
Abstract
The discovery of caspase homologs in bacteria highlighted the relationship between programmed cell death (PCD) evolution and eukaryogenesis. However, the origin of PCD genes in prokaryotes themselves (bacteria and archaea) is poorly understood and a source of controversy. Whether archaea also contain C14 peptidase enzymes and other death domains is largely unknown because of a historical dearth of genomic data. Archaeal genomic databases have grown significantly in the last decade, which allowed us to perform a detailed comparative study of the evolutionary histories of PCD-related death domains in major archaeal phyla, including the deepest branching phyla of Candidatus Aenigmarchaeota, Candidatus Woesearchaeota, and Euryarchaeota. We identified death domains associated with executioners of PCD, like the caspase homologs of the C14 peptidase family, in 321 archaea sequences. Of these, 15.58% were metacaspase type I orthologues and 84.42% were orthocaspases. Maximum likelihood phylogenetic analyses revealed a scattered distribution of orthocaspases and metacaspases in deep-branching bacteria and archaea. The tree topology was incongruent with the prokaryote 16S phylogeny suggesting a common ancestry of PCD genes in prokaryotes and subsequent massive horizontal gene transfer coinciding with the divergence of archaea and bacteria. Previous arguments for the origin of PCD were philosophical in nature with two popular propositions being the “addiction” and ‘original sin’ hypotheses. Our data support the ‘original sin’ hypothesis, which argues for a pleiotropic origin of the PCD toolkit with pro-life and pro-death functions tracing back to the emergence of cellular life—the Last Universal Common Ancestor State.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the last few decades, it has emerged that programmed cell death (PCD) played a much more significant part in the evolution of life than previously imagined (Ameisen 2002; Michod and Nedelcu 2003; Segovia et al. 2003; Blackstone 2013; Iranzo et al. 2014; Koonin and Zhang 2017; Durand 2021; Huneman 2022). PCD fulfills a critical role in the eco-evolutionary dynamics of prokaryote and unicellular eukaryote communities (Vardi et al. 2007; Bidle 2016; Durand et al. 2016; Abada and Segev 2018; Ndhlovu et al. 2021) and tissue homeostasis in multicellular organisms (Glücksmann 1951; Lockshin and Williams 1964). In addition, PCD was essential for the evolutionary transitions in individuality that characterize the history of life, such as eukaryogenesis, multicellularity, and eusociality in insects (Iranzo et al. 2014; Durand et al. 2019).
It is important to acknowledge at the outset, that the very subject of PCD in microbes has been fraught. This is largely because the term ‘PCD’ was initially discovered and subsequently interpreted as a hallmark of ontogenesis in Metazoa (Glücksmann 1951; Lockshin and Williams 1964) and the very concept of programmed microbial death in some unicellular lineages was questioned (Ratel et al. 2001; Nedelcu et al. 2011; Proto et al. 2013; Ramisetty et al. 2015). Different researchers often imply different things when referring to the various kinds of cell death, which can lead to a conflation of questions and ideas. To circumvent this, there are now both mechanistic and evolutionary definitions. The mechanistic definition we adopt is the general Berman-Frank interpretation that PCD is “an active, genetically controlled, cellular self-destruction driven by a series of complex biochemical events and specialized cellular machinery” (Berman-Frank et al. 2004). Evolutionists use a historical account of whether there was a selection for the trait or not and draw a distinction between PCD as an adaptation (true PCD) and non-adaptive PCD when the trait is a pleiotropic phenomenon (also called ersatz PCD) (Durand and Ramsey 2019). For the purposes of this study and for investigating the hypotheses below, we use the Berman-Frank mechanistic definition. The evolutionary definitions can be ignored because the ‘selection for’ and ‘selection of’ distinction are not drawn here.
There have been several hypotheses for the origin of PCD, based largely on philosophical and conceptual works, which can now be examined by comparative analyses of death domains in prokaryotes (bacteria and archaea) (Aravind et al. 1999; Asplund-Samuelsson et al. 2012; Hofmann 2019). One of the hypotheses for the emergence of PCD in the earliest cells is the ‘original sin' hypothesis, which postulates that ancestral effector genes of PCD were present at the origin of cellular life (Ameisen 2002). The argument is that PCD effectors may have been rooted in primordial non-death functions, such as the cell cycle or adaptations to environmental stress (Shrestha and Megeney 2012; Shalini et al. 2015), with subsequent pro-death functions emerging. Crosstalk between molecular pathways in extant unicellular organisms is consistent with this claim (van Creveld et al. 2018). There are other hypotheses that may be complementary and synergistic such as the ‘addiction’ hypothesis (Ameisen 2002) and the argument for life–death coevolution in the last universal common ancestor state (LUCAS) (Durand 2021). The different hypotheses may not be mutually exclusive; however, the ‘original sin’ hypothesis is much more accessible with a comparative genomics approach and is the focus here.
Genomic data have linked eukaryogenesis with PCD in bacteria (Blackstone and Green 1999; Koonin and Aravind 2002; Michod and Nedelcu 2003). Many of the ‘death domains’ have been identified and the “bacterial connection” between PCD and eukaryogenesis is well established (Koonin and Aravind 2002). While it is known that PCD played a part in the rise of eukaryotes, potentially as a conflict mediator (Blackstone and Green 1999; Michod and Nedelcu 2003; Iranzo et al. 2014), and continues to impact the social lives of microbes (Arnoult et al. 2001; Durand et al. 2011; Bayles 2014), the origin of the genetic toolkit in prokaryotes generally, is largely unknown (Koonin and Aravind 2002; Kaczanowski 2016; Klim et al. 2018). This raises some interesting questions. Were PCD genes also present in archaea, or did they emerge in bacteria after the divergence of the two lineages? One of the historical obstacles to addressing this question has been a dearth of sequence data from archaeal taxa. However, since 2012, much more data have become available (for example, Asplund-Samuelsson et al. 2012), presenting an opportunity to investigate the most ancient origins of the PCD genetic toolkit (Srivastav and Suneja 2019; Zhang et al. 2020). In addition, there has been some “tantalizing” empirical evidence for PCD genes in archaea (Asplund-Samuelsson et al. 2012; Klemenčič et al. 2019). Caspases (cysteine–aspartic proteases) (CAs) and their homologs in non-Metazoa are one of the central mediators of PCD, activating multiple enzymes that are part of the death signaling cascade (Cohen 1998). Caspase homologs refer to orthocaspases (OCAs), metacaspases (MCAs), and paracaspases (PCAs), as per the proposed unified nomenclature of the C14 peptidase family (Minina et al. 2020) (Fig. 1). There are some reports of putative caspase homologs of the C14 peptidase family, OCAs and MCAs, in archaeal phyla (Euryarchaeota, Thermoplasmatota, Thaumarchaeota, Bathyarchaeota, and Heimdallarchaeota) (see supplementary documents in Klemenčič et al. 2019).

Classification and domain organization of caspase homologs possessing the catalytic dyad in unicellular organisms according to the unified nomenclature of the C14 peptidase family (adapted from Klemenčič et al. 2015; Minina et al. 2020). Members of the family possess the catalytic p20-like region (maroon), a regulatory p10-like region (blue) and/or the linker region (thick gray line). The positions of catalytic His (H) and Cys (C) residues are shown above the p20-like region. The most common YC dyad is shown for the pseudo-orthocaspase. N-terminal pro-domains are indicated in green. Detailed classification is shown in the methodology. The figure is not drawn to scale (Color figure online)
Three homologous groups of caspases (CAs) are documented, type I, II, and III MCAs, type I and II PCAs, and OCAs (Fig. 1). All possess the p20-like region which includes the conserved Histidine–Cysteine (HC) dyad, sometimes also referred to as the Cysteine–Histidine (CH) dyad, which is situated within a characteristic caspase/haemoglobinase fold (Aravind and Koonin 2002; McLuskey and Mottram 2015) (Fig. 1). They are members of the CD clan, C14 peptidase family of the MEROPS peptidase database (Rawlings et al. 2014). Unlike CAs that are only found in Metazoa, PCAs are common in Metazoa and slime molds while MCAs are universally distributed in the three domains of life, except for the Metazoa (Uren et al. 2000). OCAs are ancestral caspase homologs present in bacteria and phytoplankton (Choi and Berges 2013; Klemenčič et al. 2015). OCAs in eukaryotes are commonly referred to as MCA-like proteases (Minina et al. 2020). MCAs, PCAs, and OCAs have different substrate specificities and N-terminal domains compared to CAs (McLuskey and Mottram 2015) but both sets of homologs correlate and are causally associated with cell survival and death functions (Vercammen et al. 2007; Shrestha and Megeney 2012; Minina et al. 2017; van Creveld et al. 2018). Cell survival functions include cell cycle regulation (Ambit et al. 2008; Lee et al. 2008), stress response (Richie et al. 2007), and virulence mediation (Proto et al. 2011; Benler and Koonin 2020). The pleiotropic nature of caspase homologs in both death and non-death-related functions (Shrestha and Megeney 2012; Klemenčič et al. 2015; Hill and Nyström 2015; Lema et al. 2021) indicate that the ‘original sin’ hypothesis is a potentially useful framework for investigating the evolution of the PCD toolkit.
The aim of this study was to investigate the potential presence of homologous caspase-like protein sequences in archaea and provide detailed comparative phylogenetic and sequence analyses of other possible death domains. Our analyses revealed type I MCAs and OCAs are surprisingly widespread in archaea. The phylogenetic reconstruction analysis and taxonomic distribution are suggestive of massive horizontal gene transfer (HGT) events between bacteria and archaea, and that at least some of the effectors of PCD were likely present prior to the diversification of prokaryotes. In addition, numerous death domains were identified in archaeal OCAs and type I MCAs, and subsequent phyletic pattern analyses inferred their putative ancestral functions. Our data provide strong support for the ‘original sin’ hypothesis of PCD. This lays the foundation for understanding the evolution and role of PCD in prokaryotic communities and as a conflict mediator in eukaryogenesis.
