
Abstract
SARS-CoV-2 is a unique event, having emerged suddenly as a highly infectious viral pathogen for human populations. Previous phylogenetic analyses show its closest known evolutionary relative to be a virus detected in bats (RaTG13), with a common assumption that SARS-CoV-2 evolved from a zoonotic ancestor via recent genetic changes (likely in the Spike protein receptor-binding domain or RBD) that enabled it to infect humans. We used detailed phylogenetic analysis, ancestral sequence reconstruction, and in situ molecular dynamics simulations to examine the Spike-RBD’s functional evolution, finding that the common ancestral virus with RaTG13, dating to no later than 2013, possessed high binding affinity to the human ACE2 receptor. This suggests that SARS-CoV-2 likely possessed a latent capacity to bind to human cellular targets (though this may not have been sufficient for successful infection) and emphasizes the importance of expanding efforts to catalog and monitor viruses circulating in both human and non-human populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Viral pathogens are a continuous and evolving challenge for human populations (Metcalf et al. 2015; Parvez and Parveen 2017). It is commonly thought that viruses maintain species-specific infectivity, often co-evolving with their host to mirror animal species trees (Huelsenbeck et al. 1997; Kaján et al. 2020). While less common, the emergence of novel viral pathogens is of particular interest because they often exhibit abnormal degrees of infectivity and/or virulence (Parrish et al. 2008), having not evolved to a natural selection balance with their new host (Brook et al. 2020).
It is known that viruses can, in some instances, spread to human populations after evolving to “cross the species barrier”(Marí Saéz et al. 2015). Yet, relatively little is known of the molecular changes that enable this evolution. One key molecular function often highlighted is “recognition” (typically mediated by protein–protein binding between viral entry protein and target host cells). Yet, while molecular recognition is clearly critical to “crossing the species barrier,” it is not clear whether it is commonly the key change enabling infectivity, or whether changes in other viral replication mechanisms are required as well. Answering these questions is critical to understand the origins of new human viral pathogens, and to assessing the capacity of other animal viruses to evolve human infectivity.
SARS-CoV-2 emerged as a human pathogen in late 2019 with high infectivity, rapidly causing a global pandemic (Andersen et al. 2020). A member of the Coronaviridae family, it is thought that SARS-CoV-2 evolved from a zoonotic origin (Gorbalenya et al. 2006; Lu et al. 2020), owing to its close known evolutionary relationship with RaTG13, a coronavirus isolated from a species of bat in 2013 (Joffrin et al. 2020; Li et al. 2020b; Schoeman and Fielding 2019), and the pangolin coronavirus, Pangolin-CoV (Zhou et al. 2020). While most of the SARS-CoV-2 genome is closely related to the RaTG13 genome, some genomic regions, including the Spike glycoprotein Receptor-Binding Domain (RBD) (which mediates “recognition” of host cells), show greater sequence similarity to the Pangolin-CoV homolog (Li et al. 2020a).
The Spike protein is a key component of the SARS-CoV-2 infection pathway (Lau et al. 2020). Knockout and overexpression studies have demonstrated that binding of the Spike-RBD to human angiotensin converting enzyme 2 (hACE2) mediates cellular entry of SARS-CoV-2 (Belouzard et al. 2012; Hoffmann et al. 2020; Lam et al. 2020; Paraskevis et al. 2020; Zhang et al. 2020). The SARS-CoV-2 Spike protein binds the hACE2 receptor with greater affinity than the SARS-CoV-1 homolog, suggesting as a possible explanation for its greater infectivity (Belouzard et al. 2012). Changes in the Spike protein have also been highlighted as a possible molecular explanation for its gain-of-function in being capable of “recognizing” and infecting human cells, implying that the Spike-RBD only recently acquired affinity for the hACE2 (Donoghue et al. 2000; Lan et al. 2020; Walls et al. 2020). This hypothesis is supported by the lower measured binding affinity between RaTG13 Spike-RBD and hACE2 (Ou et al. 2020). Given this, a critical question remains: How and when did the SARS-CoV-2 Spike protein evolve its relatively higher affinity for the hACE2? And can we directly infer whether its recent ancestor was unable to bind hACE2? With this question in mind, we robustly characterized the evolutionary changes that accompanied the emergence of SARS-CoV-2, distinguishing it from its closest zoonotic relatives, and focusing on the Spike-RBD.
