
Abstract
Plant hormone cytokinins are important regulators of plant development, response to environmental stresses and interplay with other plant hormones. Cytokinin dehydrogenases (CKXs) are proteins responsible for the irreversible break-down of cytokinins to the adenine and aldehyde. Even though plant CKXs have been extensively studied, homologous proteins from other taxa remain mainly uncharacterised. Here we present our study on the molecular evolution and divergence of the CKX from bacteria, fungi, amoeba and viridiplantae. Although CKXs are present in eukaryotes and prokaryotes, they are missing in algae and metazoan taxa. The prevalent domain architecture consists of the FAD-binding and cytokinin binding domains, whereas some bacteria appear to have only cytokinin binding domain proteins. The CKXs play important role in the various aspects of plant life including control of plant development, response to biotic and abiotic stress, influence nutrition. Results of our study suggested that CKX originates from the FAD-linked C-terminal oxidase and has a defence-oriented function. The obtained results significantly extend the current understanding of the cytokinin dehydrogenases structure–function from the relationship to homologues from other taxa and provide a starting point baseline for their future functional characterization.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cytokinins (CKs) are the class of adenine-derivative signalling molecules widely distributed in nature. Different forms of CKs have been identified in all known taxa: bacteria (Kisiala et al. 2013; Creason et al., 2014; Seo et al. 2016; Samanovic et al. 2018; Kabbara et al. 2020); fungi (Morrison et al. 2015a, b; Hinsch et al. 2015; Chanclud et al. 2016; Trdá et al. 2017); nematodes (Siddique et al. 2015); insects (Zhang et al. 2017; Body et al. 2019; Andreas et al. 2020); mammals (Seegobin et al. 2018); humans (Colombo et al. 2009; Reiter et al. 2012); algae (Stirk et al. 2003; Romanenko et al. 2016). In small amounts, CKs appear as a usual sub-product of the normal de novo biosynthesis or salvage pathways in purine metabolism (Ashihara et al. 2020). Besides, a set of cis CKs are formed as a result of tRNA degradation (reviewed in (Dabravolski 2020). In comparison with animals and fungi, plants have evolved more complex purine interconversion pathways, resulting in the production of a wide range of different CKs (reviewed in (Ashihara et al. 2020).
CKs are one of the main plant hormones, playing an essential role in the majority of plants’ physiological and metabolic reactions. Additionally, CKs control plants’ development, response to biotic and abiotic stress, influence nutrition and agronomical traits (Kieber and Schaller 2018). Modern genomics and bioinformatic resources allow us to expand our understanding of the CKs and consider them also as a universal class of signalling molecules. The CKs metabolism in plants is complex and suggested to be classified into two types: the modification of the adenine moiety and that of the side chain (Sakakibara 2006). Amongst the wide variety of plant proteins associated with CKs metabolism, the functions of phosphate-isopentenyl transferases (IPTs) and phosphoribohydrolase ‘Lonely Guy’ (LOG) in biosynthesis and Arabidopsis Histidine Kinases (AHKs), Arabidopsis Histidine phosphotransmitter (AHPs) and Response Regulators (RRs) in signalling and cytokinin dehydrogenase (CKX) in degradation have been extensively studied (Kieber and Schaller 2014) (Fig. 1). Many attempts have been made to unravel the evolution of the signalling pathway (Gruhn et al. 2014) and the entire metabolic pathway (Frebort et al. 2011). The main outcome of these studies is a commonly accepted idea that CK-related proteins originated from cyanobacteria and remained after endosymbiosis to the proto-chloroplast (Martin et al. 2015). The current model suggests that the CK regulatory mechanism originated from cyanobacteria and passed to angiosperms through Chlorophytes, Lycophytes and Bryophytes (Gruhn and Heyl 2013). It is known that all plastids (including algae) have a cyanobacterial origin (reviewed in (Keeling 2013) and (Martin et al. 2015), therefore the CKX absence in algae is intriguing.

Number of main cytokinin-related genes, presented in different taxa
CKs were shown to play an important role in the inter-species communication molecules for many bacterial, fungal and insect pathogens (Giron et al. 2013). Recently, it was shown, that the plant-pathogenic gram-positive bacteria Rhodococcus fascians (also known as Corynebacterium fascians) can produce (Pertry et al. 2009) and release a mixture of different CKs to modulate and redirect plant development (Pertry et al. 2010; Radhika et al. 2015) in favour of pathogen spreading. The human-specific gram-negative pathogen Bordetella pertussis employs a similar strategy by producing a set of CKs (Moramarco et al. 2019) that could modulate the function of the host’s immune system (Lappas 2015). Altogether, this data suggests an ancient and universal nature of the CKs (Robischon 2015).
The omnipresent FAD-binding (PF01565) domain is an important part of many catalytic proteins with wide representation in all taxa and known to be involved in immune defence, DNA repair, cellular signalling and metabolism, apoptosis, neural development and drug metabolism (reviewed in (Leys and Scrutton 2016). Moreover, recent data on organisms from extreme environments and pathogens have proved new emerging roles of FAD-binding domain-containing proteins, suggesting their adjustable and adaptive nature (reviewed in (Piano et al. 2017).
CKX is the only enzyme, responsible for the irreversible breakdown of CKs. From the time of identification in 1971 by Paces (Pačes et al. 1971), CKXs were studied exclusively in plants and their evolution was traced back only to the first primitive land conquer—Physcomitrella patens (Hedw.) (von Schwartzenberg 2006). To define the evolutionary origin of CKX, phylogenetic and structural analysis has been conducted. According to our data, we could conclude that CKX is a rather recent event of development that emerged as a part of the LOG-mediated defence mechanism, required to metabolise excess non-canonical nucleotides delivered from the environment. During growth plants are constantly interacting with other prokaryotic and eukaryotic organisms, some of them are pathogens, using CKs and CK conjugates as important virulence factors (Chanclud et al. 2016; Akhtar et al. 2020). Whilst more primitive organisms rely on simpler broad rage-specificity LOG-system (Stepchenkova et al. 2005; Sévin et al. 2017), plants require a more specific and efficient CKX-based mechanism to manage interactions with other organisms. For plants it is crucial to distinguish between two extremes: some of them could be pathogenic (should be eliminated) or symbiotic (should be promoted). The role of LOGs has remained unclear. For example, the bacterial LOG homologue (Escherichia coli, P0ADR8) was shown to catalyse irreversible hydrolysis of the N-glycosidic bond for different purine and pyrimidine substrates (AMP, dTMP, GMP, CMP, IMP, and UMP) (Sévin et al. 2017). Moreover, similar broad substrate specificity for a LOG homologue was shown for the pathogen Bordetella pertussis (Moramarco et al. 2019). Similarly, in the yeast Saccharomyces cerevisiae, the LOG homologue (YJL055W) was responsible for the detoxification of the chemotherapeutic drugs such as purine analogue 6-N-hydroxylaminopurine (HAP) and pyrimidine analogue 5-fluorouracil (5-FU) (Stepchenkova et al. 2005; Ko et al. 2008). Based on those data and known antioxidant properties of CKs (Brizzolari et al. 2016) it was suggested, that the possible function of the LOG is to protect the DNA and RNA from the incorporation of the non-canonical nucleotides (Carlsson et al. 2018). However, the suggested LOG function was neither shown nor proven in plants.