Results
Caspase Homologs in Archaea
In total, 321 caspase homologous sequences were identified spanning eleven archaeal phyla (Fig. 2 and Supplementary Table S1). Sequences identified as caspase homologs were further classified according to their structural subtypes (meta-, ortho-, and paracaspases) (Fig. 1). Fifty sequences (15.58%) possessed both the p20-like region and the p10-like region and hence classified as MCAs. The length of the linker region for the fifty sequences was 7.34 ± 27.34 amino acids (Supplementary Table S2). Following the guidelines and criteria of (Choi and Berges 2013), the presence of a short linker region (161.3 ± 32.9 aa) was used to classify the identified MCAs as type I MCAs (Supplementary Table S1) versus type II MCAs with longer linker regions. The remaining 271 sequences only contained the p20-like region and were therefore classified as OCAs (84.42%) (Supplementary Table S1).

Taxonomic distribution and abundance (%) of OCAs (green) and type I MCAs (orange) according to different phyla of archaea. Superphyla are indicated in gray boxes. Unclassified refers to sequences from environmental samples where taxonomic information was not available from the NCBI taxonomy database (Color figure online)
Archaeal OCAs and type I MCAs identified in this study showed a wide taxonomic distribution with sequences belonging to the three major superphyla (TACK, Asgard group, and DPANN) and two phyla of archaea (Thermoplasmatota and Euryarchaeota) (Fig. 2). TACK is an acronym for superphyla Thaumarchaeota, Aigarchaeota, Crenarchaeota, and Korarchaeota (Guy and Ettema 2011) and DPANN is an acronym for superphyla Aenigmarchaeota, Diapherotrites, Nanoarchaeota, Nanohaloarchaeota. Parvarchaeota and Woesearchaeota (Rinke et al. 2013; Castelle et al. 2015). The majority of OCAs and type I MCAs were identified in the phylum Euryarchaeota (51.09%) and phyla with the least number of homologs were Candidatus Aenigmarchaeota and Candidatus Woesearchaeota belonging to superphylum DPANN (Fig. 2). Unclassified organisms refer to sequences obtained from environmental samples that do not have taxonomic annotation information.
Sequence Analysis of Archaeal Orthocaspases and Type I Metacaspases
Multiple sequence alignment (MSA) and sequence logo analyses of archaeal OCAs and type I MCAs revealed the highest sequence conservation occurred in amino acid residues surrounding the catalytic HC dyad (Supplementary Fig. S1 and S2). Additional four key amino acid residues are known to have importance for the formation of the S1 pocket and enzyme specificity in eukaryotic type I MCAs (McLuskey et al. 2012). Of these, two Asp residues were conserved in archaeal type I MCAs and present in most OCAs (Supplementary Fig. S1 and S2). The remaining two amino acids, acidic Cys and Ser, showed variation in residues in both archaeal OCAs and type I MCAs (Supplementary Fig. S1 and S2). The variation in residues corresponded to the amino acids observed in CAs, PCAs, or MCAs of eukaryotes (Wei et al. 2000; Yu et al. 2011; McLuskey et al. 2012). The acidic residues important for substrate specificity in type I MCAs (Y31, numbered according to TbMCA-Ib) and PCAs (E500, numbered according to MALT1) were absent. Proline-rich repeats, which are usually found in the N-terminal region of type I MCAs (Uren et al. 2000), were identified in the linker region of archaeal type I MCAs (Supplementary Fig. S2). Archaeal type I MCAs possessed high-affinity and low-affinity Ca2+-binding sites (Supplementary Fig. S2 and S4). They were absent in archaeal OCAs.
According to the automated I-TASSER protein structure prediction, structural mimicry of archaeal type I MCAs and OCAs to TbMCA-Ib of Trypanosoma brucei (UniProt ID: Q585F3) was observed. Type I MCA of Candidatus Thorarchaeota archaeon (GenBank ID: TFG95767.1) and OCA of Candidatus Prometheoarchaeum syntrophicum (GenBank ID: QEE14607.1) were used as model templates for archaeal type I MCAs (Supplementary Fig. S4) and OCAs (GenBank ID: QEE14607.1) (Supplementary Fig. S3), respectively. The S1-binding pocket was conserved which possessed the HC dyad and the four key residues involved in substrate specificity and enzyme activity (Supplementary Figure S3 and S4). Some differences were observed in the secondary structure and the tertiary folding of archaeal OCAs and type I MCAs, including the number of the β-sheets and α-helices, as well as the missing N-terminal region in archaeal type I MCAs (Supplementary Fig. S3 and S4).
Phylogenetic Analysis
Archaeal OCAs and type I MCAs phylogenies were reconstructed using the MSA of the p20-like region and the C14 peptidase domain, respectively (Supplementary Fig. S1 and S2). Both the OCAs and type I MCAs phylogenetic distributions were incongruent with 16S rRNA phylogeny and revealed a diffuse, scattered distribution (Supplementary Fig. S1 and S2). Tree topologies revealed that the clades were clustered according to the different key residues required for the S1 pocket formation and enzyme activation, which corresponded with the residues observed in CAs, PCAs, or type I MCA of T. Brucei. Both trees were well supported with robust bootstrap values above 0.80 (Supplementary Fig. S1 and S2).
Phylogenetic reconstruction of the p20-like region of bacterial and archaeal OCAs, and eukaryotic CAs, PCAs, and type I MCAs (Fig. 3) revealed an incongruency with the species tree provided by Hug and colleagues (Hug et al. 2016). P20-like regions of PCAs and CAs of Metazoa clustered together, while eukaryotic type I MCAs and PCA of D. discoideum (UniProt ID: Q9GPM2) were placed in separate clades. CAs, PCAs, and eukaryotic type I MCAs all branch from bacterial OCAs, except for PCA of D. discoideum which branches from archaeal OCAs.

Maximum likelihood phylogenetic tree of the p20-like region of 231 amino acid sequences of prokaryotic OCAs, and eukaryotic CAs, PCAs, and type I MCAs generated using the LG + R6 model under IQ-TREE. Three metazoan CAs, H. sapiens CA 3 and CA 8, and C. elegans cell death protein 3 (UniProt IDs: Q14790, P42573, and P42574), were used as outgroup sequences. The classification is shown in different colors: eukaryotic PCAs (yellow), eukaryotic type I MCAs (orange), archaeal OCAs (red), and bacterial OCAs (blue). The tree reliability was tested using the bootstrap method with 1000 replicates, of which values below 0.50 are not shown. The tree scale bar shows substitutions per site. Corresponding Genbank IDs of archaeal OCAs, and UniProt IDs of eukaryotic CAs, PCAs, and type I MCAs, and bacterial OCAs are shown in brackets (Color figure online)
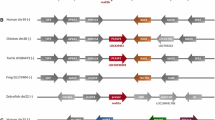
The phylogenetic relationship of type I MCAs across the three domains of life: archaea, bacteria, and eukarya were incongruent with the classic 16S rRNA phylogenetic distribution (Fig. 4). Three clades were observed that were well supported with robust bootstrap values above 0.95 (Fig. 4). For the ultrafast bootstrap used by IQ-TREE, a clade is considered reliable if its support is more than 95% (Hoang et al. 2018). Clade 1 possessed the CAs and bacterial type I MCAs, which included the alphaproteobacterium Rhizobiales bacterium (UniProt ID: A0A2A4P607). The second clade consisted of bacterial and archaeal type I MCAs. They possessed key residues resembling type I MCA as well as the low-affinity and high-affinity Ca2+-binding sites and proline-rich repeats. The third clade possessed type I MCAs of eukarya, bacteria, and archaea, which included the OCA of Asgard archaeon (Candidatus Thorarchaeota archaeon (GenBank ID: TFG95767.1)). They also possessed the key residues observed in a type I MCA as well as the low-affinity and high-affinity Ca2+-binding sites.

A Maximum likelihood phylogenetic tree of MCAs in the three domains of life generated using the LG + R5 model under IQ-TREE. The tree is based on the C14_peptidase domain of 69 protein sequences. Three metazoan CAs, H. sapiens CA 3 and CA 8, and C. elegans cell death protein 3 (UniProt IDs: Q14790, P42573, and P42574), were used as outgroup sequences. The classification according to the three domains of life is shown in different colors: eukarya (green), archaea (red), and bacteria (blue). Different clades are shown in different colors: clade 1 (orange), clade 2 (pink), and clade 3 (green). Bootstrap values based on 1000 replicates calculated by maximum likelihood. Bootstrap values below 0.50 are not shown. The tree scale bar shows substitutions per site. Corresponding Genbank IDs of archaea and UniProt IDs of eukaryotes and bacteria are shown in brackets. B Partial MSA of key residues of type I MCAs in the three domains of life under hmmalign as well as MAFFT (See Methods). The figure was generated using Jalview. Colors of the amino acid have been assigned according to the Percentage Identity color scheme on Jalview with an identity threshold above 0.50. The catalytic HC dyad is indicated with a red star. Residues involved in the formation of the S1 pocket are indicated in orange, the proline-rich repeat is shown in pink, and Ca2+-binding sites are shown in blue (high affinity) and green (low affinity). The p20-like, linker, and p10-like regions are shown below the MSA (Color figure online)
C14 Peptidase-Accompanying Domains and Domain Architectures of Archaeal OCAs and Type I MCAs
In addition to the C14 Peptidase domain, 56 different C14 peptidase-accompanying domains were identified (Supplementary Table S3). These include Peptidase_C13 (34.07%), Raptor_N (10.37%), Formylglycine-generating (FGE) sulfatase (4.44%), Polycystic Kidney Disease (PKD) (4.44%), and PKD_4 (4.44%). Thirty domains were identified only once in the sequence data. GO prediction and functional information available on the Pfam database revealed the functions of the three most abundant C14 peptidase-accompanying domains (PKD, PKD_4, and FGE_sulfatase) as cell surface proteins that protect against extreme environments (Jing et al. 2002), respond to environmental stimuli, and unknown, respectively (Supplementary Table S4). Archaeal OCAs and type I MCAs that possess C14 peptidase-accompanying domains associated with cell survival and PCD were identified. Single C14 peptidase-accompanying domains predicted to have both functions were common (Fig. 5 and Supplementary Table S4). In addition, C14 peptidase-accompanying domains implicated in interactions with the environment and neighboring cells were abundant. These included domains associated with cell adhesion, cell projection, and cell surface receptors (Supplementary Table S4). A complete list of C14 peptidase-accompanying domains, their abundance, and predicted functions is available in Supplementary Table S3 and S4.