Results
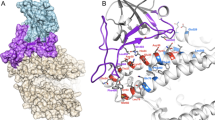
We performed phylogenetic analysis on whole viral genomic data to provide context for the evolution of the SARS-CoV-2 virus. Results supported prior studies’ conclusions, finding similar levels of nucleotide identity to the RaTG13 genome (96.0% sequence identity) and the Guangxi Pangolin-CoV genome (90.0% sequence identity) (Supplementary Fig. 1) (Belouzard et al. 2012; Zhou et al. 2020). Next, we sought to investigate the evolution of SARS-CoV-2 infectivity by performing ancestral sequence reconstruction for the Spike-RBD (Fig. 1A). Protein sequence comparisons have previously been used to investigate critical amino acid changes in the SARS-CoV-2 Spike protein (Letko et al. 2020), however, by leveraging the phylogenetic relationships between SARS-CoV-2 and its ancestors, we were able to focus on a unique subset of genetic changes specific to SARS-CoV-2’s recent evolution (Becker et al. 2008). We inferred statistically well-supported reconstructions of the Spike-RBD sequence for both the common ancestor of the SARS-CoV-2 virus (labeled “N1,” Fig. 1A, C) and the its common ancestor with the closest animal virus (labeled “N0,” Fig. 1A, C). N1 is identical to the Spike-RBD of the SARS-CoV-2 reference sequence, as expected, while the N0 Spike-RBD sequence is, to our knowledge, unique, reflecting the uniqueness of SARS-CoV-2’s viral origin (Guo et al. 2020). N0 differs from N1 at 4 positions (346, 372, 498, and 519—Fig. 1B).

Detailed examination of SARS-CoV-2 evolution. A Phylogeny illustrating the last common ancestor all SARS-CoV-2 Spike-RBDs (N1) and of SARS-CoV-2 and the RaTG13 Spike-RBD (N0). B Structural representation of the four mutations in the Spike-RBD (ribbon diagram) relative to the ACE2 receptor (Space filling model) that differs between N0 to N1. Stick models show the mutations in their N1 state. C Alignment of the Spike-RBD of SARS-CoV-2 and its ancestors for both protein (top) and DNA (bottom). Black boxes highlight the four mutations that differ from N0 to N1
The reconstruction of N1 for each of those positions is statistically well-supported, with a posterior probability (P.P.) of 1 obtained from two independent calculations (Supplemental Table 1; Methods). The reconstruction for N0 has high statistical support for positions 346, 372, and 519 (P.P. > 0.94), while position 498 was ambiguously reconstructed, with two alternate states comparable probability (Supplemental Table 1). All other positions were reconstructed with high confidence (P.P. > 0.85). Together, these four changes (t346R, t372A, h/y498Q, and n519H) differentiate the evolved SARS-Cov-2 Spike protein from the most recent common ancestor with animal viruses (Fig. 1). As such, this ancestral virus must have existed at least as early as 2013 (as one of its descendants—RaTG13—was isolated in that year), and quite likely much earlier, meaning that the branch between the N0 and N1 ancestors covers at least 7 years (and perhaps more than 50) of molecular evolution (Fig. 1A) (Ou et al. 2021).
To quantify functional differences between the N0 ancestor and the Spike-RBD sequences, we conducted 10 ns molecular dynamics (MD) simulations (Methods) of the Spike-RBD in complex with hACE2 (starting point for each simulation was modeled off crystal structures of the SARS-CoV-2 Spike-RBD/hACE2 complex) (Lam et al. 2020). We used simulation output to calculate electrostatic, polar solvation, van der Waals interactions, and solvent-accessible surface area (SASA) free energy contributions to infer the free energy of binding each Spike-RBD/hACE2 complex (Harms and Thornton 2013; Wong et al. 2020). We quantified the root-mean-squared deviation (RMSD) of the portion of the RBD closest to the hACE2 receptor (residues 397 to 512) for each of our replicates to confirm complex stability (Supplementary Fig. 2). Contrary to our expectations, the free energy of binding between the Spike-RBD and the hACE2 appears to have decreased between N0 and N1, suggesting that it did not evolve new “recognition” capacity where hACE2 is concerned. In fact, each of the 4 changes (including both alternate reconstructions at position 498 in N0) either reduced or did not significantly change the free energy of binding (Fig. 2A).