Our data suggested that D-lactate dehydrogenase, an omnipresent FAD-dependent oxidase/dehydrogenase, is the most probable source of origin for CKX. Thus, we suggested a new model of the possible neofunctionalization of this protein in multiple lineages across the tree of life. Our findings clarify and provide one possible explanation of the presence/absence of specific features of the respective proteins in algae, bacteria, metazoa and plantae.
Material and Methods
Sequences Retrieval
The sequences of 7 Arabidopsis Cytokinin dehydrogenases (CKXs) [Uniprot IDs: O22213, Q9FUJ3, Q9LTS3, Q9FUJ2, Q67YU0, Q9LY71, Q9FUJ1], Zea mais [Q9T0N8]; D-2-hydroxyglutarate dehydrogenase [O23240] and the consensus sequence of the cytokinin binding and FAD-linked oxidase (C-terminal) domains were extracted from Uniprot database and used for the following BLAST (Shiryev et al. 2007) searches in NCBI (with pBLAST, PSI and DELTA-BLAST algorithms against non-redundant protein sequences and ResSeq Select proteins databases), InterPro (Mitchell et al. 2019) (with in-build InterPro scan tool, sequence and domain architecture search modes) and Pfam (El-Gebali et al. 2019) (HMMER algorithm, sequence and domain architecture search modes) databases with P value set at ≤ 0.001 (summarised results of manually processed sequences are present in Fig. 1). All partial and fragmented sequences were eliminated. The presence of the cytokinin binding (PF09265/IPR015345) and FAD-linked oxidase (PF02913/IPR004113) domains were checked with CD-search tool (Batch mode) (NCBI) (Marchler-Bauer et al. 2017) and MOTIF search (KEGG) (Kanehisa et al. 2016) tools with E value (P ≤ 0.001). Domains, associated with the cytokinin binding domain were verified with the same tools and threshold.
Phylogenetic Analysis
Multiple sequence alignments of protein sequences were performed using MUSCLE (Edgar 2004) with default settings in Ugene software (Okonechnikov et al. 2012). Substitution models test and phylogeny analysis were carried out using the MEGA X software (Kumar et al. 2018). For the maximum likelihood tree (Whelan and Goldman 2001) the LG substitution model (Le and Gascuel 2008) was selected assuming an estimated proportion of invariant sites and four gamma-distributed rate categories to account for rate heterogeneity across sites. The gamma shape parameter was estimated directly from the data. Reliability for the internal branch was assessed using the bootstrapping method (1000 bootstrap replicates). The same settings were used in another tree reconstruction method, Neighbour-Joining (Saitou and Nei 1987), with similar results obtained.
Protein 3D Model Prediction, Search and Comparison
Structural models of the cytokinin binding domain and FAD-linked oxidase were built with SWISS-MODEL (Waterhouse et al. 2018). SWISS-MODEL is a fully automated protein homology modelling server. Effective template search algorithm combines accurate BLAST (Camacho et al. 2009) and sensitive HHblits (Remmert et al. 2012). Further, the most suitable templates are estimated based on the Global Model Quality Estimate (GMQE) (Biasini et al. 2014), describing the most likely structural similarity, and Quaternary Structure Quality Estimate (QSQE) (Bertoni et al. 2017) methods. Predicted structures were refined with the online tool 3D refine (http://sysbio.rnet.missouri.edu/3Drefine/) (Bhattacharya et al. 2016) (2020a) and verified with QMEAN (https://swissmodel.expasy.org/qmean/) (2020b) a linear combination of six structural descriptors of a given protein (Benkert et al. 2011). iPBA webserver was used for the pdb structures alignment (https://www.dsimb.inserm.fr/dsimb_tools/ipba/index.php) (2020c). The quality of the structure’s alignments was evaluated with RMSD (quality of alignment, calculated from the superimposition of protein pairs based on PB alignment) and Normalized score (the dynamic programming alignment score, calculated from the relation of the alignment score to the alignment length) (Zemla 2003; Tyagi et al. 2006, 2008). Chimera software (Pettersen et al. 2004) was used for structure visualization. To verify efficacy of our modelling approach, RaptorX (Källberg et al. 2014), Geno3D2 (Combet et al. 2002) and Robetta (Kim et al. 2004) servers were additionally used to build CKX7 structural models. Further, all generated models were compared to the original CKX7 model (2EXR). Obtained results (Supplementary table 1) suggest that SWISS-MODEL is suitable for our goals. Generated models for each tool and aligned to 2exr (pdb files) could be found in the Supplementary File 1.
Results
Sequences Identification
To track the evolutionary origin of the CKXs, we have searched for the homologous sequences from the Uniprot, Pfam, and NCBI databases using the homology-based BLAST method. Well annotated protein sequences of these proteins were used as query sequences for sequence-based homology searches (specifically, from Arabidopsis thaliana (L.) Heynh., Zea mays L., and consensus sequences from the corresponding domains, deposited in the mentioned databases).
In total, 969 sequences were identified (Supplementary Table 2) (Bacteria: 217, Eukaryota: Heterolobosea 1; Opisthokonta 3; Viridiplantae: 748). During sequence search, the truncated, partial and identical sequences were removed (Supplementary Table 3).
Interestingly, the bacterial CKXs are represented by a single-copy gene for all taxa. In contrast to bacteria, plants usually have multiple CKX genes. Moreover, only 1 protein was identified in Heterolobosea (D2V8E5) in Naegleria gruberi (Amoeba) and only 3 CKXs were identified in fungi: Basidiobolus meristosporus (A0A1Y1XXL7), Antrodiella citrinella (A0A4S4MT84) and Coprinopsis marcescibilis (A0A5C3KPW4). No homologues were found in metazoa and algae taxa.