Dual role of C14 peptidase-accompanying domains which were predicted to have cell survival and death-associated functions. Cell survival-associated functions refer to functions mentioned by Ameisen which contributed to the rise of the PCD genetic toolkit (Ameisen 2002). C14 peptidase-accompanying domain associated with cell survival or death has been classified according to appropriate categories: cell cycle (blue), cell metabolism (orange), cell differentiation (purple), and PCD (green). C14 peptidase-accompanying domains include ANAPC3 (Anaphase-promoting complex, cyclosome, subunit 3), ANAPC4_WD40 (Anaphase-promoting complex subunit 4 WD40 domain), ANF_receptor (Receptor family ligand-binding region), BiBP_C (Penicillin-Binding Protein C-terminus Family), CUB (CUB domain), DUF11 (Domain of unknown function), DZR (Double zinc ribbon), EF-hand_5 (EF hand), fn3 (Fibronectin type III domain), fn3_3 (Polysaccharide lyase family 4, domain II), Peripla_BP_5 (Periplasmic-binding protein domain), Peptidase_C13, Peptidase_M20, Peptidase_M28, PPC (Bacterial pre-peptidase C-terminal domain), TPR_1, TPR_2, TPR_11, and TPR_16 (TPR repeat), and WD_40 (WD domain, G-beta repeat). Detailed information is available in the supplementary table S4, Supplementary Materials (Color figure online)
Prediction of transmembrane domains using TMHMM revealed 127 (39.56%) sequences with varying transmembrane topologies (Supplementary Table S5). One hundred fourteen protein sequences were found to be type II membrane proteins, which are single-spanning membrane proteins that possess intracellular N-terminals and extracellular C-terminals (von Heijne 2006) and resemble signaling peptides (Krogh et al. 2001).
In total, 63 different domain architectures were identified (Figs. 6, 7). Out of 321 sequences, 183 (57.01%) sequences possessed C14 peptidase-accompanying domains of which 177 sequences were OCAs and six were type I MCAs. The most abundant domain architectures were OCAs possessing a single p20-like region (29.30%), OCAs possessing a transmembrane helix followed by a p20-like region (22.43%), and type I MCAs possessing a p20-like region followed by a p10-like region (13.40%). Forty-two domain architectures were identified only once in the sequence dataset (13.08%) and no domain architecture was unique to a specific phylum.

Domain architectures of transmembrane archaeal OCAs and type I MCAs. Domains (oval) and distances (gray line) between domains are not drawn to scale. Domains are presented with N-terminals on the left and C-terminals on the right. iTmm refers to transmembrane helices predicted to have N-terminal domains situated intracellularly and C-terminal domains extracellularly. oTMM refers to transmembrane helices predicted to have the reverse topology. Functional categories and color keys of C14 peptidase-accompanying domains and transmembrane domains are indicated below the domain architectures. Only functions associated with cell survival, death, or interactions with neighboring cells and the environment are indicated. Functions labeled ‘other’ (yellow) refer to domains associated with functions other than PCD, cell survival, or cell–cell interactions. P20- and p10-like regions of the C14 peptidase domain are colored lilac. If a domain was associated with more than one function, all the potential functions were indicated with the corresponding color (Color figure online)

Domain architectures of 195 cytosolic archaeal OCAs and type I MCAs. Domains (oval) and distances (gray line) between domains are not drawn to scale. Domains are presented with N-terminals on the left to C-terminals on the right. iTmm refers to transmembrane helices predicted to have N-terminal domains situated intracellularly and C-terminal domains extracellularly. oTMM refers to transmembrane helices predicted to have reverse topology. Functional categories associated with cell survival, death, or interaction with neighboring cells and the environment were considered, and the color key of C14 peptidase-accompanying domains and transmembrane domains is shown below the domain architectures. Functions labeled ‘Other’ refer to domains associated with functions other than PCD, cell survival, or interaction with neighboring cells and the environment. P20- and p10-like region of the C14 peptidase domain is colored lilac. If a domain was associated with more than one function, all the potential functions were indicated with the corresponding colors (Color figure online)
Transmembrane OCAs are common in archaea (39.25%). The most common domain architecture of transmembrane OCAs was an extracellular C-terminal p20-like region and domains interacting with the neighboring cells and the environment (8.41%) (Fig. 6, domain architecture 3, 5–8, 10, 14,15, 17, 18, 20–22, 25–27, 30). Compared to transmembrane OCAs, cytosolic OCAs possessed a great variety of domains, including those associated with survival (Fig. 7, domain architecture 34, 37, 38, 43, 48, 55–57, 60–62) and PCD (Fig. 7, domain architecture 43, 56, 61, and 62). The death domains (Aravind et al. 1999; Li and Roberts 2001) identified in this study include TPR_1, TPR_2, TPR_16, and TPR_11 (Tetratricopeptide repeat), and WD_40 (WD domain) and ANAPC4_WD40 (Anaphase-promoting complex subunit 4 WD40 domain) (Supplementary Table S4). Function assignments via Pfam database, dcGO, and Pfam2GO, associated these death domains with cell survival functions, such as regulation of cell growth, cell projection assembly, and cell cycle control (Supplementary Table S4).
Archaeal type I MCAs possessed simple domain architectures, of which most did not possess any C14 peptidase-accompanying domains (86.60%). Transmembrane type I MCAs were rare (3.38%). C14 peptidase-accompanying domains in archaeal type I MCAs were associated with cell adhesion and cell surface receptors (Fig. 6, domain architecture 12 and 29, and Fig. 7, domain architecture 58).
Taxonomic distribution analysis revealed that cytosolic and transmembrane OCAs and type I MCAs associated with cell survival had wide taxonomic distributions (Supplementary Table S6). They were identified in superphyla Asgard group and TACK, and phyla Thermoplasmatota and Euryarchaeota. Archaeal cytosolic OCAs possessing death domains were confined to superphyla Asgard group and TACK (Supplementary Table S6).
Discussion
Caspase homologs were identified in archaeal superphyla TACK, the Asgard group, and DPANN, and phyla Euryarchaeota and Thermoplasmatota. In total, 321 OCAs and type I MCAs were identified (Fig. 2) by stringent hmmsearch filtering, manual inspection of the HC dyad (Supplementary Fig. S1 and S2), and structural similarity (Supplementary Fig. S3 and S4). The HC dyad is a requirement for the proteolytic activity of OCAs and MCAs (Aravind and Koonin 2002; McLuskey and Mottram 2015), whereas the absence of the dyad indicates catalytic inactivity (Szallies et al. 2002). It is likely, therefore, that the OCAs and MCAs reported here are active.
The OCAs were much more abundant in archaea (84.42%) than type I MCAs (15.58%) (Fig. 2), a pattern that is similar to the findings in bacteria (Klemenčič et al. 2019). It is possible that some sequences classified as OCAs may be partial sequences, which can result in erroneous classifications, and future whole-genome sequencing analyses will resolve any potential inconsistencies. There was a bias in the taxonomic distribution of archaea (Fig. 2), which we attribute to the relative dearth of data from superphyla TACK, DPANN, and Asgard group. This will likely also change as more data become available. CAs and PCAs were not identified, which is in keeping with the previous phylogenetic predictions (Uren et al. 2000). Similarly, type II MCAs and type III MCAs were not identified. This was unsurprising because type II and III MCAs are reportedly specific to Viridiplantae and some algal species where they are associated with secondary endosymbiotic events (Choi and Berges 2013). In addition to the previously listed putative archaeal caspase homologs (Klemenčič et al. 2019), an additional 228 sequences were identified here, including previously unclassified sequences from superphyla Asgard group and TACK.
Comparative sequence and secondary structure prediction analyses of OCAs and archaeal type I MCAs confirmed the presence of key residues and structures typical of this group of homologs (Supplementary Fig. S1, S2, S3, and S4). This includes the HC dyad, and two Asp residues present in the S1 pocket that are required for substrate specificity and enzyme activity. There was some minor variation in two of the key residues. These are the acidic Cys (Supplementary Fig. S1 and S2), which is important for substrate binding (McLuskey et al. 2012), and Ser (Supplementary Fig. S1 and S2), which is important for regulation of enzyme activity (McLuskey et al. 2012). Interestingly, this variation matched those of eukaryotic type I MCAs, PCAs, or CAs (Supplementary Fig. S1 and S2) and may have some functional significance. The conservation of key residues in eukaryotic type I MCAs, PCAs, or CAs suggests that they may be derivatives of archaeal OCAs and type I MCAs. Furthermore, the structural mimicry of the automated I-TASSER predicted model of archaeal type I MCA and OCAs with the X-ray crystallography of TbMCA-Ib further suggests common ancestry and structural (and presumably functional) homology (Supplementary Fig. S4).