Characterization of SARS-CoV-2 Spike-RBD functional effects of evolution. A Table of MM/PBSA binding energies between receptor-binding domains of SARS-CoV-2 evolutionary constructs and hACE2 receptor (note that lower energy indicates tighter binding). Blue cells indicate the presence of the ancestral (N0) state and green cells (with an “x”) indicate the presence of the SARS-CoV-2 state (N1) at a given position. Two values are present for constructs with an ancestral (N0) state at position 498 (which reflect the ambiguity of its ancestral reconstruction), corresponding to h498 and y498 from left to right. Energies are shown as the mean of three replicate simulations with SEM indicated in parenthesis. B Relative effect of changes in SARS-CoV-2 receptor-binding domain from ancestral (N0) to SARS-CoV-2 (N1) state on MM/PBSA binding energies. Size of spheres indicates the relative magnitude, with red spheres indicating decreased binding affinity and blue indicating increased binding affinity. Values are averaged for h498 and y498 states (both raw values shown in parentheses). C Comparison of molecular dynamics and in vitro z-score normalized changes in binding energy for each mutation from N0 to N1. Changes are shown relative to the z-score normalized current (N1) binding energy. Positive binding energy changes indicate decreased binding affinity and negative binding energy changes indicate increased binding affinity with the given amino acid change (Color figure online)
Somewhat surprised by this reduction in affinity corresponding to recent SARS-CoV-2 evolution, we sought to confirm that our observations were not a result of an oversimplified MD-based assessment. We compared changes in binding energy from our MD data to recently released in vitro deep mutational scanning data (Starr et al. 2020). Although they did not directly measure the same ancestral N0 genotype as we did, they did directly measure the affinity for X other genotypes that cover the total sequence space of this transition. We found that they found significantly similar changes in binding energy (p = 0.042, Equivalence Test for Means) including in both alternative states for position 498 (Fig. 2C; Supplemental Table 2). In fact, the only discrepancy between the two datasets was at position 372, where the observed mutation has been shown to affect N-linked glycosylation at position 370 (Kang et al. 2021) (a phenomenon which was not directly modeled in our simulations). As a result, we can conclude our overall conclusions are not solely due to the computational methods we employed (Fig. 2C; Supplemental Table 2). However, further investigations of position 372 including N-linked glycosylation of position 370 should be carried out, as this position may have played an important role in SARS-CoV-2 infection of humans.
Discussion
Our findings on ancestral binding energies demonstrate that, contrary to expectations, recent evolutionary changes in SARS-CoV-2 did not improve the Spike-RBD’s binding with hACE2. While there are other animal coronaviruses known to bind to the hACE2 receptor with high affinity, to our knowledge, this is the first direct demonstration that an ancestor of the SARS-CoV-2 and RaTG13 lineage may have been capable of binding to hACE2. These results are in support of recent analysis which show that the ancestral lineage giving rise to SARS-CoV-2 may have been circulating undetected in bat species for over a decade (Boni et al. 2020) and that several SARS-related coronaviruses contained spike proteins already adapted to hACE2 binding (Menachery et al. 2015). This work is also in line with results demonstrating that the receptor-binding domain of SARS-CoV-2 is likely a latent ancestral trait, and not the result of substantial recent recombination or mutation (Boni et al. 2020).
This has important implications for understanding the origin of SARS-CoV-2. First, it suggests that the binding affinity between the Spike-RBD and hACE2 may not be a critical driver in the high affinity between SARS-CoV-2 and their hosts (humans). Instead, it suggests that tight hACE2 binding is a latent property of the virus, and that high affinity may instead have emerged via a other molecular changes in the SARS-CoV-2 genome (Baker et al. 2001). One such change may have occurred in the furin cleavage site, as furin activation of the Spike-RBD has recently been shown to be integral to SARS-CoV-2 infection of cells (Johnson et al. 2020; Shang et al. 2020). Second, it calls into question the presumption of a recent zoonotic potential for this disease; while other molecular components of the current SARS-CoV-2 virus may have acquired recent evolutionary changes that promoted its infectivity in humans, it appears that the high affinity for hACE2 was not among them.