Conservation of the Domain Architecture in the Cytokinin Dehydrogenase
Domain architecture plays a key role in understanding the functionality of proteins along with their evolutionary history. To evaluate the gain and loss of different domains in CKX proteins, we have checked the domain combinations in each clade.
The dominant domain architecture associated with the cytokinin dehydrogenase consists of N-terminal FAD-binding (PF01565) (app 200aa) and C-terminal cytokinin binding (pfam09265) (app 250aa) domains. However, some bacteria represent only a single cytokinin binding domain (Supplementary Table 3).
Phylogenetic Analysis
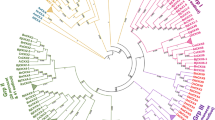
To understand the evolutionary history of the C-terminal cytokinin binding domain proteins the phylogenetic trees were inferred with the maximum likelihood and Neighbour-Joining methods. Some bacterial proteins have no FAD-binding domain, therefore only cytokinin binding domain sequences were extracted, aligned and used for the phylogenetic tree reconstruction. In general, both reconstruction methods (the maximum likelihood and Neighbour-Joining) have shown similar results (Fig. 2 and Supplementary Fig. 1). All CKX proteins delivered from the Viridiplantae have formed a separated cluster of closely related proteins. Further sub-divisions of this clade were well-correlated with the described localization of each studied protein as it was shown, for example, for Arabidopsis (Bae et al. 2007). As expected, CKXas of mosses, clubmosses and liverworts were located on separate branches (Fig. 2). Interestingly, amoeba (Naegleria gruberi) and fungi (Basidiobolus meristosporus, Antrodiella citrinella and Coprinopsis marcescibilis) proteins are closer to the bacterial protein’s clade, than to other eukaryotes.

Phylogeny estimation of the identified cytokinin binding domains. The Maximum Likelihood method and LG model were used; 1000 bootstrap replicates. Only branches with bootstrap value > 50 are shown. Fungal species highlighted in cyan, amoeba—magenta, Arabidopsis—blue, cyanobacteria—light green. Alignment length—323, Conserved site—1, Log-Likelihood -36,780.74, Discrete Rates—0.4791, 1.5209 (Color figure online)
Our results correlate with sequences similarity to the AtCKX7 cytokinin binding domain: all examined bacteria taxa (from 31 to 51%), from 39 to 43% for fungi and 52% for the amoeba. As expected, the similarity to the Viridiplantae sequences was above 60%. Thus, according to the results of the constructed phylogenetic trees, we could not prove the cyanobacterial origin of the cytokinin binding domain or any other photosynthetic bacteria taxa.
Structures Modelling and Comparison
Because our phylogenetic tree analysis could not answer the question regarding the origin of the cytokinin binding domain, we have analysed structural models of the CK-binding domains. In total, 36 models were built. The cytokinin binding domain of AtCKX7 was compared to the other cytokinin binding domains from other species (Table 1). It is not surprising that the viridiplantae and amoeba cytokinin binding domain exhibited the highest score. Bacteria from several taxa have shown similarly high matching scores (Chloroflexi, Actinobacteria, Deltaproteobacteria, Chloroflexi, Betaproteobacteria). Interestingly, fungi-derived cytokinin binding domains have exhibited rather low structural similarity.
Identification of the Origin of the Cytokinin Dehydrogenase
To identify an omnipresent FAD-dependent protein, that could be related to the cytokinin binding domain we performed a pBlast search. By application of pBlast search, we have found that on the sequence level the closest to CKX protein in Arabidopsis thaliana is the FAD-linked D-2-hydroxyglutarate dehydrogenase (D2HGDH, At4g36400) (22.88%) and its ortholog D-lactate dehydrogenase (d-LDH, At5g06580) with sequence similarity at 30.37%. The closest CKX homologue from Zea mays (1W1O) has 41.85% of sequence similarity.
To find out how D2HGDH is structurally similar to the bacterial homologues, we have built structural models from the bacterial species, with the best match for the cytokinin binding domain, and compared it to the D2HGDH (Table 2). Our analysis revealed that on the amino acid level the D2HGDH exhibited a rather low sequence similarity (about 40% for all). Our comparative analysis of the different cytokinin binding domains revealed a close structural relation of the D2HGDH to the bacterial homologous. Besides, we included 2 FAD-linked oxidase domains from the green algae proteins (A0A2K3DH85 (Chlamydomonas reinhardtii and D8U4H2 (Volvox carteri f. nagariensis). As expected, the FAD-linked oxidase domain from the Arabidopsis thaliana has shown the highest matching scores to the green algae proteins. Interestingly, Betaproteobacteria (A0A069PK84—Caballeronia glathei), PVC group (A0A1Z8SV81—Planctomycetia bacterium) and Deltaproteobacteria (A0A017TCE0—Chondromyces apiculatus) demonstrated a structural similarity level comparable to the algae (Table 2). Our results of the structure comparison are supported by the phylogenetic estimation (Fig. 3 and Supplementary Fig. 2). Similar to the structural models, D2HGDH has shown the closest relation to the green algae FAD-linked oxidases, whilst cyanobacterial protein (A0A1Z4LVD4, Calothrix parasitica) was rather distant.

Phylogeny estimation of the selected FAD-linked oxidases domains used for the structural comparison. The Maximum Likelihood method and LG model were used; 1000 bootstrap replicates. Only branches with bootstrap value > 50 are shown. Arabidopsis proteins are highlighted in blue, green algae—dark green, cyanobacteria—light green. Alignment length—265, Conserved site—6, Log-Likelihood -7931.97, Discrete Rates—0.4636, 1.5364 (Color figure online)
Taken all together, we could assume that FAD-linked oxidase is an ancient protein, rather similar in all taxa. Perhaps, this oxidase was inherited from the eukaryotes progenitor. Thus, based on our data analysis, we hypothesise, that the CKX was developed from the FAD-linked oxidase in bacteria and viridiplantae independently, as a result of an adaptation to the environmental condition. However, the suggested hypothesis needs further experimental verification.