The presence of key residues and structures required for a catalytically active C14 peptidase suggests that archaeal OCAs and type I MCAs are catalytically active. A small subset of archaeal OCAs was very similar in their key residues to an OCA found in Microcystis aeruginosa (Supplementary Fig. S1) that is known to have MCA-like activity (Klemenčič et al. 2015). This subset may, therefore, also have MCA-like activity. The conservation of some of the Ca2+-binding sites in archaeal type I MCAs suggests that Ca2+ remains important as a secondary messenger in the activation of archaeal type I MCAs. As described by (Klemenčič et al. 2015), Ca2+-binding sites were absent in archaeal OCAs.
The N-terminal domain requirement for MCA activity (McLuskey et al. 2012), and the zinc-finger domain required for cell death function (Coll et al. 2010) were absent in archaeal OCAs and type I MCAs (Supplementary Fig. S3 and S4). Tyr, which functions as a latch with Ser for regulation of enzyme activation (McLuskey et al. 2012) was also absent in both archaeal OCAs and type I MCAs. The observed differences in the secondary structure and some of the key residues required for enzyme activity and regulation (supplementary Fig. S1 and S3, Supplementary Material online) suggest there are likely subtle differences in substrate specificity or regulation of activity of archaeal OCAs and type I MCAs compared to TbMCA-Ib.
Origins of Prokaryotic Type I MCAs and OCAs
Phylogenetic reconstruction of archaeal type I MCAs and OCAs illustrates a diffuse gene distribution pattern across different phyla and is inconsistent with the species trees (Supplementary Fig. S1 and S2 and discussed further below). Both trees were well supported with robust bootstrap values above 0.80. Only one sequence belonging to the superphylum Asgard group was available (Genbank ID TFG95767.1) for type I MCA phylogenetic analysis, while none were available from superphylum DPANN, which resulted in a biased phylogeny of type I MCAs towards the phylum Euryarchaeota.
The identification of OCAs and type I MCAs in all three domains of life—archaea, bacteria, and eukarya—and their shared origins with CAs and PCAs raises new questions: were the ancestors of these PCD effectors present in the ancestors of both archaea and bacteria? Or did they emerge after the divergence of the two prokaryote lineages with subsequent HGT between them? Furthermore, since most of the eukaryotic members of the C14 peptidase family appear to have originated from the bacterial ancestors of mitochondria (Aravind et al. 2001; Koonin and Aravind 2002; Klim et al. 2018), what has been the evolutionary trajectory of the archaeal homologs? What is clear from the data here and previous publications (Uren et al. 2000) is that PCAs and CAs are limited to the so-called higher, complex organisms while MCAs are widely distributed across all lineages with the notable exception of Metazoa (Uren et al. 2000; Choi and Berges 2013). OCAs are confined to unicellular organisms (Klemenčič et al. 2015; Klemenčič and Funk 2018). The conservation of key residues, the structural similarities between archaeal OCAs and the p20-like region of PCAs and type I MCAs, and their presence in the deepest branching superphylum DPANN, phylum Euryarchaeota (Fig. 3), and the bacterial phyla Aquifex, Thermotogae, and Deinococci (Klemenčič et al. 2019) suggest that OCAs (the p20-like region) are almost certainly the most ancestral members of the peptidase C14 clan. This supports earlier predictions (Choi and Berges 2013).
Three clades of type I MCAs are neatly resolved. These are the bacterial caspase homologs (clade 1), prokaryotic type I MCAs (clade 2), and eukaryotic type I MCAs (clade 3). CAs were placed in one clade with bacterial type I MCAs, which included the Rhizobiales bacterium (UniProt ID: A0A2A4P607) bacteria (Fig. 4). This supports the origin of CAs as bacterial caspase homologs during the endosymbiotic event (Koonin and Aravind 2002) and is consistent with the previous findings tracing the evolutionary histories of CAs and MCAs across the three domains of life (Klim et al. 2018).
The origin of unicellular eukaryotic type I MCAs in clade 3 is more difficult to resolve and could have emerged in either archaea or bacteria (Fig. 4). The absence of the Ca2+-binding sites in bacterial type I MCAs in clade 1 indicates that the ancestral type I MCAs were not regulated by Ca2+, a feature that emerged later. Proline-rich repeats, which are typically found in the N-terminal domains of eukaryotic type I MCAs, were only identified in clade 2, which contains prokaryote type I MCAs. The distribution of type I MCAs across different domains indicates HGT events between prokaryotes and eukaryotes.
The incongruency between gene and species trees is most likely due to extensive HGT between archaea and bacteria (Maddison 1997; Ponting et al. 1999; Ochman et al. 2000; Aravind et al. 2001). Tracing the key residues in the S1 pocket and additional C14 peptidase-accompanying domains present in different members of the C14 peptidase family also suggest massive HGT. This is in line with what is known about large-scale gene flows between the proto-cellular organisms during the emergence of diverse prokaryotic lineages (Koonin et al. 1997; Aravind et al. 1998; Nelson et al. 1999; Nelson-sathi et al. 2015; Koonin 2016; Wagner et al. 2017; Husnik and McCutcheon 2018) and limits the identification of donor lineages (Akanni et al. 2015). Of course, there are other potential explanations. For example, one type of a C14 peptidase may have arisen de novo in an ancestral archaeon with continuation in daughter lineages and HGT and gene duplication events (Fig. 8). However, this would have to include multiple duplication events with subsequent HGT of the paralogous genes. It seems more parsimonious that the OCAs were present prior to the diversification of archaea and bacteria, suggesting that ancestral OCAs were already present in the realm of the LUCAS (Last Universal Common Ancestor State). The acronym LUCAS refers here to Koonin’s “forest of life” (FoL) concept, which was a hypothetical state prior to the existence of cells as we understand them today and in which genetic material flowed between semi-cellular compartments (Koonin 2009). Another possible explanation is that these C14 peptidases originated from eukaryotes and were transferred to prokaryotes via HGT events (Rawlings and Bateman 2019). However, their wide taxonomic distribution in all of the major phyla of archaea and bacteria suggest they were present in prokaryotes first. The mitochondrial origin of the C14 peptidase family during eukaryogenesis (Kroemer 1997; Frade and Michaelidis 1997; Mignotte and Vayssiere 1998; Blackstone and Green 1999) also supports their prokaryotic origin and is consistent with Klim and colleagues’ mitochondrial hypothesis for the origin of eukaryotic apoptosis (Klim et al. 2018).

Canonical 3-domain tree of life with caspase homologs. Only the groups of interest included in this study are indicated. HGT events between the domain eukarya and bacteria are indicated by the arrows and thickness indicates the frequency of the event (Color figure online)
C14 Peptidase-Accompanying Domains Associated with Pro-survival Outnumber Death Domains in Archaeal OCAs and Type I MCAs
The conservation of OCAs and their ancient origins raise questions about their ancestral functions. The functional inference was based on phyletic and functional pattern analysis of C14 peptidase-accompanying domains, with reference to the previous studies (Gaasterland and Ragan 1998; Aravind et al. 1999; Galperin and Koonin 2000). Of course, functional inference based on phyletic pattern analysis is predictive and awaits future empirical studies. Nevertheless, a large variety of different C14 peptidase-accompanying domains were easily identified, and these were more abundant in archaeal OCAs (97.27%) than type I MCAs (2.73%) (Supplementary Table S3,). The high abundance of the Peptidase_C13 and Raptor_N domains is spurious and explained by their sequence similarities to the C14 peptidase domain and the presence of the HC dyad (Ginalski et al. 2004; Rawlings et al. 2014). Functional assignment of the other C14 peptidase-accompanying domains using multiple GO databases indicated that domains associated with cell survival are more abundant than those associated with PCD (Fig. 5). GO functions associated with cell survival were chosen based on the Gedanken experiment by Ameisen (Ameisen 2002), which includes pro-survival functions like cell differentiation, cell cycle regulation, and cell metabolism.
Domain architecture analyses revealed archaeal OCAs resembling signaling peptides that interact with the external environment (Fig. 6). Examples include OCAs possessing a transmembrane helix followed by C14 peptidase-accompanying domains associated with cell adhesion, cell projection, and cell surface receptors. The occurrence of these membrane-bound OCAs suggests archaeal OCAs were involved in cellular responses to the surrounding environment through cell signaling mechanisms. In contrast, only a few archaeal type I MCAs possessed C14 peptidase-accompanying domains and transmembrane helices. It is acknowledged that the transmembrane components of the archaeal OCAs and type I MCAs resembling signaling peptides were predicted using the TMHMM program, which reports a ~ 78% precision in topology prediction (Krogh et al. 2001) and should be interpreted in this light.
Cytosolic archaeal OCAs possessing death domains were observed in superphylum TACK (Fig. 7 and Supplementary Table S6). However, it is uncertain if their presence implies archaeal OCAs are involved in PCD as death domains are associated with both death and pro-survival. According to the GO prediction, the death domains are also associated with cell survival functions (a quirk of the historical use of PCD terminology), such as cell cycle control, differentiation, and metabolism (Supplementary Table S4). The abundance of C14 peptidase-accompanying domains associated with cell survival in archaeal OCAs adds to this uncertainty.