If this is the case—that this viral lineage possessed the ability to bind hACE2 with high affinity for at least the past 7 years (Fig. 1B)—then why did it not emerge as a public health issue until recently? One possibility is that “recognition” via binding to hACE2 by the Spike-RBD is not sufficient, on its own, to cause human infectivity, and that other molecular components first needed to acquire new functions to do so (such as those related to viral replication inside human host cells). A second possibility is that this virus may have been capable of infecting human cells for a longer period than previously realized, but that its ancestral form either presented with far fewer symptoms (making it less disruptive and/or noticeable to those infected), was far less infectious (thereby impacting only a small number of people directly exposed to its zoonotic “source”), or was isolated to rural communities (such as those individuals showing seropositivity for SARS-related coronaviruses whom live in close proximity to bat caves in rural China Li et al. 2019; Wang et al. 2018) with population sizes insufficient for epidemic spread, in any case escaping the notice of public health monitoring (Fig. 3). To test this, a broad and concerted effort to sequence the range of coronaviruses across human populations would need to be conducted, to test whether a closely related virus may also be circulating (Ge et al. 2013; Turoňová et al. 2020; Yamamoto and Bauer 2020).

Schematic of two possible evolutionary scenarios stemming from the observed evolutionary SARS-CoV-2 Spike-RBD function. In Scenario 1, it is postulated that a zoonotic ancestral SARS-CoV-2 strain possessed the ability to effectively bind hACE2 but was unable to effectively enter human cells, requiring the presence of subsequent molecular changes to infect humans. In Scenario 2, an ancestral SARS-CoV-2 strain was actively infecting humans prior to the outbreak at low levels or in rural communities, thus escaping public health detection until subsequent molecular changes or contacts with more individuals lead to increased infectivity and/or severity. In Scenario 3, a high severity, ancestral SARS-CoV-2 strain capable of infecting humans was circulating in zoonotic reservoirs, requiring increased exposure or amplification to human hosts to result in broad transmission
Naturally, as an in-silico study, these results should be interpreted with some caution. Insofar as they can be validated, however, with exception to position 372, the results are consistent with direct in vitro functional measurements (Starr et al. 2020). Ideally, combinatorial libraries could be constructed and functionally screened (Nsubuga et al. 2006; Wang et al. 2018) to glean more detailed insights into the molecular mechanisms underlying the recent evolution of this virus. Combinatorial libraries should focus on position 372 where discrepancies were observed between our data and observations given by Starr et al. (2020) and Kang et al. (2021).
Predicting the emergence of highly infectious and virulent diseases, while difficult, is vital for human population health (Li et al. 2019). To do so, we must take steps to understand how pandemic diseases—such as SARS-CoV-2—emerged as they did, and to understand when they acquired the novel molecular functions that enabled their infectivity. In this case, it appears that the SARS-CoV-2 Spike-RBD did not recently evolve binding affinity to a human-specific protein to enable its “recognition” thereof. Instead, that function appears to have been latent, making it clear that the evolution of this disease—along with so many other aspects of its etiology—is more complex than expected.
Methods
Confirmation of SARS-CoV-2 Etiology
A phylogenetic analysis of 26 viral genomes was performed to confirm known SARS-CoV-2 ancestors. 24 known enzootic and endemic viruses and the SARS-CoV-2 reference genome and the Pangolin-CoV genome were downloaded from the National Center for Biotechnology Information (NCBI 2018) and Lam et al. (2020) respectfully. Selected sequences were aligned using the Multiple Alignment using Fast Fourier Transform Version 7 (MAFFT) FFT-NS-2 algorithm (Katoh et al. 2002; Katoh and Standley 2013). MAFFT default parameters were used in our alignment, with gap penalties of 1.53. PhyML 3.0 was employed to construct a phylogeny of aligned genomes (Guindon et al. 2010; Guindon and Gascuel 2003). Bayes values ≥ 0.90 were considered statistically significant. The output tree was visualized using the online tool, Interactive Tree of Life (iTOL) (Letunic and Bork 2019), and statistically significant clades were examined to validate current knowledge surrounding SARS-CoV-2 evolution.