Discussion
We have summarised the distribution of the main CK regulating genes in different taxa (Fig. 1). The dominant bacterial signalling mechanism comprises a two-component regulatory system, that could include a wide range of sensor domain/s, kinase (Ser/Thr/Tyr kinase or histidine kinase) and RRs (Stock et al. 2000). In contrast to bacteria, in the eukaryotes, this signalling system is rare and very likely to be inherited, from the endosymbiotic organelles (mitochondria and chloroplasts) (Capra and Laub 2012). The CHASE domain is the only known sensor for the CKs (Mougel and Zhulin 2001) that was suggested to emerge from the chemicals sensing pathway (Bilwes et al. 1999; Wang et al. 2017). Whilst the RR domain is ubiquitous for all taxa, the HPT domain is missing in red algae and the CHASE domain—in red algae and metazoan (Fig. 1).
CKXs are involved in irreversible degradation of the wide range of CKs, releasing adenine (or its derivative) and aldehyde. CKXs belong to FAD-dependent enzymes, containing FAD-binding N-terminal domain and C-terminal cytokinin binding domain. Due to the inhibitor properties of adenine for several metabolic pathways, the natural CKX concentration is usually low (Ashihara et al. 2018). Thus, it makes an additional complication for the study of CKX in vivo. CKXs have been studied only in viridiplantae (reviewed in (Kieber and Schaller 2014), whilst they have been also identified in bacteria, Naegleria gruberi (Percolozoa, Amoeba) and 3 fungi species (Basidiobolus meristosporus, Antrodiella citrinella and Coprinopsis marcescibilis) (Supplementary Table 2). In comparison to LOG and IPT genes (their representation in different taxa is similar), CKX is rather under-represented in all living organisms (Fig. 1). Notably, that bacteria and fungi with CKXs exhibit mainly pathogenic (Pertry et al. 2010) or closely associate with plants or animals (Mondo et al. 2017) lifestyle.
Cytokinin binding (pfam09265) domain was identified as a key part in the irreversible degradation of the wide range of the CKS in plants (Bilyeu et al. 2001). The amino acid “signature” of the adenine binding site exhibits a strong influence on substrate recognition and turnover rates (Malito et al. 2004). The proved crystal structures of the CKXs from Zea mais (Malito et al. 2004) and Arabidopsis thaliana (Bae et al. 2007) suggested a conserved catalytic mechanism for those two species. However, no functional data are available from non-plant species.
The phylogenetic data of the CKX proteins could not provide a definitive conclusion about the evolutionary origin of the CKXs. Also, our data could not support the relation to the cyanobacteria, as it was shown for other CK-related genes (Pils and Heyl 2009) or the amoeba Naegleria gruberi, where CKX genes have been delivered via the Chlamydia, that was recently reported by another group (Wang et al. 2020). The proposed role of Chlamydia is based on the close relationship between the human pneumonia-causing bacteria Protochlamydia naegleriophila with amoeba Naegleria gruberi (Casson et al. 2008) and several shared genes between Chlamydia and plant progenitor. There are several studies, proving participation of Chlamydia in the evolution of the photoautotrophic eukaryotes: 39 proteins have been identified by Becker et al. 2008 (Becker et al. 2008); 53 by Collingro et al. 2011 (Collingro et al. 2011) and 21 by Huang and Gogarten 2007 (Huang and Gogarten 2007). None of the identified genes is associated with CK signalling or CK metabolism. In comparison to the massive transfer of genes from cyanobacterial endosymbiont (about 4500 (Martin et al. 2002), many of which are associated with CK, the role of Chlamydia in CKX transfer remains very doubtful. On the contrary, it appears that CKXs in prokaryotes and eukaryotes may have developed independently. Due to the low number of available sequence samples (only one for amoeba and three for fungi), it is not possible to make any solid conclusion or assumption regarding their evolution pathway based only on the close relation to the prokaryotes on the phylogenetic tree. On the other hand, phylogeny and structural comparison of the FAD-linked oxidases from different taxa provides a strong link to a close relation between bacterial, algal and viridiplantae proteins. In this study, we have identified that bacterial, fungal, amoebal and viridiplantae CKXs are orthologs, delivered from FAD-linked oxidase. Our structural analysis reveals that FAD-linked oxidase was, most probably, inherited in the eukaryotes progenitor (Fig. 4, pathway 1). Furthermore, our data are in strong agreement with the previous report (Cristescu and Egbosimba 2009) investigating the evolutional history of the D-Lactate Dehydrogenases, where authors have defined CKX as a new clade on the common tree of the FAD-binding enzymes.

Model for the evolutionary origin of the CKX from FAD-linked oxidase
In total, based on the phylogenetic analysis and comparison of the structures we could conclude that the plant cytokinin binding domain is not originated from the cyanobacteria as was previously suggested (Gruhn and Heyl 2013) for the CK signalling pathway.
In Arabidopsis thaliana D-2-hydroxyglutarate dehydrogenase (D2HGDH, At4g36400) is a protein with mitochondrial localization, and D-lactate dehydrogenase (d-LDH, At5g06580) was identified in both, chloroplasts and mitochondria. As it was shown, both enzymes have wide substrate specificity (Engqvist et al. 2009) and participate in the dark-induced senescence and starvation by catabolism of the amino acids and chlorophyll (Engqvist et al. 2011). Until now no data available indicating the role of some CKs as substrates for the D2HGDH or d-LDH.
Another possible scenario of the FAD-linked oxidase evolution could be from the pro-mitochondrial endosymbiont, as it was suggested to be Proteobacteria (Yang et al. 1985; Martin et al. 2015). In addition to the mitochondrial subcellular localization of the D2HGDH and d-LDH, D2HGDH has shown close structural similarity to several Proteobacterial FAD-linked oxidases (Table 2), pointing to horizontal gene transfer from the endosymbiont (Fig. 4, pathway 2).Cyanobacteria have both genes (CKXs and FAD-linked oxidase) identified (Table 2 and Supplementary Table 4), suggesting the possibility that both genes could be transferred with pro-chloroplastic endosymbiont (Fig. 4, pathway 3) but CKX was lost during green algae evolution. On the next step, mitochondria- and plastid-delivered FAD-linked oxidases could be inherited by the land plants independently (Fig. 4, pathway 4)—which would explain the weak similarity between sequences. Based on the structural similarity of the FAD-linked oxidase and its omnipresent nature, evolutionary pathway 1 is more likely. Similarly, CKX is more likely to emerge independently in every taxon (Fig. 4, pathway 5), which is supported by an investigation of the evolution of the FAD-binding enzyme (Cristescu and Egbosimba 2009).