The pleiotropic nature of MCAs and the association of these effector enzymes with both PCD and cell survival functions are established (Shrestha and Megeney 2012; Hill and Nyström 2015). Their pleiotropic nature and the higher abundance of C14 peptidase-accompanying domains associated with pro-survival, therefore, raise questions about the role of ancestral OCAs and type I MCAs in archaea. Did they evolve because of PCD-associated cell death functions, or were they co-opted from ancestral proteins with pro-survival functions? Intuitively, it seems more parsimonious that these cytosolic archaeal OCAs were initially involved in pro-survival activities and were subsequently co-opted into death-related functions.
Archaeal OCAs and Eukaryogenesis
According to the Entangle-Engulf-Enslave (E3) model, the physical interaction between a Candidatus Prometheoarchaeum syntrophicum MK-D1 archaeon and an alphaproteobacterium involved cellular protrusions and phagocytosis-independent engulfment (Imachi et al. 2020). This is because phagocytic machinery is absent in Asgard group, the proposed archaeal clade involved in archaea–alphaproteobacteria symbiosis (Moreira and Lopez-Garcia 1998). Using this hypothetical framework, which is now one of the most updated hypotheses for eukaryogenesis (Imachi et al. 2020), we wished to investigate the potential role of these archaeal OCAs in the rise of FECA (First Eukaryotic Common Ancestor). Two OCAs found in Candidatus Prometheoarchaeum syntrophicum strain MK-D1 archaeon possessed simple domain architectures with no C14 peptidase-accompanying domains (Fig. 6, domain architecture 1 and Fig. 7, domain architecture 32). In contrast, OCAs of other Asgard archaea (Fig. 6, domain architecture 2, 3, 7, 14, 17, 19, and 24 and Fig. 7, domain architecture 36 and 60) were all transmembrane proteins with domains associated with cell metabolism, cell adhesion, cell cycle control, cell projection, cell differentiation, and cell surface proteins/receptors implicated in stress responses. The abundance of cell surface and receptor-like OCAs in the Asgard group suggests that these archaeal OCAs may be candidate PCD-related genes that were also important in the E3 model of eukaryogenesis (Fig. 6).
The ‘Original Sin’ Hypothesis and the PCD Toolkit
The phyletic pattern analysis of archaeal OCAs revealed that they are not only the most ancestral peptidase C14 members, but they also contain an abundance of pro-survival domains (Figs. 6, 7). This is especially true of the deep-branching phyla Euryarchaeota and Thermoplasmatota, linking these ancestral OCAs with cell survival prior to eukaryogenesis. Although OCAs of DPANN only possessed the p20-like region, it is possible that additional genomic data will identify some of the C14 peptidase-accompanying domains. Although we are unsure if ancestral OCAs and type I MCAs were associated with PCD, it is clear that the ancestral effectors of PCD were already present prior to the diversification of bacteria and archaea. Type I MCAs appeared later in Thermoplasmatota and Euryarchaeota (Fig. 9) and acquired Ca2+-binding sites to refine their activity (Figs. 4, 9). In addition, OCAs and type I MCAs of superphylum TACK acquired death domains and other C14 peptidase-accompanying domains via HGT (Figs. 6, 7, 9). It seems that this pre-adaptation allowed for the emergence of pro-death processes in prokaryotes and eukaryotes (Aravind et al. 1999; Ameisen 2002). Klemenčič and Funk have suggested before that PCD evolved from effectors involved in physiological cellular processes (Klemenčič and Funk 2018). The findings presented here enhance these assertions and support the ‘original sin’ hypothesis which is a philosophical argument put forward by Ameisen in 2002 that argues for diverse pro-survival functions for the PCD toolkit with subsequent refinement for essential death-related functions (Ameisen 2002).

Proposed acquisitions of the different C14 peptidase domains and their associated pro-survival or pro-death domains in archaea. A simple C14 peptidase distribution is shown on the tree of life: archaea (red), bacteria (blue), and eukarya (green). The acquisitions of domains are shown in lines of different colors: p20-like region (red), p10-like region (light blue), C14 peptidase-accompanying domains associated with cell survival (yellow), and death domains (green). The important regions specific to type I MCAs, the Ca2+-binding sites (blue circle) and proline-rich repeats (pink circle), are indicated. LUCAS refers to the Last Universal Common Ancestor State. The point of endosymbiosis leading to eukarya is shown with a star. Horizontal gene transfer events between the domain eukarya and bacteria are indicated by the arrows (Color figure online)
Conclusion
OCAs and type I MCAs are present in archaea and have a wide taxonomic distribution in the three domains of life. The phylogeny of OCAs and type I MCAs suggest that these are the ancestral peptidase C14 members and were present since LUCAS. Their strong similarity with other members in bacteria and eukarya further supports this and the incongruency with the species tree proposes massive HGT of domains between prokaryotes and proto-cellular compartments prior to their divergence. The phyletic and functional pattern analyses of C14 peptidase-accompanying domains and death domains provide strong support for the ‘original sin’ hypothesis that OCAs in the Asgard group were perhaps first involved in pro-survival functions with subsequent co-option for PCD functions in prokaryotes and the earliest eukaryotes.
Materials and Methods
Sequence Retrieval and Identification of Caspase Homologs
The NCBI Protein database (https://www.ncbi.nlm.nih.gov/protein/) (Accessed 2 June 2021) was queried using search terms: “C14,” “caspase,” “metacaspase,” and “orthocaspase” in organism “archaea” under default parameters. Retrieved protein sequences were identified as caspase homologs by the presence of the Peptidase_C14 domain (PF00656) (http://pfam.xfam.org/) (Mistry et al. 2021) using the hmmsearch tool from the HMMER v3.0 package (http://hmmer.org/) (Eddy 1998) under a domain independent E-value ≤ 0.0001 and score/bias ratio ≥ 10. Hmmsearch uses profile hidden Markov Models (HMM) to detect sequence similarity and homology (Eddy 1998). The raw Peptidase_C14 HMM profile (PF00656) (206 amino acids) obtained from the Pfam database was used. The aligned MSA file of 321 sequences was aligned to Peptidase_C14 HMM profile (PF00656) using the hmmalign tool under the-trim option. The presence of the conserved histidine and cysteine (HC) dyad was identified by visual inspection under Jalview v2.0 (Waterhouse et al. 2009) according to the reference position (H220 and C276) provided on the MEROPS database (Rawlings et al. 2014) as well as using the reference sequence of TbMCA-Ib of T. brucei. Sequences not possessing the HC dyad or pseudo-dyads (Eyers and Murphy 2016) were removed.
Classification and Taxonomy Assignment of Caspase Homologs
A p20-like and p10-like region profile HMM was constructed with HMMER v3.0 hmmbuild, according to the approach followed by Klemenčič and colleagues (Klemenčič et al. 2019). The first 457 and the last 211 columns of Pfam Peptidase_C14 (PF00656) seed alignment (112 sequences with gaps) were used as the input for the p20-like and the p10-like regions, respectively. To identify PCAs, hmmsearch was performed using the Immunoglobulin (Ig) (PF00047), Ig_2 (PF13895), or Ig_3 (PF13927) HMM profiles (http://pfam.xfam.org/), commonly found N-terminal pro-domains in PCAs, and the unaligned 321 protein sequence file under a domain independent E-value ≤ 0.0001 and score/bias ratio ≥ 10 using the-domtblout parameter to produce the domain hits table as the output. MCAs were classified according to their subtypes based on sequence data of the amino acid length of the linker region between p20-like and p10-like regions (Uren et al. 2000; Choi and Berges 2013) (Fig. 1). Linker region length was determined from the alignment coordinates in the domain hits table obtained from the hmmsearch hit.
Taxonkit, an NCBI taxonomic classification toolkit (Shen and Ren 2021) was then used to convert taxonomy names to their respective taxonomy ID (TaxId) and assign the full taxonomy to 321 sequences. If no taxonomic information was available, it was assigned as ‘unclassified.’
Sequence Analysis and Tertiary Model Prediction
Sequence analysis of archaeal OCAs and type I MCAS was performed by aligning respective unaligned sequences to the constructed p20-like region HMM profiles and the Peptidase_C14 HMM profile (PF00656) using hmmalign under-trim option. A more reliable alignment was produced by realigning the produced MSA using MAFFT v.7.470 (Katoh and Standley 2013) under default parameters. Sequences were filtered by removing sequences with no residues at the site of four key residues required for the correct formation of the S1 pocket (Cys, Asp, Ser, and Asp) (Wei et al. 2000; Yu et al. 2011; McLuskey et al. 2012). Three metazoan CAs, Homo sapiens CASP8, H. sapiens CASP3, and Caenorhabditis elegans cell death protein 3 (UniProt IDs: Q14790, P42573, and P42574), PCAs of H. sapiens (MALT1), C. elegans (MALT1), Amphiprion percula (MALT PCA3) and Dictyostelium discoideum (PCP_DICDI) (UniProt IDs: Q9UDY8, G5EG87, A0A3P8TIJ8, and Q9GPM2), and type I MCA of T. brucei (TbMCA-Ib) (UniProt ID: Q585F3) were included for sequence comparison. PCAs of Dictyostelium discoideum (UniProt ID: Q9GPM2) and Amphiprion percula (UniProt ID: A0A3P8TIJ8) were only used for the sequence comparison with archaeal type I MCAs as they possessed the p10-like region. Secondary structure prediction was performed using JPred (Protein Secondary Structure Prediction Server) (Drozdetskiy et al. 2015) under Jalview with default parameters. To generate sequence logos of archaeal OCAs and type I MCAs, MSAs were transformed to HMMs using hmmbuild and submitted to Skylign5 (https://www.skylign.org/) (Wheeler et al. 2014). The full alignment of OCAs (271 sequences) or type I MCAs (50 sequences) was used using the parameter ‘information above background.’ Putative protein tertiary structures of archaeal OCAs and type I MCAs were predicted using I-TASSER under default parameters (https://zhanglab.dcmb.med.umich.edu/I-TASSER/) (Yang and Zhang 2015). I-TASSER uses a hierarchical approach to predict protein structures by multiple threading approach LOMETS (Local Meta-Threading Server, version 3). Protein structures were visualized on PYMOL (The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC). The protein sequence of an archaeal OCA (GenBank ID: QEE14607.1) and type I MCA (GenBank ID: TFG95767.1) was selected as the representative OCA and type I MCA model with the TbMCA-Ib of T. brucei (PDB ID: 4AFV) as the template.
