Rautsaw RM, Hofmann EP, Margres MJ, Holding ML, Strickland JL, Mason AJ, Rokyta DR, Parkinson CL (2019) Intraspecific sequence and gene expression variation contribute little to venom diversity in sidewinder rattlesnakes (Crotalus cerastes). Proc R Soc B: Biol Sci 286:20190810
Rautsaw RM, Jiménez-Velázquez G, Hofmann EP, Alencar LRV, Grünwald CI, Martins M, Carrasco P, Doan TM, Parkinson CL (2022) VenomMaps: Updated species distribution maps and models for New World pitvipers (Viperidae: Crotalinae). Sci Data 9:232
Robinson MD, McCarthy DJ, Smyth GK (2010) Edger: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Rokyta DR, Lemmon AR, Margres MJ, Aronow K (2012) The venom-gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genom 13:312
Rokyta DR, Margres MJ, Calvin K (2015) Post-transcriptional mechanisms contribute little to phenotypic variation in snake venoms. G3 Genes, Genomes, Genet 5:2375–2382
Rokyta DR, Wray KP, Lemmon AR, Lemmon EM, Caudle SB (2011) A high-throughput venom-gland transcriptome for the Eastern Diamondback Rattlesnake (Crotalus adamanteus) and evidence for pervasive positive selection across toxin classes. Toxicon 57:657–671
Rokyta DR, Wray KP, Margres MJ (2013) The genesis of an exceptionally lethal venom in the timber rattlesnake (Crotalus horridus) revealed through comparative venom-gland transcriptomics. BMC Genom 14:394
Rokyta DR, Wray KP, McGivern JJ, Margres MJ (2015) The transcriptomic and proteomic basis for the evolution of a novel venom phenotype within the timber rattlesnake (Crotalus horridus). Toxicon 98:34–48. https://doi.org/10.1016/j.toxicon.2015.02.015
Rotenberg D, Bamberger ES, Kochva E (1971) Studies on ribonucleic acid synthesis in the venom glands of Vipera palaestinae (Ophidia, Reptilia). Biochem J 121:609–612
Rübsamen K, Breithaupt H, Habermann B (1971) Biochemistry and pharmacology of the crotoxin complex. Naunyn-Schmiedebergs Arch. Pharmak 270:274–288. https://doi.org/10.1007/BF00997027
Sasa M (1997) Cerrophidion godmani in Costa Rica: a case of extremely low allozyme variation? J Herpetol 31:569
Saviola AJ, Pla D, Sanz L, Castoe TA, Calvete JJ, Mackessy SP (2015) Comparative venomics of the Prairie Rattlesnake (Crotalus viridis viridis) from Colorado: Identification of a novel pattern of ontogenetic changes in venom composition and assessment of the immunoreactivity of the commercial antivenom CroFab®. J Proteom 121:28–43
Schield DR, Perry BW, Adams RH, Holding ML, Nikolakis ZL, Gopalan SS, Smith CF, Parker JM, Meik JM, DeGiorgio M, Mackessy SP, Castoe TA (2022) The roles of balancing selection and recombination in the evolution of rattlesnake venom. Nat Ecol Evol. https://www.nature.com/articles/s41559-022-01829-5
Schramer TD, de la Torre-Loranca MA, Salazar-Saavedra M, Kalki Y, Wylie DB (2018) Addiotional diet information for Cerrophidion godmani (Günther, 1863) and a dietary synopsis of the genus Cerrophidion Campbell and Lamar, 1992. Revista Latinoamericana de Herpetología 1:35
Sievers F, Higgins DG (2018) Clustal Omega for making accurate alignments of many protein sequences. Protein Sci 27:135–145
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. https://doi.org/10.1093/bioinformatics/btv351
Strickland JL, Mason AJ, Rokyta DR, Parkinson CL (2018) Phenotypic variation in Mojave rattlesnake (Crotalus scutulatus) venom is driven by four toxin families. Toxins 10:1–23
Strickland JL, Smith CF, Mason AJ, Schield DR, Borja M, Castañeda-Gaytán G, Spencer CL, Smith LL, Trápaga A, Bouzid NM, Campillo-García G, Flores-Villela OA, Antonio-Rangel D, Mackessy SP, Castoe TA, Rokyta DR, Parkinson CL (2018) Evidence for divergent patterns of local selection driving venom variation in Mojave Rattlesnakes (Crotalus scutulatus). Sci Rep 8:17622
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tange O (2020) GNU Parallel 20201122 (’Biden’). GNU Parallel is a general parallelizer to run multiple serial command line programs in parallel without changing them. https://doi.org/10.5281/zenodo.4284075
Tasoulis T, Isbister G (2017) A review and database of snake venom proteomes. Toxins 9:290
Torre-Loranca MAL, Grünwald CI, Valdenegro-Brito AE, Cervantes-Burgos RI, García-Vázquez UO (2019) New distributional records for the rare Mexican pitviper Cerrophidion petlalcalensis (Squamata: Viperidae) from Veracruz and Oaxaca, Mexico, with comments on distribution and the natural history. Revista Latinoamericana de Herpetologia 2:71–77
Tumescheit C, Firth AE, Brown K (2020) Cialign—a highly customisable command line tool to clean, interpret and visualise multiple sequence alignments. bioRxiv. https://doi.org/10.1101/2020.09.14.291484