Thus, according to our results, we hypothesise that CKX is an evolutionarily recent protein that emerged from the FAD-linked oxidase. Many CKX-encoding genes were identified in bacteria, however, none of them was characterized and their substrate specificity remains unknown. It would be interesting to examine the difference in substrate preferences between the single cytokinin binding domain and the fused to the N-terminal FAD-binding domain. The natural environment of the algae does not support close interaction with any bacteria or provide a condition for the exchange of the CKs. Several plant-parasitic algae with a wide range of hosts have been described (Brooks 2004) whilst their genomes were not examined. Thus, the possibility of the CKX existence in algae remains, however further research is required to answer this question.
The CK degradation system of the evolutionarily primitive land plant Physcomitrella patens (Hedw.) is relatively simple, with high preferences in substrates to natural cis-zeatin (von Schwartzenberg et al. 2007). On contrary, the CKXs of vascular land plants (especially angiosperms) similarly to the CK signalling pathway (Kaltenegger et al. 2018), have undergone several duplication events, resulting in a high level of complexity and specificity (Niemann et al. 2018; Czajkowska et al. 2019). Such complexity could explain a high number of different CK forms, presented and metabolised in plants (Galuszka et al. 2007). We could notice a positive correlation between the complexity of the CKX and the number of potential pathogens (Czajkowska et al. 2019). From one side plants have to metabolise divers forms of non-canonical nucleotides, delivered from soil microflora and pathogens. On the other side, some forms of CKs could be toxic for bacterial or fungal invaders, so representing a defence mechanism. Until now no research has been conducted to evaluate the antibacterial/antifungal potential of the different CK conjugates. Therefore, it would be a good option for future studies.
Conclusion
Our results suggest that CKX is a recently evolved gene that resulted from the specialization of the duplicated FAD-linked oxidase. We assume CKX has emerged as a result of close interaction between different organisms (bacteria, fungi, plants and animals) probably as a pathogen defence mechanism. For the higher plants, this interaction began with land colonisation, for bacteria and fungi this interaction began as a pathogenic or commensal lifestyle. Our results suggest that FAD-linked oxidase is the most probable source of origin for the CKXs. Our findings also explain the possible reason for the absence of CKXs in algae.
Data Availability
Used data available upon request.
References
Akhtar SS, Mekureyaw MF, Pandey C, Roitsch T (2020) Role of cytokinins for interactions of plants with microbial pathogens and pest insects. Front Plant Sci 10:1777. https://doi.org/10.3389/fpls.2019.01777
Andreas P, Kisiala A, Emery RJN et al (2020) Cytokinins are abundant and widespread among insect species. Plants 9:208. https://doi.org/10.3390/plants9020208
Ashihara H, Stasolla C, Fujimura T, Crozier A (2018) Purine salvage in plants. Phytochemistry 147:89–124. https://doi.org/10.1016/j.phytochem.2017.12.008
Ashihara H, Ludwig IA, Crozier A (2020) Plant nucleotide metabolism - biosynthesis, degradation, and alkaloid formation, 1st edn. Wiley
Bae E, Bingman CA, Bitto E et al (2007) Crystal structure of Arabidopsis thaliana cytokinin dehydrogenase. Proteins Struct Funct Bioinforma 70:303–306. https://doi.org/10.1002/prot.21678
Becker B, Hoef-Emden K, Melkonian M (2008) Chlamydial genes shed light on the evolution of photoautotrophic eukaryotes. BMC Evol Biol 8:203. https://doi.org/10.1186/1471-2148-8-203
Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27:343–350. https://doi.org/10.1093/bioinformatics/btq662
Bertoni M, Kiefer F, Biasini M et al (2017) Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci Rep 7:10480. https://doi.org/10.1038/s41598-017-09654-8
Bhattacharya D, Nowotny J, Cao R, Cheng J (2016) 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Res 44:W406–W409. https://doi.org/10.1093/nar/gkw336
Biasini M, Bienert S, Waterhouse A et al (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. https://doi.org/10.1093/nar/gku340
Bilwes AM, Alex LA, Crane BR, Simon MI (1999) Structure of CheA, a Signal-Transducing Histidine Kinase. Cell 96:131–141. https://doi.org/10.1016/S0092-8674(00)80966-6
Bilyeu KD, Cole JL, Laskey JG et al (2001) Molecular and biochemical characterization of a cytokinin oxidase from Maize. Plant Physiol 125:378–386. https://doi.org/10.1104/pp.125.1.378
Body MJA, Appel HM, Edger PP, Schultz JC (2019) A gall-forming insect manipulates hostplant phytohormone synthesis, concentrations, and signaling. Plant Biol
Brizzolari A, Marinello C, Carini M et al (2016) Evaluation of the antioxidant activity and capacity of some natural N 6 -substituted adenine derivatives (cytokinins) by fluorimetric and spectrophotometric assays. J Chromatogr B 1019:164–168. https://doi.org/10.1016/j.jchromb.2015.12.047
Brooks FE (2004) Plant-parasitic algae (Chlorophyta: Trentepohliales) in American Samoa. Pac Sci 58:419–428. https://doi.org/10.1353/psc.2004.0026
Camacho C, Coulouris G, Avagyan V et al (2009) BLAST+: architecture and applications. BMC Bioinformatics 10:421. https://doi.org/10.1186/1471-2105-10-421
Capra EJ, Laub MT (2012) Evolution of two-component signal transduction systems. Annu Rev Microbiol 66:325–347. https://doi.org/10.1146/annurev-micro-092611-150039
Carlsson M, Hu G-Z, Ronne H (2018) Gene dosage effects in yeast support broader roles for the LOG1, HAM1 and DUT1 genes in detoxification of nucleotide analogues. PLoS ONE 13:e0196840. https://doi.org/10.1371/journal.pone.0196840