Phylogenetic Analysis
Four MSAs (archaeal type I MCA and OCA, and type I MCA and OCA across three domains of life) were created for the reconstruction of respective phylogenies. Archaeal OCA and type I MCA MSAs created for the sequence analysis were used, excluding sequences with no taxonomic classification. For the sequence selection of type I MCAs across the three domains of life, all 33 filtered archaeal type I MCA sequences were used. UniProt IDs of an identical number of type I MCAs of eukarya belonging to unicellular phyla and bacteria were obtained from supplementary documents (Klemenčič et al. 2019). Respective protein sequences were obtained from the UniProt database (The UniProt Consortium 2021). For the sequence selection of the p20-like region, 143 filtered p20-like regions of archaeal OCA sequences were used. An identical number of UniProt IDs of bacterial OCAs were obtained from supplementary documents (Klemenčič et al. 2019) and the respective sequences were obtained from the UniProt database. The p20-like region of eukaryotic CAs (H. sapiens CASP8, H. sapiens CASP3, and C. elegans cell death protein 3), PCAs (H. sapiens (MALT1), C. elegans (MALT1), and D. discoideum (PCP_DICDI)), and eleven eukaryotic type I MCAs with known functions (Tsiatsiani et al. 2011) were included. Eukaryotic OCAs were not considered as the aim was to determine the origin and investigate the evolutionary history of the p20-like region from prokaryotic OCAs to eukaryotic CAs, type I MCAs, and PCAs. An identical protocol was followed for the construction of all four MSAs. Unaligned MSA file of acquired protein sequences was aligned to the p20-like region HMM profile for OCAs and the entire Peptidase_C14 HMM profile using hmmalign under-trim option. Gaps were removed using seqmagick 0.7.0 at default parameters as gaps are treated as unknown characters in IQ-TREE (Nguyen et al. 2015). The resulting trimmed MSA file was then aligned using MAFFT under default parameters. Sequences were filtered by removing sequences with no residues at the site of four key residues required for the correct formation of the S1 pocket (McLuskey et al. 2012). The best substitution model for phylogenetic reconstruction was chosen using IQ-TREE to correct for multiple changes at the same site under a maximum likelihood (ML) approach, including all sites (including gaps) (Kalyaanamoorthy et al. 2017). Considered substitution models included a wide range of models that are supported by IQ-TREE, including advanced partition and mixture models (Minh et al. 2020). Rate heterogeneity across sites was considered as well (Minh et al. 2020). The phylogenetic tree was reconstructed using IQ-TREE using an ML approach (Nguyen et al. 2015). The reliability of the reconstructed tree was assessed using the bootstrap method with 1000 replicates in IQ-TREE (Hoang et al. 2018). The phylogenetic tree was visualized using the Interactive Tree of Life (iTOL) v5.0 online service (https://itol.embl.de/) (Letunic and Bork 2019).
Identification of C14 Peptidase-Accompanying Domains
C14 peptidase-accompanying domains refer to Pfam domains identified within an OCA or type I MCA sequence, adjacent to the C14 peptidase domain. The entire database of HMM profiles was retrieved from Pfam (ftp://ftp.ebi.ac.uk/pub/databases/Pfam/releases/Pfam33.0/) and the HMM profile database was prepared for usage using hmmpress in the HMMER package. The identified, full-length 321 amino acid sequences of caspase homologs were subjected to a hmmsearch against the produced HMM profile database under a threshold E-value of ≤ 0.0001, independent E-value ≤ 0.0001, and score bias ratio ≥ 10, and the output was produced using-domtblout option (Eddy 1998).
Identification of Transmembrane Domains
Transmembrane domains in caspase-like protein sequences were predicted using the TMHMM Online Server v2.0 under default values (http://www.cbs.dtu.dk/services/TMHMM/) (Sonnhammer et al. 1998) with the unaligned MSA of the 321 sequences as the input file. Transmembrane domains were classified according to their topology (von Heijne 2006). If the C14 peptidase domain was located C-terminally to a single N-terminal transmembrane helix, it was regarded as a signal peptide, following the approach of Krogh et al. (Krogh et al. 2001).
Functional Prediction and Classification of C14 Peptidase-Accompanying Domains
Putative functions (biological processes) of identified C14 peptidase-accompanying domains were predicted using the gene ontology (GO) information using the Pfam2Go (https://rdrr.io/github/missuse/ragp/man/pfam2go.html) and dcGO Enrichment approach (https://supfam.org/SUPERFAMILY/dcGO/), as well as the information available on Pfam website (https://pfam.xfam.org/). The Pfam release used for the functional annotation was v34.0 (Mistry et al. 2021). For dcGO GO assignment, False Discovery Rate (FDR) threshold of 0.05 was chosen as the cut-off value and the remaining setting was set as default. Multiple GO databases were used for functional prediction (Forslund and Sonnhammer 2008). Predicted functions were further classified: cell survival, PCD, and interaction with neighboring cells and the environment. Cell survival-associated functions refer to the functions listed by Ameisen (Ameisen 2002). The classification of the predicted functions (biological processes) used is as follows: cell differentiation, cell cycle (DNA replication, transcription, and recombination, DNA repair mechanisms, cell membrane repair mechanisms, cell division, chromatin organization, remodeling or dissolution of the nuclear membrane and chromosomal migration for cell division), and cell metabolism (regulation of ionic channels). The term ‘PCD-associated’ refers to the death domains that act as ligands or adaptors of the PCD molecular pathway domains (Aravind et al. 1999; Li and Roberts 2001). Interaction with neighboring cells and the environment refers to biological processes: cell adhesion, cell projection, cell surface protein or receptor, response to stress, and interaction with neighboring cells. The remaining functions were classified under “other.” If no function was predicted by the GO databases, the function of the domain was assigned as “unknown.”
Domain Architecture Analysis
Protein domain architectures of archaeal caspase homologs were created by using a custom R script (https://github.com/Asplund-Samuelsson/caspases.git) and an input file (Sequence ID, Domain, alignment coordinates). Domain refers to the C14_peptidase domain, C14 peptidase-accompanying domains, and transmembrane helices. Domains were placed in sequential order according to the alignment coordinates. An overlap of two domains was calculated by their predicted alignment coordinate end and start position of domain1 and domain2. If two domains on a sequence overlapped more than 25%, the domain with the lower E-value was accepted. C14 peptidase domain took precedence if other domains were found. If a transmembrane helix occupied the same region as a C14 peptidase-accompanying domain, the C14 peptidase-accompanying domain took precedence. Consecutive repeats of identical domains were identified as a single domain.
Data Availability
The data underlying this article are available at https://github.com/laso415/C14peptidase as well as in the article and its online supplementary material. The datasets were derived from sources: NCBI Protein database, UniProt database, Pfam database, and GO databases (dcGO and Pfam2Go), as indicated in the Materials and Methods section and supplementary data. The specific accession numbers are provided in the respective Materials and Methods sections and the supplementary materials.