Construction of Spike Glycoprotein Ancestral Sequence
nBLASTx (Altschul et al. 1990), run using a BLOSUM 62 matrix, a gap opening penalty of 11 and a gap extension penalty of 1, was employed to extract the Spike glycoprotein from the 479 SARS-CoV-2 sequences obtained from GISAID (Elbe and Buckland-Merrett 2017; Shu and McCauley 2017) selecting for one sequence per day per country from December 30, 2019 to March 25, 2020 (see Supplemental Data for accession IDs), and the Pangolin-CoV genome (Lam et al. 2020). Additional, Spike sequences, including the RaTG13 Spike protein, were obtained directly from NCBI (2018). Protein sequences were initially aligned using the Multiple Sequence Alignment by Log-Expectation (MUSCLE) program (Edgar 2021). The optimal parameters for phylogenetic reconstruction analysis were taken from the best-fit evolutionary model selected using the Akaike Information Criterion (AIC) implemented in the PROTTEST3 software (Darriba et al. 2011), and were inferred to be the Jones–Taylor–Thornton (JTT) model (Le and Gascuel 2008) with gamma-distributed among-site rate variation and empirical state frequencies. Phylogeny was inferred from these alignments using the RaXML v8.2.9 software (Stamatakis 2014) and results were visualized using FigTree v1.4.4 (https://github.com/rambaut/figtree/releases). Ancestral sequence reconstruction was performed with the FastML software (Ashkenazy et al. 2012) and further validated independently using the Graphical Representation of Ancestral Sequence Predictions (GRASP) software (Foley et al. 2020). Statistical confidence in each position’s reconstructed state for each ancestor was determined from posterior probability; any reconstructed positions with less than 95% posterior probability was considered ambiguous, and alternate states were also tested.
Mutagenesis of Ancestral Proteins
To understand the evolutionary importance of sequence changes observed between ancestral, zoonotic, and SARS-CoV-2 spike protein sequences, in-silico mutagenesis and binding energy studies were performed. A previously constructed x-ray crystallography structure for the complex between the receptor-binding domain (RBD) of the SARS-CoV-2 spike protein and the human hACE2 receptor were obtained from RCSB (accession number 6M0J). Utilizing PyMOL (https://pymol.org/2/) mutagenesis wizard, the four missense mutations (R346t, A372t, Q498h or Q498y, H519n) identified between the N0 and N1 sequences were introduced into the SARS-CoV-2 RBD sequence, replicating the sequence of the putative ancestral zoonotic (N0) sequence. In addition to the N1 and N0 structures, additional structures were developed in a similar fashion, selectively including each of the 4 mutations to represent all the possible combinations that these mutations may have existed throughout evolutionary time.
Simulation of ACE2 Interactions Using Molecular Docking
Molecular interactions were characterized with molecular dynamics simulations using GROMACS (Van Der Spoel et al. 2005), TIP3P waters, and CHARM07 force-field parameters for proteins. For each condition, three replicate 10 ns simulations were run, starting from crystal structures or structural models. Historical mutations were introduced and energy-minimized before MD simulation. Each system was solvated in a cubic box with a 10 Å margin, then neutralized and brought to 150 mM ionic strength with sodium and chloride ions. This was followed by energy minimization to remove clashes, assignment of initial velocities from a Maxwell distribution, and 1 ns of solvent equilibration in which the positions of heavy protein and DNA atoms were restrained. Production runs were 50 ns, with the initial 10 ns excluded as burn-in. The trajectory time step was 2 fs, and final analyses were performed on frames taken every 12.5 ps. We used TIP3P waters and the CHARM07 FF03 parameters for proteins, as implemented in GROMACS 4.5.5; these parameters were selected as they are appropriate for modeling inter-protein interactions. (Duan et al. 2003; González 2011) Analyses were performed using VMD 1.9.1. (Humphrey et al. 1996) GROMACS output was uploaded into Visual Molecular Dynamics (VMD) for Root-Mean Squared Deviation (RMSD) Analysis using the RMSD trajectory tool. After discovering large deviations in RMSD values for the full RBD, which we attributed to noise at the ends of the RBD, we isolated our analysis to residues 397 to 512 of the RBD.
Measurement of Binding Energies
Next, we measured the binding energies between residues 397 to 512 and the ACE2 receptor using g_mmpbsa (Kumari et al. 2014), a program which employs Molecular mechanics Poisson–Boltzmann surface area (MMPBSA) calculations to determine binding energy. Polar solvation energy, apolar solvation energy, van der Waals forces, and SASA energy were calculated every 0.25 ns using a grid space of 0.5 and a solute dielectric constant of 2. The output of the three replicates was amalgamated and binding energy was calculated using the bootstrap analysis (n = 2000 bootstraps) published by Kumari et al. (2014) We then characterize the genetic effect of each mutation (on average) and assessed whether there were any statistically significant epistatic interactions using established methods (Anderson et al. 2015; Yang et al. 2019).