Casson N, Michel R, Müller K-D et al (2008) Protochlamydia naegleriophila as etiologic agent of pneumonia. Emerg Infect Dis 14:168–172. https://doi.org/10.3201/eid1401.070980
Chanclud E, Kisiala A, Emery NRJ et al (2016) Cytokinin production by the rice blast fungus is a pivotal requirement for full virulence. PLOS Pathog 12:e1005457. https://doi.org/10.1371/journal.ppat.1005457
Collingro A, Tischler P, Weinmaier T et al (2011) Unity in variety-the pan-genome of the chlamydiae. Mol Biol Evol 28:3253–3270. https://doi.org/10.1093/molbev/msr161
Colombo F, Falvella FS, De Cecco L et al (2009) Pharmacogenomics and analogues of the antitumour agent N 6 -isopentenyladenosine. Int J Cancer 124:2179–2185. https://doi.org/10.1002/ijc.24168
Combet C, Jambon M, Deleage G, Geourjon C (2002) Geno3D: automatic comparative molecular modelling of protein. Bioinformatics 18:213–214. https://doi.org/10.1093/bioinformatics/18.1.213
Creason AL, Vandeputte OM, Savory EA et al (2014) Analysis of genome sequences from plant pathogenic rhodococcus reveals genetic novelties in virulence loci. PLoS ONE 9:e101996. https://doi.org/10.1371/journal.pone.0101996
Cristescu ME, Egbosimba EE (2009) Evolutionary history of d-lactate dehydrogenases: a phylogenomic perspective on functional diversity in the fad binding oxidoreductase/transferase type 4 family. J Mol Evol 69:276–287. https://doi.org/10.1007/s00239-009-9274-x
Czajkowska BI, Finlay CM, Jones G, Brown TA (2019) Diversity of a cytokinin dehydrogenase gene in wild and cultivated barley. PLoS ONE 14:e0225899. https://doi.org/10.1371/journal.pone.0225899
Dabravolski S (2020) Multi-faceted nature of the tRNA isopentenyltransferase. Funct Plant Biol 47:475. https://doi.org/10.1071/FP19255
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. https://doi.org/10.1186/1471-2105-5-113
El-Gebali S, Mistry J, Bateman A et al (2019) The Pfam protein families database in 2019. Nucleic Acids Res 47:D427–D432. https://doi.org/10.1093/nar/gky995
Engqvist M, Drincovich MF, Flügge U-I, Maurino VG (2009) Two d-2-hydroxy-acid dehydrogenases in Arabidopsis thaliana with catalytic capacities to participate in the last reactions of the methylglyoxal and β-oxidation pathways. J Biol Chem 284:25026–25037. https://doi.org/10.1074/jbc.M109.021253
Engqvist MKM, Kuhn A, Wienstroer J et al (2011) Plant d-2-hydroxyglutarate dehydrogenase participates in the catabolism of lysine especially during senescence. J Biol Chem 286:11382–11390. https://doi.org/10.1074/jbc.M110.194175
Frebort I, Kowalska M, Hluska T et al (2011) Evolution of cytokinin biosynthesis and degradation. J Exp Bot 62:2431–2452. https://doi.org/10.1093/jxb/err004
Galuszka P, Popelková H, Werner T et al (2007) Biochemical characterization of cytokinin oxidases/dehydrogenases from arabidopsis thaliana expressed in Nicotiana tabacum L. J Plant Growth Regul 26:255–267. https://doi.org/10.1007/s00344-007-9008-5
Giron D, Frago E, Glevarec G et al (2013) Cytokinins as key regulators in plant–microbe–insect interactions: connecting plant growth and defence. Funct Ecol 27:599–609. https://doi.org/10.1111/1365-2435.12042
Gruhn N, Heyl A (2013) Updates on the model and the evolution of cytokinin signaling. Curr Opin Plant Biol 16:569–574. https://doi.org/10.1016/j.pbi.2013.09.001
Gruhn N, Halawa M, Snel B et al (2014) A Subfamily of putative cytokinin receptors is revealed by an analysis of the evolution of the two-component signaling system of plants. PLANT Physiol 165:227–237. https://doi.org/10.1104/pp.113.228080
Hinsch J, Vrabka J, Oeser B et al (2015) De novo biosynthesis of cytokinins in the biotrophic fungus C laviceps purpurea: De novo cytokinin synthesis by a fungal pathogen. Environ Microbiol 17:2935–2951. https://doi.org/10.1111/1462-2920.12838
Huang J, Gogarten J (2007) Did an ancient chlamydial endosymbiosis facilitate the establishment of primary plastids? Genome Biol 8:R99. https://doi.org/10.1186/gb-2007-8-6-r99
iPBA webserver (2020c) https://www.dsimb.inserm.fr/dsimb_tools/ipba/index.php. Accessed 1 Dec 2020
Kabbara S, Bidon B, Kilani J et al (2020) Cytokinin sensing in bacteria. Biomolecules 10:186. https://doi.org/10.3390/biom10020186
Källberg M, Margaryan G, Wang S et al (2014) RaptorX server: a resource for template-based protein structure modeling. In: Kihara D (ed) Protein structure prediction. Springer, New York, New York, NY, pp 17–27
Kaltenegger E, Leng S, Heyl A (2018) The effects of repeated whole genome duplication events on the evolution of cytokinin signaling pathway. BMC Evol Biol 18:76. https://doi.org/10.1186/s12862-018-1153-x
Kanehisa M, Sato Y, Kawashima M et al (2016) KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44:D457–D462. https://doi.org/10.1093/nar/gkv1070
Keeling PJ (2013) The number, speed, and impact of plastid endosymbioses in eukaryotic evolution. Annu Rev Plant Biol 64:583–607. https://doi.org/10.1146/annurev-arplant-050312-120144
Kieber JJ, Schaller GE (2014) Cytokinins. Arab Book 12:e0168. https://doi.org/10.1199/tab.0168
Kieber JJ, Schaller GE (2018) Cytokinin signaling in plant development. Development 145:149344. https://doi.org/10.1242/dev.149344
Kim DE, Chivian D, Baker D (2004) Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32:W526–W531. https://doi.org/10.1093/nar/gkh468
Kisiala A, Laffont C, Emery RJN, Frugier F (2013) Bioactive cytokinins are selectively secreted by Sinorhizobium meliloti nodulating and nonnodulating strains. Mol Plant Microbe Interact 26:1225–1231. https://doi.org/10.1094/MPMI-02-13-0054-R
Ko N, Nishihama R, Pringle JR (2008) Control of 5-FOA and 5-FU resistance by Saccharomyces cerevisiae YJL055W. Yeast 25:155–160. https://doi.org/10.1002/yea.1554
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Lappas CM (2015) The plant hormone zeatin riboside inhibits T lymphocyte activity via adenosine A2A receptor activation. Cell Mol Immunol 12:107–112. https://doi.org/10.1038/cmi.2014.33