References
Abada A, Segev E (2018) Multicellular features of phytoplankton. Front Mar Sci 5:144. https://doi.org/10.3389/fmars.2018.00144
Akanni WA, Siu-ting K, Creevey CJ et al (2015) Horizontal gene flow from Eubacteria to Archaebacteria and what it means for our understanding of eukaryogenesis. Philos Trans R Soc B Biol Sci 370:20140337. https://doi.org/10.1098/rstb.2014.0337
Ambit A, Fasel N, Coombs GH, Mottram JC (2008) An essential role for the Leishmania major metacaspase in cell cycle progression. Cell Death Differ 15:113–122. https://doi.org/10.1038/sj.cdd.4402232
Ameisen J-C (2002) On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ 9:367–393. https://doi.org/10.1038/sj/cdd/4400950
Aravind L, Koonin EV (2002) Classification of the caspase-hemoglobinase fold: detection of new families and implications for the origin of the eukaryotic separins. Proteins Struct Funct Genet 46:355–367. https://doi.org/10.1002/prot.10060
Aravind L, Tatusov RL, Wolf YI et al (1998) Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet 14:442–444. https://doi.org/10.1016/S0168-9525(98)01553-4
Aravind L, Dixit VM, Koonin EV (1999) The domains of death. Elsevier Sci Ltd 0004:47–53. https://doi.org/10.1016/s0968-0004(98)01341-3
Aravind L, Dixit VM, Koonin EV (2001) Apoptotic molecular machinery: vastly increased complexity in vertebrates revealed by genome comparisons. Science. https://doi.org/10.1126/science.291.5507.1279
Arnoult D, Tatischeff I, Estaquier J et al (2001) On the evolutionary conservation of the cell death pathway: mitochondrial release of an apoptosis-inducing factor during Dictyostelium discoideum cell death. Mol Biol Cell 12:3016–3030. https://doi.org/10.1091/mbc.12.10.3016
Asplund-Samuelsson J, Bergman B, Larsson J (2012) Prokaryotic caspase homologs: phylogenetic patterns and functional characteristics reveal considerable diversity. PLoS ONE 7:1–14. https://doi.org/10.1371/journal.pone.0049888
Bayles KW (2014) Bacterial programmed cell death: Making sense of a paradox. Nat Rev Microbiol 12:63–69. https://doi.org/10.1038/nrmicro3136
Benler S, Koonin EV (2020) Phage lysis-lysogeny switches and programmed cell death: Danse macabre. BioEssays 42:2000114. https://doi.org/10.1002/bies.202000114
Berman-Frank I, Bidle KD, Haramaty L, Falkowski PG (2004) The demise of the marine cyanobacterium, Trichodesmium spp., via an autocatalyzed cell death pathway. Limnol Oceanogr 49:997–1005. https://doi.org/10.4319/lo.2004.49.4.0997
Bidle KD (2016) Programmed cell death in unicellular phytoplankton. Curr Biol 26:R594–R607. https://doi.org/10.1016/j.cub.2016.05.056
Blackstone NW (2013) Why did eukaryotes evolve only once? Genetic and energetic aspects of conflict and conflict mediation. Philos Trans R Soc Lond B Biol Sci 368:20120266. https://doi.org/10.1098/rstb.2012.0266
Blackstone NW, Green DR (1999) The evolution of a mechanism of cell suicide. BioEssays 21:84–88
Castelle CJ, Wrighton KC, Thomas BC et al (2015) Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol 25:690–701. https://doi.org/10.1016/j.cub.2015.01.014
Choi CJ, Berges JA (2013) New types of metacaspases in phytoplankton reveal diverse origins of cell death proteases. Cell Death Dis 4:e490. https://doi.org/10.1038/cddis.2013.21
Cohen GM (1998) Caspases: the executioners of apoptosis. Biochem J 95:1–16. https://doi.org/10.1016/s0378-4274(98)80028-3
Coll NS, Vercammen D, Smidler A et al (2010) Arabidopsis type I metacaspases control cell death. Science 330:1393–1397. https://doi.org/10.1126/science.1194980
Consortium TU (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49:D480–D489. https://doi.org/10.1093/nar/gkaa1100
Drozdetskiy A, Cole C, Procter J, Barton GJ (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Res 43:W389–W394. https://doi.org/10.1093/nar/gkv332
Durand PM (2021) The Evolutionary Origins of Life and Death. The University of Chicago Press, Chicago
Durand PM, Ramsey G (2019) The nature of programmed cell death. Biol Theory 14:30–41. https://doi.org/10.1007/s13752-018-0311-0
Durand PM, Rashidi A, Michod RE (2011) How an organism dies affects the fitness of its neighbors. Am Nat 177:224–232. https://doi.org/10.1086/657686
Durand PM, Sym S, Michod RE (2016) Programmed cell death and complexity in microbial systems. Curr Biol 26:R587–R593. https://doi.org/10.1016/j.cub.2016.05.057
Durand PM, Barreto Filho MM, Michod RE (2019) Cell death in evolutionary transitions in individuality. Yale J Biol Med 92:1–12
Eddy SR (1998) Profile hidden Markov models. Bioinformatics 14:755–763. https://doi.org/10.1093/bioinformatics/14.9.755
Eyers PA, Murphy JM (2016) The evolving world of pseudoenzymes: proteins, prejudice and zombies. BMC Biol 14:98. https://doi.org/10.1186/s12915-016-0322-x
Forslund K, Sonnhammer ELL (2008) Predicting protein function from domain content. Bioinformatics 24:1681–1687. https://doi.org/10.1093/bioinformatics/btn312
Frade JM, Michaelidis TM (1997) Origin of eukaryotic programmed cell death: a consequence of aerobic metabolism? BioEssays 19:827–832. https://doi.org/10.1002/bies.950190913
Gaasterland T, Ragan MA (1998) Microbial genescapes: phyletic and functional patterns of ORF distribution among prokaryotes. Microb Comp Genomics 3:199–217. https://doi.org/10.1089/omi.1.1998.3.199
Galperin MY, Koonin EV (2000) Who’s your neighbor? New computational approaches for functional genomics. Nat Biotechnol 18:609–613. https://doi.org/10.1038/76443
Ginalski K, Zhang H, Grishin NV (2004) Raptor protein contains a caspase-like domain. Trends Biochem Sci 29:522–524. https://doi.org/10.1016/j.tibs.2004.08.006
Glücksmann A (1951) Cell deaths in normal vertebrate ontogeny. Biol Rev 26:59–86. https://doi.org/10.1111/j.1469-185X.1951.tb00774.x
Guy L, Ettema TJG (2011) The archaeal “TACK” superphylum and the origin of eukaryotes. Trends Microbiol 19:580–587. https://doi.org/10.1016/j.tim.2011.09.002
Hill SM, Nyström T (2015) The dual role of a yeast metacaspase: what doesn’t kill you makes you stronger. BioEssays 37:525–531. https://doi.org/10.1002/bies.201400208
Hoang DT, Chernomor O, von Haeseler A et al (2018) UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol 35:518–522. https://doi.org/10.1093/molbev/msx281
Hofmann K (2019) The evolutionary origins of programmed cell death signaling. Cold Spring Harb Perspect Biol 12:a036442. https://doi.org/10.1101/cshperspect.a036442
Hug LA, Baker BJ, Anantharaman K et al (2016) A new view of the tree of life. Nat Microbiol 1:16048. https://doi.org/10.1038/nmicrobiol.2016.48
Huneman P (2022) Philosophical sketches of death in biology: an historical and analytic investigation. Palgrave Macmillan, London
Husnik F, McCutcheon JP (2018) Functional horizontal gene transfer from bacteria to eukaryotes. Nat Rev Microbiol 16:67–79. https://doi.org/10.1038/nrmicro.2017.137
Imachi H, Nobu MK, Nakahara N et al (2020) Isolation of an archaeon at the prokaryote–eukaryote interface. Nature 577:519–525. https://doi.org/10.1038/s41586-019-1916-6
Iranzo J, Lobkovsky A, Wolf YI, Koonin E (2014) Virus-host arms race at the joint origin of multicellularity and programmed cell death. Cell Cycle 13:3083–3088. https://doi.org/10.4161/15384101.2014.949496
Jing H, Takagi J, Liu J et al (2002) Archaeal surface layer proteins contain β Propeller, PKD, and β Helix domains and are related to metazoan cell surface proteins. Structure 10:1453–1464. https://doi.org/10.1016/S0969-2126(02)00840-7
Kaczanowski S (2016) Apoptosis: its origin, history, maintenance and the medical implications for cancer and aging. Phys Biol 13:31001. https://doi.org/10.1088/1478-3975/13/3/031001
Kalyaanamoorthy S, Minh BQ, Wong TKF et al (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589. https://doi.org/10.1038/nmeth.4285
Katoh K, Standley DM (2013) MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability Article Fast Track. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Klemenčič M, Funk C (2018) Structural and functional diversity of caspase homologues in non-metazoan organisms. Protoplasma 255:387–397. https://doi.org/10.1007/s00709-017-1145-5
Klemenčič M, Novinec M, Dolinar M (2015) Orthocaspases are proteolytically active prokaryotic caspase homologues : the case of Microcystis aeruginosa. Mol Microbiol 98:142–150. https://doi.org/10.1111/mmi.13110
Klemenčič M, Asplund-Samuelsson J, Dolinar M, Funk C (2019) Phylogenetic distribution and diversity of bacterial pseudo-orthocaspases underline their putative role in photosynthesis. Front Plant Sci 14:293. https://doi.org/10.3389/fpls.2019.00293
Klim J, Gładki A, Kucharczyk R et al (2018) Ancestral state reconstruction of the apoptosis machinery in the common ancestor of eukaryotes. G3 (Bethesda) 8:2121–2134. https://doi.org/10.1534/g3.118.200295
Koonin EV (2009) On the origin of cells and viruses: primordial virus world scenario. Ann N Y Acad Sci 1178:47–64. https://doi.org/10.1111/j.1749-6632.2009.04992.x
Koonin EV (2016) Horizontal gene transfer: essentiality and evolvability in prokaryotes, and roles in evolutionary transitions. F1000Research 5:1805. https://doi.org/10.12688/f1000research.8737.1
Koonin E, Aravind L (2002) Origin and evolution of eukaryotic apoptosis: the bacterial connection. Cell Death Differ 9:394–404. https://doi.org/10.1038/sj/cdd/4400991
Koonin EV, Zhang F (2017) Coupling immunity and programmed cell suicide in prokaryotes: Life-or-death choices. BioEssays 39:1–9. https://doi.org/10.1002/bies.201600186
Koonin EV, Mushegian AR, Galperin MY, Walker DR (1997) Comparison of archaeal and bacterial genomes: computer analysis of protein sequences predicts novel functions and suggests a chimeric origin for the archaea. Mol Microbiol 25:619–637. https://doi.org/10.1046/j.1365-2958.1997.4821861.x