Comparison to In Vitro Data
In vitro changes in binding energy for the four mutations were obtained from Starr et al. (2020). These data and our binding energies for the N1, 346_372_498, 346_372_519, 346_498_519, and 372_498_519 were each standardized using Z-scores. Changes in binding energy to the N1 state for each standardized score were calculated by subtracting the N1 energy from the mutant energy. An equivalence test for means was performed on the standardized changes in binding energy.
Data Availability
All data are publicly available through NCBI. See attached supplementary data for accession IDs.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med 26:450–452
Anderson DW, McKeown AN, Thornton JW (2015) Intermolecular epistasis shaped the function and evolution of an ancient transcription factor and its DNA binding sites. Elife. https://doi.org/10.7554/eLife.07864
Ashkenazy H, Penn O, Doron-Faigenboim A, Cohen O, Cannarozzi G, Zomer O, Pupko T (2012) FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res. https://doi.org/10.1093/nar/gks498
Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98:10037–10041
Becker MM, Graham RL, Donaldson EF, Rockx B, Sims AC, Sheahan T, Pickles RJ, Corti D, Johnston RE, Baric RS, Denison MR (2008) Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc Natl Acad Sci USA 105:19944–19949
Belouzard S, Millet JK, Licitra BN, Whittaker GR (2012) Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4:1011–1033
Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry BW, Castoe TA, Rambaut A, Robertson DL (2020) Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol 5:1408–1417
Brook CE, Boots M, Chandran K, Dobson AP, Drosten C, Graham AL, Grenfell BT, Müller MA, Ng M, Wang LF, van Leeuwen A (2020) Accelerated viral dynamics in bat cell lines, with implications for zoonotic emergence. Elife. https://doi.org/10.7554/eLife.48401
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S (2000) A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. https://doi.org/10.1161/01.res.87.5.e1
Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 24:1999–2012
Edgar RC (2021) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Elbe S, Buckland-Merrett G (2017) Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob Chall 1:33–46
Foley G, Mora A, Ross CM, Bottoms S, Sützl L, Lamprecht ML, Zaugg J, Essebier A, Balderson B, Newell R, Thomson RES, Kobe B, Barnard RT, Guddat L, Schenk G, Carsten J, Gumulya Y, Rost B, Haltrich D, Sieber V, Gillam EMJ, Bodén M (2020) Identifying and engineering ancient variants of enzymes using graphical representation of ancestral sequence predictions (GRASP). BioRxiv. https://doi.org/10.1101/2019.12.30.891457
Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C, Zhang YJ, Luo CM, Tan B, Wang N, Zhu Y, Crameri G, Zhang SY, Wang LF, Daszak P, Shi ZL (2013) Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503:535–538
González MA (2011) Force fields and molecular dynamics simulations. Collect SFN 12:169–200
Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ (2006) Nidovirales: evolving the largest RNA virus genome. Virus Res 117:17–37
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Guo H, Hu BJ, Yang XL, Zeng LP, Li B, Ouyang S, Shi ZL (2020) Evolutionary arms race between virus and host drives genetic diversity in bat severe acute respiratory syndrome-related coronavirus spike genes. J Virol. https://doi.org/10.1128/JVI.00902-20
Harms MJ, Thornton JW (2013) Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat Rev Genet 14:559–571
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Muller MA, Drosten C, Pohlmann S (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280
Huelsenbeck JP, Rannala B, Yang Z (1997) Statistical tests of host-parasite cospeciation. Evolution 51:410–419
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38
Joffrin L, Goodman SM, Wilkinson DA, Ramasindrazana B, Lagadec E, Gomard Y, Minter GL, Dos Santos A, Schoeman MC, Sookhareea R, Tortosa P, Julienne S, Gudo ES, Mavingui P, Lebarbenchon C (2020) Bat coronavirus phylogeography in the western Indian ocean. Sci Rep. https://doi.org/10.1101/742866