Whittington AC, Mason AJ, Rokyta DR (2018) A single mutation unlocks cascading exaptations in the origin of a potent pitviper neurotoxin. Mol Biol Evol 35:887–898
Wooldridge BJ, Pineda G, Banuelas-Ornelas JJ, Dagda RK, Gasanov SE, Rael ED, Lieb CS (2001) Mojave rattlesnakes (Crotalus scutulatus scutulatus) lacking the acidic subunit DNA sequence lack Mojave toxin in their venom. Compar Biochem Physiol B Biochem Mol Biol 130:169–179
Wright S (1949) The genetic structure of populations. Ann Eugen 15:323–354
Yamazaki Y, Matsunaga Y, Tokunaga Y, Obayashi S, Saito M, Morita T (2009) Snake venom vascular endothelial growth factors (VEGF-Fs) exclusively vary their structures and functions among species. J Biol Chem 284:9885–9891
Zancolli G, Calvete JJ, Cardwell MD, Greene HW, Hayes WK, Hegarty MJ, Herrmann HW, Holycross AT, Lannutti DI, Mulley JF, Sanz L, Travis ZD, Whorley JR, Wüster CE, Wüster W (2019) When one phenotype is not enough: divergent evolutionary trajectories govern venom variation in a widespread rattlesnake species. Proc R Soc B Biol Sci. https://doi.org/10.1098/rspb.2018.2735
Zelanis A, Andrade-Silva D, Rocha MM, Furtado MF, Serrano SMT, Junqueira-de Azevedo ILM, Ho PL (2012) A transcriptomic view of the proteome variability of newborn and adult Bothrops jararaca snake venoms. PLoS Neglected Trop Dis 6:e1554
Zelanis A, Menezes MC, Kitano ES, Liberato T, Tashima AK, Pinto AF, Sherman NE, Ho PL, Fox JW, Serrano SM (2016) Proteomic identification of gender molecular markers in Bothrops jararaca venom. J Proteom 139:26–37. https://doi.org/10.1016/j.jprot.2016.02.030
Zhang C, Rabiee M, Sayyari E, Mirarab S (2018) Astral-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinfo 19:15–30. https://doi.org/10.1186/s12859-018-2129-y
Zhang J, Kobert K, Flouri T, Stamatakis A (2014) PEAR: a fast and accurate illumina paired-end reAd mergeR. Bioinformatics 30:614–620
Acknowledgements
We want to thank Jason Jones, Leo Badillo, and Cristobal Moreno for help in the field. We greatly appreciate Tristan Schramer’s assistance with illustrations, and digital photography. Additionally, he and N. Jade Mellor are thanked for the many great discussions and comments on the manuscript. We greatly appreciate Carl Whittington for his comments on the PLA\(_2\) model analysis. Parallel computing resources were provided by the Clemson Palmetto High-Performance Computing Cluster and in part, with support from the Clemson University Genomics and Bioinformatics Facility, which receives support from two Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant numbers P20GM109094 and P20GM139767.
Funding
This work was supported by the National Science Foundation Grants DUE 1161228, DEB 1638879, and DEB 1822417 to C.L.P., DEB 1638902 to D.R.R, and a Fulbright García Robles graduate fellowship to R.A.R.G.
Author information
Authors and Affiliations
Contributions
RARG, EPH, and CLP conceived the study. CIG, HFC, ITAC, RRC, MADL, MB, GCG, MS, MLH, and RMR collected samples used in this study. RMR, EPH, MLH, JLS, AJM, and RARG generated data, RARG, RMR, and EPH processed data and performed analyses. RARG drafted the manuscript. JLS, AJM, DRR, EAM, MLH, and CLP provided analytical and conceptual input. RMR, EAM, EPH, AJM, JLS, MLH, and CLP reviewed and edited the manuscript. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Consent for Publication
Not applicable
Ethics Approval
All protocols involving live snakes followed the ASIH guidelines and were approved by the University of Central Florida and Clemson University Animal Care and Use Committees (16-17W: UCF and 2017-067: CU). The sample (LIAP244) was collected in and exported from Costa Rica under Investigation Permit SINAC-ACC-OSJ-re-428-2021. Samples (CHFCB-0271, CHFCB-0272, CHFCB-0274, CHFCB-0290, CHFCB-0291, CHFCB-0300, CHFCB-0236, CHFCB-0238, CHFCB-0239, CHFCB-0276, CHFCB-027, CHFCB-0296, CHFCB-0471) were collected in and exported from Mexico under SEMARNAT:SGPA/DGVS/01090/17;SGPA/DGVS/002288/18;SGPA/DGVS/08831/20.
Additional information
Handling editor: Keith Crandall.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rosales-García, R.A., Rautsaw, R.M., Hofmann, E.P. et al. Sequence Divergence in Venom Genes Within and Between Montane Pitviper (Viperidae: Crotalinae: Cerrophidion) Species is Driven by Mutation–Drift Equilibrium. J Mol Evol 91, 514–535 (2023). https://doi.org/10.1007/s00239-023-10115-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-023-10115-2