Kroemer G (1997) Mitochondrial implication in apoptosis. Towards an endosymbiont hypothesis of apoptosis evolution. Cell Death Differ 4:443–456. https://doi.org/10.1038/sj.cdd.4400266
Krogh A, Larsson B, Von Heijne G, Sonnhammer ELL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. https://doi.org/10.1006/jmbi.2000.4315
Lee REC, Puente LG, Kaern M, Megeney LA (2008) A non-death role of the yeast metacaspase: Yca1p alters cell cycle dynamics. PLoS ONE 3:e2956. https://doi.org/10.1371/journal.pone.0002956
Lema AS, Klemenčič M, Völlmy F et al (2021) The role of Pseudo-Orthocaspase (SyOC) of Synechocystis sp. PCC 6803 in attenuating the effect of oxidative stress. Front Microbiol 12:82. https://doi.org/10.3389/fmicb.2021.634366
Letunic I, Bork P (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259. https://doi.org/10.1093/nar/gkz239
Li D, Roberts R (2001) WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci 58:2085–2097. https://doi.org/10.1007/pl00000838
Lockshin A, Williams M (1964) Programmed cell death—II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol 10:643–649. https://doi.org/10.1016/0022-1910(64)90034-4
Maddison WP (1997) Gene trees in species trees. Syst Biol 46:523–536. https://doi.org/10.1093/sysbio/46.3.523
McLuskey K, Mottram JC (2015) Comparative structural analysis of the caspase family with other clan CD cysteine peptidases. Biochem J 466:219–232. https://doi.org/10.1042/BJ20141324
McLuskey K, Rudolf J, Proto WR et al (2012) Crystal structure of a Trypanosoma brucei metacaspase. Proc Natl Acad Sci U S A 109:7469–7474. https://doi.org/10.1073/pnas.1200885109
Michod RE, Nedelcu AM (2003) Cooperation and conflict during the unicellular—multicellular and prokaryotic—eukaryotic transitions. In: Moya A, Font E (eds) Evolution: From molecules to ecosystems, 1st edn. Oxford University Press, Oxford, pp 195–208
Mignotte B, Vayssiere J-L (1998) Mitochondria and apoptosis. Eur J Biochem 252:1–15. https://doi.org/10.1046/j.1432-1327.1998.2520001.x
Minh BQ, Schmidt HA, Chernomor O et al (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534. https://doi.org/10.1093/molbev/msaa015
Minina EA, Coll NS, Tuominen H, Bozhkov PV (2017) Metacaspases versus caspases in development and cell fate regulation. Cell Death Differ 24:1314–1325. https://doi.org/10.1038/cdd.2017.18
Minina EA, Staal J, Alvarez VE et al (2020) Classification and nomenclature of metacaspases and paracaspases: no more confusion with caspases. Mol Cell 77:927–929. https://doi.org/10.1016/j.molcel.2019.12.020
Mistry J, Chuguransky S, Williams L et al (2021) Pfam: The protein families database in 2021. Nucleic Acids Res 49:D412–D419. https://doi.org/10.1093/nar/gkaa913
Moreira D, Lopez-Garcia P (1998) Symbiosis between methanogenic archaea and delta-proteobacteria as the origin of eukaryotes: the syntrophic hypothesis. J Mol Evol 47:517–530. https://doi.org/10.1007/pl00006408
Ndhlovu A, Durand P, Ramsey G (2021) Programmed cell death as a black queen in microbial communities. Mol Ecol 30:1110–1119. https://doi.org/10.1111/mec.15757
Nedelcu AM, Driscoll WW, Durand PM et al (2011) On the paradigm of altruistic suicide in the unicellular world. Evolution 65:3–20. https://doi.org/10.1111/j.1558-5646.2010.01103.x
Nelson KE, Clayton RA, Gill SR et al (1999) Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399:323–329. https://doi.org/10.1038/20601
Nelson-sathi S, Sousa FL, Röttger M et al (2015) Origins of major archaeal clades correspond to gene acquisitions from bacteria. Nature 517:77–80. https://doi.org/10.1038/nature13805.Origins
Nguyen L, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Ochman H, Lawrence JG, Groisman EA (2000) Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304. https://doi.org/10.1038/35012500
Ponting CP, Aravind L, Schultz J et al (1999) Eukaryotic signalling domain homologues in archaea and bacteria. Ancient ancestry and horizontal gene transfer. J Mol Biol 289:729–745. https://doi.org/10.1006/jmbi.1999.2827
Proto WR, Castanys-Munoz E, Black A et al (2011) Trypanosoma brucei Metacaspase 4 Is a Pseudopeptidase and a Virulence Factor. J Biol Chem 286:39914–39925. https://doi.org/10.1074/jbc.M111.292334
Proto WR, Coombs GH, Mottram JC (2013) Cell death in parasitic protozoa: regulated or incidental? Nat Rev Microbiol 11:58–66. https://doi.org/10.1038/nrmicro2929
Ramisetty BCM, Natarajan B, Santhosh RS (2015) MazEF-mediated programmed cell death in bacteria: “What is this?” Crit Rev Microbiol 41:89–100. https://doi.org/10.3109/1040841X.2013.804030
Ratel D, Boisseau S, Nasser V et al (2001) Programmed cell death or cell death programme? That is the question. J Theor Biol 208:385–386. https://doi.org/10.1006/jtbi.2000.2218
Rawlings N, Bateman A (2019) Origins of peptidases. Biochimie 166:4–18. https://doi.org/10.1016/j.biochi.2019.07.026
Rawlings ND, Waller M, Barrett AJ, Bateman A (2014) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 42:D503–D509. https://doi.org/10.1093/nar/gkt953
Richie DL, Miley MD, Bhabhra R et al (2007) The Aspergillus fumigatus metacaspases CasA and CasB facilitate growth under conditions of endoplasmic reticulum stress. Mol Microbiol 63:591–604. https://doi.org/10.1111/j.1365-2958.2006.05534.x
Rinke C, Schwientek P, Sczyrba A et al (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499:431–437. https://doi.org/10.1038/nature12352
Segovia M, Haramaty L, Berges JA, Falkowski PG (2003) Cell death in the unicellular chlorophyte Dunaliella tertiolecta. A hypothesis on the evolution of apoptosis in higher plants and metazoans. Plant Physiol 132:99–105. https://doi.org/10.1104/pp.102.017129
Shalini S, Dorstyn L, Dawar S, Kumar S (2015) Old, new and emerging functions of caspases. Cell Death Differ 22:526–539. https://doi.org/10.1038/cdd.2014.216
Shen W, Ren H (2021) TaxonKit: a practical and efficient NCBI taxonomy toolkit. J Genet Genomics. https://doi.org/10.1016/j.jgg.2021.03.006
Shrestha A, Megeney L (2012) The non-death role of metacaspase proteases. Front Oncol 2:78. https://doi.org/10.3389/fonc.2012.00078
Sonnhammer EL, von Heijne G, Krogh A (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proceedings Int Conf Intell Syst Mol Biol 6:175–182
Srivastav R, Suneja G (2019) Recent Advances in Microbial Genome Sequencing. In: Tripathi V, Kumar P, Tripathi P (eds) Microbial Genomics in Sustainable Agroecosystems, 1st edn. Springer Singapore, Singapore, pp 131–144
Szallies A, Kubata BK, Duszenko M (2002) A metacaspase of Trypanosoma brucei causes loss of respiration competence and clonal death in the yeast Saccharomyces cerevisiae. FEBS Lett 517:144–150. https://doi.org/10.1016/s0014-5793(02)02608-x
Tsiatsiani L, Van Breusegem F, Gallois P et al (2011) Metacaspases. Cell Death Differ 18:1279–1288. https://doi.org/10.1038/cdd.2011.66
Uren AG, O’Rourke K, Aravind L et al (2000) Identification of Paracaspases and Metacaspases. Mol Cell 6:961–967. https://doi.org/10.1016/s1097-2765(05)00086-9
van Creveld SG, Ben-Dor S, Mizrachi A, et al (2018) A redox-regulated type III metacaspase controls cell death in a marine diatom. bioRxiv 444109. https://doi.org/10.1101/444109
Vardi A, Eisenstadt D, Murik O et al (2007) Synchronization of cell death in a dinoflagellate population is mediated by an excreted thiol protease. Environ Microbiol 9:360–369. https://doi.org/10.1111/j.1462-2920.2006.01146.x
Vercammen D, Declercq W, Vandenabeele P, Van Breusegem F (2007) Are metacaspases caspases? J Cell Biol 179:375–380. https://doi.org/10.1083/jcb.200705193
von Heijne G (2006) Membrane-protein topology. Nat Rev Mol Cell Biol 7:909–918. https://doi.org/10.1038/nrm2063
Wagner A, Whitaker RJ, Krause DJ et al (2017) Mechanisms of gene flow in archaea. Nat Rev Microbiol 15:492–501. https://doi.org/10.1038/nrmicro.2017.41
Waterhouse AM, Procter JB, Martin DMA et al (2009) Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. https://doi.org/10.1093/bioinformatics/btp033
Wei Y, Fox T, Chambers SP et al (2000) The structures of caspases-1, -3, -7 and -8 reveal the basis for substrate and inhibitor selectivity. Chem Biol 7:423–432. https://doi.org/10.1016/S1074-5521(00)00123-X
Wheeler TJ, Clements J, Finn RD (2014) Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinformatics 15:7. https://doi.org/10.1186/1471-2105-15-7
Yang J, Zhang Y (2015) Protein structure and function prediction using I-TASSER. Curr Protoc Bioinforma. https://doi.org/10.1002/0471250953.bi0508s52
Yu JW, Jeffrey PD, Ha JY et al (2011) Crystal structure of the mucosa-associated lymphoid tissue lymphoma translocation 1 (MALT1) paracaspase region. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1111708108
Zhang Z, Wang J, Wang J et al (2020) Estimate of the sequenced proportion of the global prokaryotic genome. Microbiome 8:134. https://doi.org/10.1186/s40168-020-00903-z
Acknowledgements
This work was supported by Paleontological Scientific Trust (PAST) Africa (https://past.org.za/) and Genus: DSI-NRF Centre of Excellence in Palaeosciences (https://genus.africa/).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Handling editor: Kerry Geiler-Samerotte.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
La, S.R., Ndhlovu, A. & Durand, P.M. The Ancient Origins of Death Domains Support the ‘Original Sin’ Hypothesis for the Evolution of Programmed Cell Death. J Mol Evol 90, 95–113 (2022). https://doi.org/10.1007/s00239-021-10044-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-021-10044-y