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320. https://doi.org/10.1093/molbev/msn067
Leys D, Scrutton NS (2016) Sweating the assets of flavin cofactors: new insight of chemical versatility from knowledge of structure and mechanism. Curr Opin Struct Biol 41:19–26. https://doi.org/10.1016/j.sbi.2016.05.014
Malito E, Coda A, Bilyeu KD et al (2004) Structures of michaelis and product complexes of plant cytokinin dehydrogenase: implications for flavoenzyme catalysis. J Mol Biol 341:1237–1249. https://doi.org/10.1016/j.jmb.2004.06.083
Marchler-Bauer A, Bo Y, Han L et al (2017) CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45:D200–D203. https://doi.org/10.1093/nar/gkw1129
Martin W, Rujan T, Richly E et al (2002) Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc Natl Acad Sci USA 99:12246–12251. https://doi.org/10.1073/pnas.182432999
Martin WF, Garg S, Zimorski V (2015) Endosymbiotic theories for eukaryote origin. Philos Trans R Soc B Biol Sci 370:20140330. https://doi.org/10.1098/rstb.2014.0330
Mitchell AL, Attwood TK, Babbitt PC et al (2019) InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 47:D351–D360. https://doi.org/10.1093/nar/gky1100
Mondo SJ, Dannebaum RO, Kuo RC et al (2017) Widespread adenine N6-methylation of active genes in fungi. Nat Genet 49:964–968. https://doi.org/10.1038/ng.3859
Moramarco F, Pezzicoli A, Salvini L et al (2019) A LONELY GUY protein of Bordetella pertussis with unique features is related to oxidative stress. Sci Rep 9:17016. https://doi.org/10.1038/s41598-019-53171-9
Morrison EN, Emery RJN, Saville BJ (2015a) Phytohormone involvement in the ustilago maydis: zea mays pathosystem: relationships between abscisic acid and cytokinin levels and strain virulence in infected cob tissue. PLoS ONE 10:e0130945. https://doi.org/10.1371/journal.pone.0130945
Morrison EN, Knowles S, Hayward A et al (2015b) Detection of phytohormones in temperate forest fungi predicts consistent abscisic acid production and a common pathway for cytokinin biosynthesis. Mycologia 107:245–257. https://doi.org/10.3852/14-157
Mougel C, Zhulin IB (2001) CHASE: an extracellular sensing domain common to transmembrane receptors from prokaryotes, lower eukaryotes and plants. Trends Biochem Sci 26:582–584. https://doi.org/10.1016/S0968-0004(01)01969-7
Niemann MCE, Weber H, Hluska T et al (2018) The cytokinin oxidase/dehydrogenase CKX1 is a membrane-bound protein requiring homooligomerization in the endoplasmic reticulum for its cellular activity. Plant Physiol 176:2024–2039. https://doi.org/10.1104/pp.17.00925
Okonechnikov K, Golosova O, Fursov M (2012) Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28:1166–1167. https://doi.org/10.1093/bioinformatics/bts091
Pačes V, Werstiuk E, Hall RH (1971) Conversion of N 6 -(Δ 2 -Isopentenyl)adenosine to adenosine by enzyme activity in tobacco tissue. Plant Physiol 48:775–778. https://doi.org/10.1104/pp.48.6.775
Pertry I, Václavíková K, Depuydt S et al (2009) Identification of Rhodococcus fascians cytokinins and their modus operandi to reshape the plant. Proc Natl Acad Sci 106:929–934. https://doi.org/10.1073/pnas.0811683106
Pertry I, Václavíková K, Gemrotová M et al (2010) Rhodococcus fascians impacts plant development through the dynamic fas-mediated production of a cytokinin mix. Mol Plant Microbe Interact 23:1164–1174. https://doi.org/10.1094/MPMI-23-9-1164
Pettersen EF, Goddard TD, Huang CC et al (2004) UCSF chimera?A visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
Piano V, Palfey BA, Mattevi A (2017) Flavins as covalent catalysts: new mechanisms emerge. Trends Biochem Sci 42:457–469. https://doi.org/10.1016/j.tibs.2017.02.005
Pils B, Heyl A (2009) Unraveling the evolution of cytokinin signaling. Plant Physiol 151:782–791. https://doi.org/10.1104/pp.109.139188
QMEAN (2020b) https://swissmodel.expasy.org/qmean/. Accessed 1 Dec 2020
Radhika V, Ueda N, Tsuboi Y et al (2015) Methylated cytokinins from the phytopathogen Rhodococcus fascians mimic plant hormone activity. Plant Physiol 169:1118–1126. https://doi.org/10.1104/pp.15.00787
Reiter V, Matschkal DMS, Wagner M et al (2012) The CDK5 repressor CDK5RAP1 is a methylthiotransferase acting on nuclear and mitochondrial RNA. Nucleic Acids Res 40:6235–6240. https://doi.org/10.1093/nar/gks240
Remmert M, Biegert A, Hauser A, Söding J (2012) HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 9:173–175. https://doi.org/10.1038/nmeth.1818
Robischon M (2015) Do cytokinins function as two-way signals between plants and animals?: cytokinins may not only mediate defence reactions via secondary compounds, but may directly interfere with developmental signals in insects. BioEssays 37:356–363. https://doi.org/10.1002/bies.201400099
Romanenko EA, Kosakovskaya IV, N.G. Kholodny Institute of Botany NAS of Ukraine, 2, Tereshchenkovskaya St., Kiev 01004, Ukraine, et al (2016) Phytohormones of microalgae: biological role and involvement in the regulation of physiological processes. Pt II. Cytokinins and gibberellins. Algologia 26:203–229. https://doi.org/10.15407/alg26.02.203
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sakakibara H (2006) Cytokinins: activity, biosynthesis, and translocation. Annu Rev Plant Biol 57:431–449. https://doi.org/10.1146/annurev.arplant.57.032905.105231
Samanovic MI, Hsu H-C, Jones MB et al (2018) Cytokinin signaling in Mycobacterium tuberculosis. Mbio 9:e00989-e1018. https://doi.org/10.1128/mBio.00989-18
Schwartzenberg K (2006) Moss biology and phytohormones—cytokinins in physcomitrella. Plant Biol 8:382–388. https://doi.org/10.1055/s-2006-923962
Seegobin M, Kisiala A, Noble A et al (2018) Canis familiaris tissues are characterized by different profiles of cytokinins typical of the tRNA degradation pathway. FASEB J 32:6575–6581. https://doi.org/10.1096/fj.201800347