Johnson BA, Xie X, Kalveram B, Lokugamage KG, Muruato A, Zou J, Zhang X, Juelich T, Smith JK, Zhang L, Bopp N, Schindewolf C, Vu M, Vanderheiden A, Swetnam D, Plante JA, Aguilar P, Plante KS, Lee B, Weaver SC, Suthar MS, Routh AL, Ren P, Ku Z, An Z, Debbink K, Shi PY, Freiberg AN, Menachery VD (2020) Furin cleavage site is key to SARS-CoV-2 pathogenesis. BioRxiv. https://doi.org/10.1101/2020.08.26.268854
Kaján GL, Doszpoly A, Tarján ZL, Vidovszky MZ, Papp T (2020) Virus-host coevolution with a focus on animal and human DNA viruses. J Mol Evol 88:41–56
Kang L, He G, Sharp AK, Wang X, Brown AM, Michalak P, Weger-Lucarelli J (2021) A selective sweep in the spike gene has driven SARS-CoV-2 human adaptation. BioRxiv. https://doi.org/10.1101/2021.02.13.431090
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Kumari R, Kumar R, Lynn A (2014) g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54:1951–1962
Lam TT, Shum MH, Zhu HC, Tong YG, Ni XB, Liao YS, Wei W, Cheung WY, Li WJ, Li LF, Leung GM, Holmes EC, Hu YL, Guan Y (2020) Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature. https://doi.org/10.1038/s41586-020-2169-0
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. https://doi.org/10.1038/s41586-020-2180-5
Lau SKP, Luk HKH, Wong ACP, Li KSM, Zhu L, He Z, Fung J, Chan TTY, Fung KSC, Woo PCY (2020) Possible bat origin of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis. https://doi.org/10.3201/eid2607.200092
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320
Letko M, Marzi A, Munster V (2020) Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol 5:562–569
Letunic I, Bork P (2019) Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz239
Li H, Mendelsohn E, Zong C, Zhang W, Hagan E, Wang N, Li S, Yan H, Huang H, Zhu G, Ross N, Chmura A, Terry P, Fielder M, Miller M, Shi Z, Daszak P (2019) Human-animal interactions and bat coronavirus spillover potential among rural residents in Southern China. Biosaf Health 1:84–90
Li C, Yang Y, Ren L (2020a) Genetic evolution analysis of 2019 novel coronavirus and coronavirus from other species. Infect Genet Evol. https://doi.org/10.1016/j.meegid.2020.104285
Li X, Zai J, Zhao Q, Nie Q, Li Y, Foley BT, Chaillon A (2020b) Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J Med Virol 92:602–611
Lu H, Stratton CW, Tang YW (2020) Outbreak of pneumonia of unknown etiology in Wuhan, China: the mystery and the miracle. J Med Virol 92:401–402
Marí Saéz A, Weiss S, Nowak K, Lapeyre V, Zimmermann F, Düx A, Kühl HS, Kaba M, Regnaut S, Merkel K, Sachse A, Thiesen U, Villányi L, Boesch C, Dabrowski PW, Radonić A, Nitsche A, Leendertz SA, Petterson S, Becker S, Krähling V, Couacy-Hymann E, Akoua-Koffi C, Weber N, Schaade L, Fahr J, Borchert M, Gogarten JF, Calvignac-Spencer S, Leendertz FH (2015) Investigating the zoonotic origin of the West African Ebola epidemic. EMBO Mol Med 7:17–23
Menachery VD, Yount JBL, Debbink K, Agnihothram S, Gralinski LE, Plante JA, Graham RL, Scobey T, Ge X-Y, Donaldson EF, Randell SH, Lanzavecchia A, Marasco WA, Shi Z-L, Baric RS (2015) A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat Med 21:1508–1513
Metcalf CJ, Birger RB, Funk S, Kouyos RD, Lloyd-Smith JO, Jansen VA (2015) Five challenges in evolution and infectious diseases. Epidemics 10:40–44
NCBI (2018) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx1095
Nsubuga P, White ME, Thacker SB, Anderson MA, Blount SB, Broome CV, Chiller TM, Espitia V, Imtiaz R, Sosin D, Stroup DF, Tauxe RV, Vijayaraghavan M, Trostle M (2006) Public health surveillance: a tool for targeting and monitoring interventions. In: Jamison DT, Breman JG, Measham AR, Alleyne G, Claeson M, Evans DB, Jha P, Mills A, Musgrove P (eds) Disease control priorities in developing countries. The International Bank for Reconstruction and Development/The World Bank Oxford University Press, Copyright © 2006, The International Bank for Reconstruction and Development/The World Bank Group, New York
Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, Guo L, Guo R, Chen T, Hu J, Xiang Z, Mu Z, Chen X, Chen J, Hu K, Jin Q, Wang J, Qian Z (2020) Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 11:1620
Ou J, Zhou Z, Dai R, Zhang J, Zhao S, Wu X, Lan W, Ren Y, Cui L, Lan Q, Lu L, Seto D, Chodosh J, Wu J, Zhang G, Zhang Q (2021) V367F mutation in SARS-CoV-2 spike RBD emerging during the early transmission phase enhances viral infectivity through increased human ACE2 receptor binding affinity. J Virol. https://doi.org/10.1128/jvi.00617-21
Paraskevis D, Kostaki EG, Magiorkinis G, Panayiotakopoulos G, Sourvinos G, Tsiodras S (2020) Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect Genet Evol 79:104212
Parrish CR, Holmes EC, Morens DM, Park EC, Burke DS, Calisher CH, Laughlin CA, Saif LJ, Daszak P (2008) Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol Mol Biol Rev 72:457–470
Parvez MK, Parveen S (2017) Evolution and emergence of pathogenic viruses: past, present, and future. Intervirology 60:1–7
Schoeman D, Fielding BC (2019) Coronavirus envelope protein: current knowledge. Virol J 16:69
Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, Li F (2020) Cell entry mechanisms of SARS-CoV-2. Natl Acad Sci. https://doi.org/10.1073/pnas.2003138117
Shu Y, McCauley J (2017) GISAID: global initiative on sharing all influenza data—from vision to reality. Eurosurveillance 22:13
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, Navarro MJ, Bowen JE, Tortorici MA, Walls AC, King NP, Veesler D, Bloom JD (2020) Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. https://doi.org/10.1016/j.cell.2020.08.012
Turoňová B, Sikora M, Schürmann C, Hagen WJH, Welsch S, Blanc FEC, von Bülow S, Gecht M, Bagola K, Hörner C, van Zandbergen G, Landry J, de Azevedo NTD, Mosalaganti S, Schwarz A, Covino R, Mühlebach MD, Hummer G, Krijnse Locker J, Beck M (2020) In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 370:20–208
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26:1701–1718
Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. https://doi.org/10.1016/j.cell.2020.02.058
Wang N, Li SY, Yang XL, Huang HM, Zhang YJ, Guo H, Luo CM, Miller M, Zhu G, Chmura AA, Hagan E, Zhou JH, Zhang YZ, Wang LF, Daszak P, Shi ZL (2018) Serological evidence of bat SARS-related coronavirus infection in humans. China Virol Sin 33:104–107
Wong MC, Javornik Cregeen SJ, Ajami NJ, Petrosino JF (2020) Evidence of recombination in coronaviruses implicating pangolin origins of nCoV-2019. BioRxiv. https://doi.org/10.1101/2020.02.07.939207
Yamamoto N, Bauer G (2020) Apparent difference in fatalities between Central Europe and East Asia due to SARS-COV-2 and COVID-19: Four hypotheses for possible explanation. Med Hypotheses. https://doi.org/10.1016/j.mehy.2020.110160
Yang G, Anderson DW, Baier F, Dohmen E, Hong N, Carr PD, Kamerlin SCL, Jackson CJ, Bornberg-Bauer E, Tokuriki N (2019) Higher-order epistasis shapes the fitness landscape of a xenobiotic-degrading enzyme. Nat Chem Biol 15:1120–1128
Zhang T, Wu Q, Zhang Z (2020) Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol 30:1346
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, Chen H-D, Chen J, Luo Y, Guo H, Jiang R-D, Liu M-Q, Chen Y, Shen X-R, Wang X, Zheng X-S, Zhao K, Chen Q-J, Deng F, Liu L-L, Yan B, Zhan F-X, Wang Y-Y, Xiao G-F, Shi Z-L (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. https://doi.org/10.1016/j.cub.2020.03.02
Funding
Support provided by DWA startup funds from Alberta Children’s Hospital Research Institute and the Research Computing Services at University of Calgary.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Handling Editor: Belinda Chang.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Brintnell, E., Gupta, M. & Anderson, D.W. Phylogenetic and Ancestral Sequence Reconstruction of SARS-CoV-2 Reveals Latent Capacity to Bind Human ACE2 Receptor. J Mol Evol 89, 656–664 (2021). https://doi.org/10.1007/s00239-021-10034-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-021-10034-0