Seo H, Kim S, Sagong H-Y et al (2016) Structural basis for cytokinin production by LOG from corynebacterium glutamicum. Sci Rep 6:31390. https://doi.org/10.1038/srep31390
Sévin DC, Fuhrer T, Zamboni N, Sauer U (2017) Nontargeted in vitro metabolomics for high-throughput identification of novel enzymes in Escherichia coli. Nat Methods 14:187–194. https://doi.org/10.1038/nmeth.4103
Shiryev SA, Papadopoulos JS, Schaffer AA, Agarwala R (2007) Improved BLAST searches using longer words for protein seeding. Bioinformatics 23:2949–2951. https://doi.org/10.1093/bioinformatics/btm479
Siddique S, Radakovic ZS, De La Torre CM et al (2015) A parasitic nematode releases cytokinin that controls cell division and orchestrates feeding site formation in host plants. Proc Natl Acad Sci 112:12669–12674. https://doi.org/10.1073/pnas.1503657112
Stepchenkova EI, Kozmin SG, Alenin VV, Pavlov YI (2005) Genome-wide screening for genes whose deletions confer sensitivity to mutagenic purine base analogs in yeast. BMC Genet 6:31. https://doi.org/10.1186/1471-2156-6-31
Stirk WA, Novák O, Strnad M, van Staden J (2003) Cytokinins in macroalgae. Plant Growth Regul 41:13–24. https://doi.org/10.1023/A:1027376507197
Stock AM, Robinson VL, Goudreau PN (2000) Two-Component signal transduction. Annu Rev Biochem 69:183–215. https://doi.org/10.1146/annurev.biochem.69.1.183
Trdá L, Barešová M, Šašek V et al (2017) Cytokinin metabolism of pathogenic fungus leptosphaeria maculans involves isopentenyltransferase, adenosine kinase and cytokinin oxidase/dehydrogenase. Front Microbiol 8:1374. https://doi.org/10.3389/fmicb.2017.01374
Tyagi M, Gowri VS, Srinivasan N et al (2006) A substitution matrix for structural alphabet based on structural alignment of homologous proteins and its applications. Proteins Struct Funct Bioinforma 65:32–39. https://doi.org/10.1002/prot.21087
Tyagi M, de Brevern AG, Srinivasan N, Offmann B (2008) Protein structure mining using a structural alphabet. Proteins Struct Funct Bioinforma 71:920–937. https://doi.org/10.1002/prot.21776
von Schwartzenberg K, Nunez MF, Blaschke H et al (2007) Cytokinins in the bryophyte physcomitrella patens: analyses of activity, distribution, and cytokinin oxidase/dehydrogenase overexpression reveal the role of extracellular cytokinins. Plant Physiol 145:786–800. https://doi.org/10.1104/pp.107.103176
Wang F-F, Cheng S-T, Wu Y et al (2017) A bacterial receptor pcrk senses the plant hormone cytokinin to promote adaptation to oxidative stress. Cell Rep 21:2940–2951. https://doi.org/10.1016/j.celrep.2017.11.017
Wang X, Ding J, Lin S et al (2020) Evolution and roles of cytokinin genes in angiosperms 2: Do ancient CKXs play housekeeping roles while non-ancient CKXs play regulatory roles? Hortic Res 7:29. https://doi.org/10.1038/s41438-020-0246-z
Waterhouse A, Bertoni M, Bienert S et al (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. https://doi.org/10.1093/nar/gky427
Whelan S, Goldman N (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18:691–699. https://doi.org/10.1093/oxfordjournals.molbev.a003851
Yang D, Oyaizu Y, Oyaizu H et al (1985) Mitochondrial origins. Proc Natl Acad Sci 82:4443–4447. https://doi.org/10.1073/pnas.82.13.4443
Zemla A (2003) LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res 31:3370–3374. https://doi.org/10.1093/nar/gkg571
Zhang H, Guiguet A, Dubreuil G et al (2017) Dynamics and origin of cytokinins involved in plant manipulation by a leaf-mining insect: Insects as a source of cytokinins. Insect Sci 24:1065–1078. https://doi.org/10.1111/1744-7917.12500
3Drefine (2020a) http://sysbio.rnet.missouri.edu/3Drefine/. Accessed 1 Dec 2020
Acknowledgements
The authors thank Dr. Jillian Higgins from the University of York, UK for the critical reading and evaluation of the manuscript.
Funding
This research was not supported by any funding agency.
Author information
Authors and Affiliations
Contributions
SAD conceived and designed the analysis, collected data, performed the analyses, contributed to the interpretation of the results, and wrote the manuscript. SNI supervises the project. SAD and SNI contributed to the mastering of data and analysis. All authors discussed the results, commented on the manuscript, and contributed to the final version for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Code Availability
Not applicable.
Additional information
Handling editor: Bojian Zhong.
Supplementary Information
Below is the link to the electronic supplementary material.
239_2021_10035_MOESM2_ESM.xlsx
Supplementary Table 2. List of the CKX sequences used in this study. Taxonomy was simplified. Supplementary Table 3. Sequences used for the reconstruction of the phylogenetic tree and structures modelling. Taxonomy was simplified. Supplementary Table 4. The amino acid similarity of AtCKX7 protein to the CKX domain from other species (XLSX 29 KB)
239_2021_10035_MOESM3_ESM.pdf
Supplementary Figure 1. Phylogeny estimation of the identified cytokinin binding domains. The Neighbor-Joining method and JTT model were used; 1000 bootstrap replicates. Only branches with bootstrap value>50 are shown (PDF 41 KB)
239_2021_10035_MOESM4_ESM.pdf
Supplementary Figure 2. Phylogeny estimation of the selected FAD-linked oxidases domains used for the comparison of structures. The Neighbor-Joining method and JTT model were used; 1000 bootstrap replicates. Only branches with bootstrap value>50 are shown. Arabidopsis proteins are highlighted in blue, green algae – dark green, cyanobacteria – light green (PDF 50 KB)
Rights and permissions
About this article

Cite this article
Dabravolski, S.A., Isayenkov, S.V. Evolution of the Cytokinin Dehydrogenase (CKX) Domain. J Mol Evol 89, 665–677 (2021). https://doi.org/10.1007/s00239-021-10035-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-021-10035-z